
Abstract
Background
Chronic and oral administration of benzylamine improves glucose tolerance. Picolylamine is a selective functional antagonist of the human adenosine A2B receptor. Phosphonic diamide derivatives enhance the cellular permeability and in turn their biological activities.
Methods
A series of heteroaryl phosphonicdiamide derivatives were designed as therapeutics to control and manage type2 diabetes. Initially defined Lipinski parameters encouraged them as safer drugs. Molecular docking of these compounds against Protein tyrosine phosphatase (PTP), the potential therapeutic target of type 2 diabetes, revealed their potential binding ability explaining their anti-diabetic activity in terms of PTP inhibition. Human intestinal absorption, Caco-2 cell permeability, MDCK cell permeability, BBB penetration, skin permeability and plasma protein binding abilities of the title compounds were calculated by PreADMET server. A convenient method has been developed for the synthesis of title compounds through the formation of 1-ethoxy-N,N’-bis(4-fluorobenzyl/pyridin-3-ylmethyl)phosphinediamine by the reaction of 4-fluorobenzylamine/ 3-picolylamine with ethyldichlorophosphite, subsequently reacted with heteroaryl halides using lanthanum(III) chloride as a catalyst.
Results
All the compounds exhibited significant in vitro anti-oxidant activity and in vivo evaluation in streptozotocin induced diabetic rat models revealed that the normal glycemic levels were observed on 12th day by 9a and 20th day by 5b, 5c, 9e and 9f. The remaining compounds also exhibited normal glycemic levels by 25th day.
Conclusion
The results from molecular modeling, in vitro and in vivo studies are suggesting them as safer and effective therapeutic agents against type2 diabetes.

Development of PTPs inhibitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
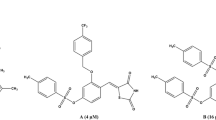
The stipulation of anti-diabetic drugs is snowballing hastily, due to millions of people is distressing about diabetes. Several budding essential mechanisms for diabetes are characterized by elevation of blood glucose levels caused by decreased production of the hormone insulin and/or increased resistance to the action of insulin by certain cells. Tyrosine phosphorylation is associated with a group of enzymes which are mainly involved in the negative regulation of insulin signaling and intertwined in the insulin resistance, complementary to type 2 diabetes [1],[2]. Protein tyrosine phosphatase-1B (PTP-1B) is one of the PTP enzymes a major negative regulator in both insulin and leptin signaling. It has been observed to serve as an outstanding target for the treatment of cancer, diabetes and obesity [3]. Mice lacking the PTP-1B have enhanced insulin sensitivity which certifies that the inhibition activity of PTP-1B could be a novel way of treating type 2 diabetes and obesity [1],[2]. Thus insulin action will be enhanced by persuading the activity of cellular PTPases and glucose production can be reduced [4],[5]. This study created an interest in designing the new drugs by structural modification of existing drugs (Figures 1 and 2).

A few anti-diabetic drugs.

The study of the reported drugs i-vii reveals that they are ideal for anti-diabetic activity due to the thiazolidine-2,4-dione (i, ii, iii), pyridinyl (i,ii), quinolone (iv), urea and amide (v, vii), Flouro substituted, heteroaryl pyrazine (vi) and benzyl amine (vii). Compound xiii is a α-aminophosphonate with established anti-diabetic property which gave an idea to focus on phosphorus containing drugs.
Benzylamine is used to treat diabetes in traditional medicine. Chronic and oral administration of benzylamine improves glucose tolerance and the circulating lipid profile without increasing oxidative stress in overweight and pre-diabetic mice [12]. The stipulation of picolylamine was attested in the synthesis of various pharmacological compounds such as 99mTc(I)-complexs [13] and selective functional antagonists of the human adenosine A2B receptor [14]. When compared to normal benzyl amine analogues, picolylamine analogues are exhibiting the potential pharmacological activity [15]. Among the 2-picolyl, 3-picolyl and 4-picolyl amines, the performance of 3-picolyl amines are virtuous [16].
Phosphonic diamide derivatives enhance the cellular permeability and in turn their activities akin to the analogous phosphoric diamide prodrugs of 3′-azido-3′-deoxythymidine (AZT) monophosphate with AZT [17], glycine methyl ester phosphonic diamide of a 9-[2-(phosphonomethoxy)ethyl]-adenine (PMEA) analogue [18], and diamides of 9-[2-(phosphonomethoxy)ethyl]-N6-(cyclopropyl)-2-aminoadenine [19]. If phosphonic diamides hydrolyze in vivo to produce phosphonic acids benzyl amine itself act as antidiabetic agent [12]. Phosphonic diamide derivatives are used as prodrugs to improve the membrane permeability of drugs. P-C bond is playing an important role in preserving so many syndromes and in the synthesis of numerous anticancer [20], antiviral [21], antimicrobial [22], anti-diabetic [23], and antioxidant agents [24]. If the carbon in the P-C bond is aromatic, it acts better than the aliphatic carbon. Quinolines are expressed as LXR mediate disease inhibitors [25]. Quinoline phosphonicdiesters are known for preventing hypercholesterolemia and diabetes [26]. There are number of patents which are dependent on this type of drugs.
Lipinski parameters help in preclinical trials to avoid the tedious and costly procedures that can define them as drugs and to avoid the failure rates. Lipinski parameters suggest the potency of the compounds with a variety of molecular descriptors [27]. The in silico studies involving construction, optimization and molecular dynamics will generate the stable conformations of the molecules. It is also an important task to find out the structure based intermolecular interactions of the compounds with the biologically meaningful and effective targets at specified conditions [28]. This helps to predict the inhibitory activity and the strength of the molecule to form a stable complex with the target. The identification of binding orientations of the compounds in the binding site of target will provide fruitful information on their reactivity. Hence, in the present study we applied Lipinski parameters and molecular docking studies to predict the drug likeliness and binding ability of the compounds to the protein tyrosine phosphatase.
Although several synthetic methods are described for the preparation of such P-C bond containing compounds, one of them is the Michaelis–Arbuzov reaction. Unfortunately, it has some drawbacks when use classical conditions such as length of reaction time, high temperature and removal of the trialkyl phosphite used in a large excess. These drastic conditions may be responsible for side reactions, low yields and limits the application of such reactions to sensitive substrates. Recently, researchers focused on rare earth elemental catalysts due to their high catalytic properties and also act as Lewis acids. In this connection, we selected Lanthanum (III) chloride as an efficient catalyst for nucleophilic substitution on hetero aromatic ring for the synthesis of heteroaryl phosphonicdiamide derivatives via Michaelis-Arbuzov reaction.
The improved production and ineffective scavenging of reactive oxygen species (ROS) cause chemical changes in virtually all cellular components, leading to lipid peroxidation. The enhanced production of free radicals and oxidative stress is central event to the development of diabetic complications. This was supported by demonstration of increased levels of indicators of oxidative stress in diabetic individuals suffering from complications [29]. Oxidative stress is involved in the pathogenesis of diabetes and its complications. Use of antioxidants reduces oxidative stress and alleviates diabetic complications [30]. There are many reports on effects of antioxidants in the management of diabetes [31],[32]. So the in vitro antioxidant activity was carried out as preliminary test for all the title compounds. The results of antioxidant activity supported for the reduction of oxidative stress. Finally, title compounds were screened for their in vivo anti-diabetic activity on mice. Most of the title compounds are effective and satisfactory in reducing glucose levels in both the tests.
Materials and methods
Chemistry
Chemicals were procured from Sigma–Aldrich and Merck were used as such without further purification. All solvents used for spectroscopic and other physical studies were reagent grade and were further purified by literature methods [33]. Melting points (m p) were determined by Guna Digital Melting Point apparatus using a calibrated thermometer. They expressed in degrees centigrade (°C) and are uncorrected. Infrared spectra (IR) were obtained on a Perkin-Elmer Model 281-B spectrophotometer. Samples were analyzed as potassium bromide (KBr) disks. Absorptions were reported in wave numbers (cm−1). 1H and 13C NMR spectra were recorded as solutions in DMSO-d 6 on a Bruker AMX 400 MHz spectrometer operating at 400 MHz for 1H, 100 MHz for 13C and 161.9 MHz for 31P NMR. The 1H and 13C chemical shifts were expressed in parts per million (ppm) with reference to tetramethylsilane (TMS) and 31P chemical shifts to 85% H3PO4. LCMS mass spectra were recorded on a Jeol SX 102 DA/600 Mass spectrometer.
Synthesis of N,N’-di(4-fluorobenzyl)(2-pyrazinyl)phosphonic diamide (5a)
To a stirred solution of 4-fluorobenzylamine (0.002 mol) in dry tetrahydrofuran (THF) (10 mL), ethyldichlorophosphite (0.001 mol) was added at 0°C in the presence of triethylamine (TEA) (0.002 mol) under N2 atmosphere. After completion of the addition, the reaction mixture was heated to 30°C and stirred for 2 h to form the intermediate 1-ethoxy-N,N’-bis(4-fluorobenzyl)phosphinediamine (3). The reaction progress was monitored by thin layer chromatography (TLC) using ethyl acetate: hexane (1:1) as mobile phase. After completion of the reaction, it was filtered to remove triethylamine hydrochloride. 2-Chloropyrazine (4a) (0.001 mol) in dry THF (10 mL) was added to the filtrate under N2 atmosphere in the presence of La(III)Cl3.7H2O (20 mol%) and the reaction mixture was refluxed for 3 h. The progress of the reaction was monitored by TLC using ethyl acetate: hexane (1:1) as mobile phase. After completion of the reaction, catalyst was removed by filtration and the filtrate was concentrated in vacuum to afford the crude product. It was purified by silica gel column chromatography eluting with ethyl acetate: hexane (1:2) mixture to afford the title compound, N,N’-di(4-fluorobenzyl)(2-pyrazinyl)phosphonic diamide (5a). The same experimental procedure was adopted for the preparation of the remaining title compounds 5b-f (Scheme 1).

Synthesis of substituted heteroaryl-N,N’-di(4-fluorobenzyl) phosphonicdiamides (5a-f).
Spectral data
N,N’-Di(4-fluorobenzyl)(2-pyrazinyl)phosphonic diamide (5a)
Yield: 72%; mp: 162-164°C; IR (KBr): ῡ 3378 (N-H), 1252 (P = O), 1018 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.52-6.84 (11H, m, Ar), 5.12 (2H, brs, H-8), 4.08-3.83 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 161.3 (C-4), 154.3 (C-1), 152.2 (C-1′), 148.9 (C-6′), 145.6 (C-3′), 147.9 (C-4′), 122.4-121.5 (C-2 & C-6), 117.4-116.2 (C-3 & C-5), 31.2 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 28.9; LC MS (%): m/z 375.7 (100%) [MH+•]; Anal. Calcd. for C18H17N4F2OP: C 57.76; H 4.58; N 14.97; Found: C 57.63; H 4.39; N 14.77.
2,4-Dioxo-1,2,3,4-tetrahydro-5-pyrimidinyl-N,N’-di(4-fluorobenzyl)phosphonic diamide (5b)
Yield: 70%; mp: 189-191°C; IR (KBr): ῡ 3386 (N-H), 1238 (P = O), 992 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 9.06 (1H, brs, H-3′), 8.32-6.74 (9H, m, Ar), 5.73-5.68 (1H, s, H-5′), 5.15 (2H, brs, H-8), 3.83-4.08 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 169.6 (C-2′), 161.6 (C-4), 161.3 (C-4′), 158.2 (C-1′), 155.4 (C-1), 141.9 (C-6′), 122.6-121.8 (C-2 & C-6), 117.8-116.8 (C-3 &C-5), 30.9 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 27.6; LC MS (%): m/z 407.8 (100%) [MH+•]; Anal. Calcd. for C18H17N4F2O3P: C 53.21; H 4.22; N 13.79; Found: C 53.08; H 4.12; N 13.55.
N,N’-Di(4-fluorobenzyl)[5-(trifluoromethyl)-2-pyridyl]phosphonic diamide (5c)
Yield: 69%; mp: 202-204°C; IR (KBr): ῡ 3354 (N-H), 1261 (P = O), 1010 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 7.65-6.50 (11H, m, Ar), 5.12 (2H, brs, H-8), 4.06-3.83 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 159.1 (C-4), 155.9 (C-1′), 154.8 (C-3′), 154.0 (C-1), 133.6 (C-5′), 133.5 (C-4′), 122.2-121.1 (C-2 & C-6), 119.6 (C-6′), 118.9 (C-7′), 117.7-116.6 (C-3 & C-5), 30.9 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 28.7; LC MS (%): m/z 442.8 (100%) [MH+•]; Anal. Calcd. for C20H17F5N3OP: C 54.43; H 3.88; N 9.52; Found: 54.15; H 3.51; N 9.22.
N,N’-Di(4-fluorobenzyl)(2-pyrimidinyl)phosphonic diamide (5d)
Yield: 75%; mp: 167-169°C; IR (KBr): ῡ 3346 (N-H), 1268 (P = O), 996 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.55-6.79 (11H, m, Ar), 5.14 (2H, brs, H-8), 4.08-3.83 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 162.5 (C-1′), 161.9 (C-4), 155.1 (C-1), 154.3 (C-3′ & C-5′), 124.4 (C-4′), 121.8-121.1 (C-2 & C-6), 117.6-116.4 (C-3 & C-5), 31.4 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 28.2; LC MS (%): m/z 375.4 (100%) [MH+•]; Anal. Calcd. for C18H17F2N4OP: C 57.76; H 4.58; N 14.97; Found: 57.62; H 4.41; N 14.82.
N,N’-Di(4-fluorobenzyl)(4-isoquinolyl)phosphonic diamide (5e)
Yield: 75%; mp: 221-224°C; IR (KBr): 3364 (N-H), 1274 (P = O), 986 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.65-6.76 (14H, m, Ar), 5.16 (2H, brs, H-8), 4.06-3.84 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 161.6 (C-4), 156.2 (C-4′), 155.2 (C-1), 142.6 (C-2′), 135.9 (C-9′), 129.5 (C-7′), 129.2 (C-10′), 127.5 (C-5′), 127.1 (C-8′), 126.9 (C-6′), 126.2 (C-1′), 122.3-121.5 (C-2 & C-6), 117.7-116.6 (C-3 & C-5), 30.7 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 27.8; LC MS (%): m/z 424.5 (100%) [MH+•]; Anal. Calcd. for C23H20F2N3OP: C 65.25; H 4.76; N 9.92; Found: C 65.09; H 4.57; N 9.71.
N,N’-Di(4-fluorobenzyl)(3-quinolyl)phosphonic diamide (5f)
Yield: 72%; mp: 179-181°C; IR (KBr): 3372 (N-H), 1259 (P = O), 1012 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.63-6.92 (14H, m, Ar), 5.13 (2H, brs, H-8), 4.08-3.83 (4H, m, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 161.5 (C-4), 157.2 (C-9′), 154.5 (C-1), 148.4 (C-2′), 136.8 (C-3′), 131.7 (C-6′), 128.5 (C-4′), 127.3 (C-7′), 126.8 (C-10′), 126.7 (C-5′), 123.6 (C-8′), 122.1-121.3 (C-2 & C-6), 117.8-116.2 (C-3 & C-5), 30.5 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 28.9; LC MS (%): m/z 424.6 (100%) [MH+•]; Anal. Calcd. for C23H20F2N3OP: C 65.25; H 4.76; N 9.92; Found: C 65.08; H 4.51; N 9.84.
Synthesis of 4,6-dimethoxy-1,3,5-triazin-2-yl-N,N’-di(3-pyridylmethyl)phosphonicdiamide (9a)
To a stirred solution of 3-picolylamine (6) (0. 002 mol) in dry tetrahydrofuran (THF) (10 mL), ethyldichlorophosphite (2) (0.001 mol) was added at 0°C in the presence of triethylamine (TEA) (0.002 mol) under N2 atmosphere. After completion of the addition, the reaction mixture was raised to 30°C and stirred for 2 h to form the intermediate 1-ethoxy-N,N’-bis(pyridin-3-ylmethyl)phosphinediamine (7). The reaction progress was monitored by thin layer chromatography (TLC) using ethyl acetate: hexane (1:1) as mobile phase. After completion of the reaction, it was filtered to remove triethylamine hydrochloride. 2-Chloro-4,6-dimethoxy-1,3,5-triazine (8a) (0.001 mol) in dry THF (10 mL) was added to the filtrate under N2 atmosphere at 20°C in the presence of La(III)Cl3.7H2O (20 mol%) and the reaction mixture was refluxed for 3 h. The progress of the reaction was monitored by TLC using ethyl acetate: hexane (1:1). After completion of the reaction, catalyst was removed by filtration and the filtrate was concentrated in rota-evaporator to afford the crude product. It was purified by silica gel column chromatography eluting with ethyl acetate: hexane (1:2) mixture to afford the title compound, 4,6-dimethoxy-1,3,5-triazin-2-yl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9a). The same experimental procedure was adopted for the preparation of the remaining title compounds 9b-f (Scheme 2).

Synthesis of substituted heteroaryl-N,N’-di(3-pyridylmethyl) phosphonicdiamides (9a-f).
Spectral data
4,6-Dimethoxy-1,3,5-triazin-2-yl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9a)
Yield: 70%; mp: 198-200°C; IR (KBr): ῡ 3371 (N-H), 1242 (P = O), 989 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.92-7.26 (8H, m, Ar), 5.63 (2H, brs, H-8), 4.39-4.35 (4H, d, H-7), 3.72 (6H, s, -OMe); 13C NMR (100 MHz, DMSO-d 6 ): δ 178.3 (C-3′ & C-5′), 169.2 (C-1′), 149.5 (C-2), 148.1 (C-4), 145.8 (C-1), 135.3 (C-6), 121.5 (C-5), 51.9 (C-OMe), 38.6 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 21.6; LC MS (%): m/z 402.5 (100%) [MH+•]; Anal. Calcd. for C17H20N7O3P: C 50.87; H 5.02; N 24.43; Found: C 50.66; H 4.81; N 24.23.
N,N’-Di(3-pyridylmethyl)(3-quinolyl)phosphonic diamide (9b)
Yield: 74%; mp: 206-208°C; IR (KBr): 3376 (N-H), 1247 (P = O), 1005 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.61-7.46 (14H, m, Ar), 5.64 (2H, brs, H-8), 4.35-4.31 (4H, d, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 149.4 (C-2), 148.9 (C-4), 147.5 (C-2′), 145.2 (C-9′), 135.3 (C-6), 134.6 (C-1), 132.3 (C-3′), 131.7 (C-6′), 128.9 (C-4′), 127.5 (C-5′), 127.3 (C-10′), 126.1 (C-7′), 122.5 (C-8′), 121.7 (C-5), 38.8 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 20.2; LC MS (%): m/z 390.3 (100%) [MH+•]; Anal. Calcd. for C21H20N5OP: C 64.77; H 5.18; N 17.99; Found: C 64.51; H 5.03; N 17.81.
4-Isoquinolyl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9c)
Yield: 73%; mp: 166-169°C; IR (KBr): ῡ 3379 (N-H), 1253 (P = O), 995 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.63-7.45 (14H, m, Ar), 5.62 (2H, brs, H-8), 4.37-4.34 (4H, d, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 152.5 (C-4′), 149.1 (C-2), 148.5 (C-4), 145.2 (C-1), 143.9 (C-2′), 135.1 (C-6), 134.5 (C-9′), 130.2 (C-10′), 129.5 (C-7′), 128.2 (C-6′), 127.4 (C-5′), 127.1 (C-8′), 125.9 (C-1′), 121.3 (C-5), 38.4 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 19.1; LC MS (%): m/z 390.5 (100%) [MH+•]; Anal. Calcd. for C21H20N5OP: C 64.77; H 5.18; N 17.99; Found: C 64.68; H 5.12; N 17.85.
1,3-Benzothiazol-2-yl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9d)
Yield: 66%; mp: 175-177°C; IR (KBr): ῡ 3383 (N-H), 1245 (P = O), 1013 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.48-7.44 (12H, m, Ar), 5.62 (2H, brs, H-8), 4.35-4.31 (4H, d, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 162.2 (C-1′), 155.3 (C-8′), 149.2 (C-2), 148.9 (C-4), 145.5 (C-1), 135.8 (C-6), 134.3 (C-9′), 129.5 (C-4′), 127.1 (C-5′), 125.9 (C-6′), 125.7 (C-3′), 121.2 (C-5), 38.3 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 21.6; LC MS (%): m/z 396.5 (100%) [MH+•]; Anal. Calcd. for C19H18N5OPS: C 57.71; H 4.59; N 17.71; Found: C 57.62; H 4.41; N 17.58.
3-Cyano-2-pyridyl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9e)
Yield: 68%; mp: 172-174°C; IR (KBr): ῡ 3388 (N-H), 1258 (P = O), 1018 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.66-7.41 (11H, m, Ar), 5.63 (2H, brs, H-8), 4.32-4.30 (4H, d, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 157.3 (C-1′), 156.3 (C-3′), 149.3 (C-2), 148.8 (C-4), 145.5 (C-1), 138.4 (C-5′), 135.9 (C-4′), 135.8 (C-6), 121.4 (C-5), 118.3 (C-7′), 113.6 (C-6′), 38.2 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 22.7; LC MS (%): m/z 365.7 (100%) [MH+•]; Anal. Calcd. for C18H17N6OP: C 59.34; H 4.70; N 23.07; Found: C 59.19; H 4.48; N 22.91.
1-Isoquinolyl-N,N’-di(3-pyridylmethyl)phosphonic diamide (9f)
Yield: 73%; mp: 185-187°C; IR (KBr): ῡ 3387 (N-H), 1261 (P = O), 1010 (P-CAr) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 8.61-7.48 (14H, m, Ar), 5.64 (2H, brs, H-8), 4.34-4.31 (4H, d, H-7); 13C NMR (100 MHz, DMSO-d 6 ): δ 159.3 (C-1′), 149.1 (C-2), 148.5 (C-4), 145.9 (C-1), 144.8 (C-3′), 135.9 (C-10′), 135.3 (C-6), 129.7 (C-6′), 129.5 (C-9′), 129.1 (C-8′), 128.5 (C-7′), 127.3 (C-5′), 122.6 (C-4′), 121.7 (C-5), 38.4 (C-7); 31P NMR (161.9 MHz, DMSO-d 6 ): δ 18.9; LC MS (%): m/z 390.2 (100%) [MH+•]; Anal. Calcd. for C21H20N5OP: C 64.77; H 5.18; N 17.99; Found: C 64.55; H 5.02; N 17.75.
Molecular modeling
All the in silico studies were carried out in the Molecular Operating Environment (MOE) software tool [34].
Protein preparation and processing
The three dimensional X-Ray Crystallographic structure of Protein tyrosine phosphatase (PTP) was retrieved from Protein Data Bank (PDB ID: 2F71). The structure was loaded into the MOE working environment ignoring the water molecules and hetero atoms. Polar hydrogens were added to the protein and subjected protonation followed by energy minimization in the implicit solvated environment in MMFF94x force field at a gradient cut off value of 0.05. A stabilized conformation of the protein was obtained after energy minimization and it was used for docking study.
Molecular docking
The above obtained stable conformation of the protein was preceded with molecular docking process. The binding site was defined with Arg 24, Asp 181, Ser 216, Ala 217, Gly 220, Arg 221 and Arg 254 residues. These are all the residues that were found to be interacting with the previously reported sulfamic acid inhibitor and hence considered for the docking of library of the present novel compounds. All the ligands were docked into the specified binding site using alpha triangle placement methodology where the Poses are generated by superposition of ligand atom triplets and triplets of receptor site points. A random triplet of ligand atoms and a random triplet of alpha sphere centers are used to determine the binding pose at each interaction. The free energy of binding of each compound from each pose generated after docking process is determined by London dG scoring function. A total of 30 conformations were generated for each compound and they were refined and rescored again using the same scoring function. The pose with lowest binding score was selected for further analysis and to analyze the binding mode orientations of the ligands in the binding site.
ADMET study
Pharmacokinetic parameters like absorption, distribution, metabolism and excretion of compounds designates their disposition. Such parameters influence the pharmacokinetics of the drug in the body and in turn influence their performance and pharmacological activity [35]. In that sequence we have predicted some ADMET properties for the designed compounds to define them as drug candidates at their significant conditions. The parameters such as Caco-2 (colon adeno carcinoma) cell permeability, MDCK (Madin-Darby canine kidney) cell permeability, BBB (blood-brain barrier) penetration, HIA (human intestinal absorption), skin permeability and plasma protein binding ability were predicted by submitting the structures to PreADMET online software tool (http://preadmet.bmdrc.org/index.php?option=com_content&view=frontpage&Itemid=1) a web-based application server for predicting ADMET.
Pharmacology
Compounds 5a-f and 9a-f were screened for in vitro antioxidant activity by DPPH (2,2-diphenyl-1-picrylhydrazyl), NO and H2O2 methods where Ascorbic acid and BHT (Butylated hydroxytoluene) as standards. Subsequently all the title compounds were screened for their in vivo antihyperglycemic activity in twenty five days period and examined for every four days. The experimental procedures are described below.
Antioxidant activity
DPPH radical scavenging activity
The DPPH radical scavenging activity was measured from the bleaching of the purple colored methanol solution of 2,2-diphenyl-1-picrylhydrazyl (DPPH). Initially 1 mL of various concentrations of test compounds (50, 75, 100 and 150 μg/ mL) in methanol were added to 4 mL of 0.004% (w/v) methanol solution of DPPH. The resultant test solutions were incubated for 30 min period at room temperature and absorbance was read against blank at 517 nm. All the tests were carried out in triplicate. The % of inhibition (I%) of free radical production from DPPH was calculated by following equation.
Nitric oxide (NO) scavenging activity
NO scavenging activity action was measured by slightly modified method of Green et al. and Marcocci et al. [36]. The mixture of 1 mL of sodium nitro prusside (10 mM) and 1.5 mL of phosphate buffer saline (0.2 M, pH 7.4) were tested to different concentrations (50, 75, 100 and 150 μg/mL) of the test compounds and incubated for 150 min at 25°C and treated with 1 mL of Griess reagent and absorbance of the chromophore was measured at 546 nm. Butylated hydroxyl toluene was used as the standard in the present method. Tests were carried out in triplicate. Nitric oxide scavenging activity was calculated by the following equation.
Hydrogen peroxide (H2O2) scavenging activity
Radical scavenging activity of the title compounds was screened against H2O2 through the method of Ruch et al. [37]. A solution of H2O2 (40 mM) in phosphate buffer (PH 7.4) was prepared, 0.6 mL of prepared H2O2 solution was added to the test compounds at different concentrations (50, 75, 100 and 150 μg/mL) and the absorbance value for the reaction mixture was recorded at 230 nm for every test sample in average of triplicate. Tests were carried out in triplicate. The per cent of scavenging of H2O2 was calculated by the following equation.
Where Acontrol is the absorbance of the control reaction (containing all reagents except the test compound) and Asample is the absorbance of the test compound and Acetate buffer as Ablank.
In vivo antihyperglycemic activity
Induction of diabetes
Male wistar albino rats (body weight 180-200 grams) were subjected to intra-peritoneal administration of Streptozotocin dissolved in freshly prepared 0.01M ice-cold citrate buffer (PH 4.3) at a dose of 50 mg/Kg body weight. After 72 hours, the animals with fasting blood glucose levels ≥350 mg/dL were used to evaluate the anti-diabetic activity of title compounds. Blood glucose levels were measured with the help of Accuchec Glucometer (Glucose oxidase method). All the animals were maintained in ventilated cages provided with standard pellet diet and water in light/dark cycle of (12h/12h) [38]. All of animal experiments were carried out according to the guidelines of the Sri Venkateswara University’s Institutional Animal Care and Use Committee (No./02(i)/a/CPCSCA/IAEC/SVU/TV).
Experimental design
The animals were divided into fifteen groups and each group maintained six rats. Group 1 as normal rats Untreated, Group 2 as diabetic rats Untreated, Group 3 as Diabetic rats treated with standard Glibenclamide (25 mg/kg b.w.) and Group 4-15 as Diabetic rats treated with title compounds (25 mg/kg b.w.) from 5a-f and 9a-f respectively for each group. After an overnight fast, the drug dissolved in DMSO (25 mg/kg b.w.) was fed to 4-15 group rats by gastric intubation using force feeding needle. Normal untreated and diabetic untreated rats were fed with normal diet and distilled water alone. Group 3 diabetic rats were treated with Glibenclamide 25 mg/kg b.w. Blood samples were collected to measure blood glucose levels from the tail vein on 1st, 4th, 8th, 12th, 16th, 20th and 25th days after the administration of drug and blood glucose levels were determined by glucose oxidase–peroxidase method [39].
Results
Chemistry
The IR spectra of 5a-f showed the expected absorption bands at 998-1008, 3350–3330 and 1255–1233 cm−1 for the P-C(Ar), NH and P = O stretching vibrations respectively [40]. The signals in δ 5.12-5.16 of 5a-f and δ 5.63-5.64 of 9a-f are representing the NH protons attached to the phosphorus atom. All 13C signal of aromatic carbon attached to the phosphorus is observed in between the range of δ 128-139 and δ 152-169. 31P NMR signals appeared in the range of 27.3 to 28.6 ppm as expected for the P = O group of the title compounds.
Prediction of Lipinski parameters
The three dimensional structures were constructed for all the compounds and their stable conformations were obtained after optimization. These conformations were used to study their Lipinski parameters and the results showed that all of them are showing best properties with good agreement to Lipinski rule suggesting them as safer drugs. All the compounds have the molecular weight less than 500 Da, the lowest molecular weight of 364 Da was found with 9e and the highest molecular weight of 441 Da was found with 5c. The number of hydrogen bond donors is found to be less than 5 and the hydrogen bond acceptors is less than 10 for all the compounds. The logP values observed below 5 are themselves indicating that they are all non-toxic to the host system. The molar refractivity is also found to be in the optimal range of 40-150. The remaining descriptors like surface area, volume, hydration energy, polarizability and energy levels are also encouraging them with suitable features to bind and inhibit the target, there by better results can be expected and promotes them as safer and effective drugs (Table 1).
Molecular docking
A total of 30 binding pose conformations were generated for each compound from docking simulations using MOE dock system. The free energy of binding of each ligand was ranked and assessed by London dG scoring function. The binding energies and hydrogen bond interactions of each Receptor-Ligand complexes were studied and the information is tabulated in Table 2. The best lowest docking score -11.810 Kcal/mol was observed for the compound 9e and second the highest docking score of -9.813 Kcal/mol was observed for 9a. The remaining compounds are also showing better docking scores indicating the good affinity levels between the receptor and compounds. The binding mode orientations of 9e ligand-receptor complex are showing that the ligand is interacting with the binding site with the help of a single arene cat ionic interaction with Arg24 residue. Hydrogen bond interactions were not seen for 9e in the complex. It was observed from all docking complexes that Arg24 residue is playing a major role in interacting with almost all of the compounds. In addition with 9e, the arene cat ionic interaction was also observed with the compounds 5d, 9b, 9c, 9d and 9f. More over in all of these complexes the arene cat ionic interaction was contributed by Arg24 residue only. This indicates that the aromatic rings of the compounds are highly influencing them to interact with the Arginine residue. The hydrogen bond interactions were not observed for 9d and 9e where as such bonds were observed in the remaining docking complexes (Figure 3 and Additional file 1: Figure S1-S12). However, 9d and 9e are also showing satisfactory docking scores along with remaining compounds. So, finally it can be predicted from these studies that all these compounds have the ability to bind with PTP-1B and inhibits its activity.

Molecular docking complexes of 5a, 5f and 9b and 9e with PTP (PDB ID: 2F71)
ADMET results
Human intestinal absorption, Caco-2 cell permeability, MDCK cell permeability, BBB penetration, skin permeability and plasma protein binding abilities of the title compounds were calculated by PreADMET server and the results presented in Figure 4.

ADMET results of title compounds 5a-f and 9a-f.
The HIA results demonstrate the best absorption of the title compounds 5a-f and 9a-f into Human Intestine. Weak plasma protein binding results represent their virtuous properties such as diffusion or transport across cell membranes, interaction with a pharmacological target and excretion. This is due to, generally the drugs less bound to plasma protein exist freely for diffusion or transport across cell membranes and also for interaction with a pharmacological target. The title compounds 9a-j, altogether showed moderate cellular permeability against Caco-2 cells. The compound 5b exhibited medium MDCK cellular permeability. In turn all the above parameters represent their good excretion, disposition and efficacy values in the human body.
The Blood-Brain Barrier (BBB) penetration is represented as BB = [Brain]/[Blood], where [Brain] and [Blood] are the steady-state concentration of radio labeled compound in brain and peripheral blood. Predicting BBB penetration helps to know whether the compounds able to pass across the blood-brain barrier or not. This parameter expresses the BBB penetration capacity and absorption rate of compound to CNS. All the compounds were observed to be having moderate absorption to CNS.
The skin permeability is a crucial parameter that can define the transdermal delivery of the compound as the risk assessment during accidental contact with the skin. The skin permeability values are defined as logKp, cm/hr for all the compounds, where Kp = Km*D/h. Km is distribution coefficient between stratum corneum and vehicle, D is average diffusion coefficient (cm2/h) and h is thickness of skin (cm) [41].
Pharmacology
The title compounds were assessed for anti-oxidant and anti-hyperglycemic activity. The detailed discussion regarding the assessment method is demonstrated as follows.
Antioxidant activity
Free radical 2,2-diphenyl-1-picrylhydrazyl (DPPH) scavenging activity
DPPH is usually used as a reagent to evaluate free radical scavenging activity of antioxidants [42]. DPPH is a stable free radical and accepts an electron or hydrogen radical to become a stable diamagnetic molecule [43]. The reduction capability of DPPH radical is determined by the decrease in absorbance at 517 nm induced by antioxidants. In the present study, Ascorbic acid was used as a standard, the title compounds 5a-f and 9a-f were able to reduce the stable radical DPPH to the yellow-colored diphenylpicrylhydrazine. The compounds were evaluated at four different concentrations of 50 μg/mL, 75 μg/mL, 100 μg/mL and 150 μg/mL and the IC50 values were determined from these evaluations. The scavenging effect of title compounds as compared to standard with the DPPH radical is in the following order 5f >5e >9f > Ascorbic acid >5c >9c and the remaining compounds showed less effect than these compounds. The compounds showed almost all same order at all concentrations and the complete results are given in Additional file 1. The IC50 value of each compound was considered as the concentration (μg/mL) of the compound at which 50% of DPPH reduction was observed. These results are presented in Figure 5 and in Additional file 1: Table S5.

IC 50 values in μg/mL of the title compounds on DPPH scavenging activity, Nitric oxide radical scavenging activity and superoxide radical (O 2 − ) scavenging activity.
Assay of Nitric oxide radicals scavenging activity
In the current investigation, newly synthesized compounds exhibited an excellent NO radicals scavenging activity. The compounds were evaluated at four different concentrations of 50 μg/mL, 75 μg/mL, 100 μg/mL and 150 μg/mL and the IC50 values were determined from these evaluations. Amongst the title compounds 5e > BHT >5f >5a >5c >9f have exerted significant inhibitory activity and the remaining compounds exhibited less effect than these compounds on radicals that are generated in vitro and the complete results are given in Additional file 1. The IC50 value of each compound was considered as the concentration (μg/mL) of the compound at which 50% of NO reduction was observed. These results are presented in Figure 5 and in Additional file 1: Table S6.
Assay of superoxide radical (O2−) scavenging activity
Superoxide radical is known to be a very harmful species to cellular components as a precursor of more reactive oxygen species [44]. The superoxide radical is known to be produced in vivo and can result in the formation of H2O2via dismutation reaction. Moreover, the conversion of superoxide and H2O2 into more reactive species, for instance, the hydroxyl radical, has been thought to be one of the unfavorable effects caused by superoxide radicals [45]. The newly synthesized compounds are efficient scavengers for the superoxide radical generated in riboflavin–NBT–light system in vitro and their activity is in comparable to that of Ascorbic acid. The compounds were evaluated for their scavenging effects at four different concentrations of 50 μg/mL, 75 μg/mL, 100 μg/mL and 150 μg/mL and the IC50 values were determined from these evaluations. The scavenging effects of the compounds are in the following order 5e >5f >9f >9c > Ascorbic acid and the remaining compounds exhibited less scavenging effect than these compounds on radicals that are generated in vitro. The compounds exhibited almost all same order at all concentrations and the complete results are presented in Additional file 1. This result clearly indicates that the tested compounds have a noticeable effect on scavenging superoxide radical. The IC50 value of each compound was considered as the concentration (μg/mL) of the compound at which 50% of NO reduction was observed. These results are presented in Figure 5 and in Additional file 1: Table S7.
In over view of observation, the compounds 5e, 5f, 9c, 9f are showing the better antioxidant activity, it may be due to the presence of quinolone group in the structures of the title compounds. These results are supported by the previous reports of Shridhar et al. of the antioxidant activity of eight substituted quinolines [46].
The proposed mechanism for the DPPH radical scavenging activity with the title compounds
All the title compounds are containing atleast two N-H functional groups in their structure. These N-H groups are playng the main role in the abridging of free radicals, there by oxidative stress decreases. This mechanisim was supported by previous reports. This reduction of oxidative stress is the basis for mechanism for the antihyperglycemic actitivity (Figure 6).

The proposed mechanism for the DPPH radical scavenging activity with the title compounds [[47]].
Anti-hyperglycemic activity
All the title compounds showed significant anti-diabetic activity in the diabetes-induced rats when compared with the standard Glibenclamide. All the rats were kept in the observation for 25 days and seasoned the glycemic levels (mg/dL) for every four days after administration of drug. The diabetic rats showing glucose levels ≥350 mg/dL were taken for the experiment on the first day. On the fourth day, the glucose levels were almost all decreased i.e 221 ± 3.25 (9a) <242 ± 3.42 (9f) <244 ± 2.35 (5f) <245 ± 2.56 (Standard) <245 ± 4.86 (9b) <250 ± 3.24 (5c). On the eighth day the compounds 9a (152 ± 3.46) and 9f (195 ± 3.48) showed the least glycemic levels than the other compounds. On the 12th day, the compound 9a induced rats showed the normal glycemic levels (120 ± 3.15) and compound 9b (146 ± 1.95), 9f (153 ± 2.32), 5c (165 ± 2.76), Glibenclamide (166 ± 2.46) and 9e (168 ± 2.68) induced rats gave moderate glycemic levels. On the 16th day only the compound 9a (103 ± 1.47) gave the good result. The remaining compounds gave the moderate glycemic levels except 5a, 5d, 5e and 9c. On the 20th day all the compounds showed normal glycemic levels but 9d gave moderate and 5a, 5d, 5e and 9c gave the high glycemic levels. Finally on the last day, all the title compounds gave excellent results with glycemic levels in between 82 ± 1.58 (9a) to 109 ± 1.23 (9d) apart from 5a, 5d, 5e and 9c. The detailed observation of results of all title compounds are reproduced in Table 3 (Figure 7).

Anti-diabetic activity of compounds 5a-f and 9a-f in STZ induced diabetic rats.
The molecules which were bound at Arg24 of Protein tyrosine phosphatase (PTP) gave the potential anti-hyperglycemic properties against Diabetic rats. From these results, compound 9a can be stated as an effective anti-hyperglycemic compound among all as it exerted its effect in the earlier days among all. This may be due to the presence of two methoxy groups on the triazine moiety which are binding at Arg24 of PTP that can make it more reactive and effective. On the 20th day 5b, 5c, 9a, 9e and 9f compounds gave normal glycemic levels, it may be due to the presence of structural moieties like uracil, trifluoromethyl, dimethoxytriazine, nicotinonitrile and quinoline moieties respectively. On the othe other hand the same ligand groups are binding with PTP at Arg24. But a few compounds exhibited moderate results though they contain the quinoline and isoquinoline structures. On an overall, all the compounds have shown good anti-diabetic activities by 25th day except 5a, 5d, 5e and 9c. Over again, these results from molecular docking studies and in vivo assays of the title compounds supporting for previous reports that development of PTPs inhibitors is very ease for diabetes prevention [48].
Conclusion
Lanthanum (III) chloride is stated as an efficient catalyst for the Michaelis-Arbuzov reaction and for the synthesis of the title compounds 5a-f and 9a-f by two-step reaction. The molecular descriptors of all the compounds from Lipinski parameters explained their drug likeliness and suggesting them as safer drugs. The descriptors also indicate that they are all not harmful to the host system because of their optimal logP values. The molecular docking study revealed their strong ability to interact with the target and inhibits its activity, there by predicting their anti-diabetic activity. PreADMET results demonstrated that the title compounds exhibit good absorption, permeability, penetration abilities in the human body. This prediction is confirmed by in vivo screening of these compounds in the diabetic induced rat models where the test compounds exhibited significant antihyperglycemic activity comparative to the standard Glibenclamide. Almost all the compounds brought the glycemic levels to normal on the 25th day. Especially, 9a was shown normal glycemic levels on 12th day. On the 20th day 5c, 5b, 9a, 9e and 9f were shown normal glycemic levels. All the compounds exhibited good diabetic levels on 25th day except 5a, 5d, 5e and 9c.
Supporting information
Supporting Tables of the antioxidant activity and supporting Figures are given in the supporting information as Additional file 1.
Additional file
References
Kennedy BP, Ramachandran C: Protein tyrosine phosphatase-1B in diabetes. Biochem Pharmacol. 2000, 60: 877-883. 10.1016/S0006-2952(00)00305-1.
Johnson TO, Ermolieff J, Jirousek MR: Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Disc. 2002, 1: 696-709. 10.1038/nrd895.
Wang Q, Gao J, Liu Y, Liu C: Molecular dynamics simulation of the interaction between protein tyrosine phosphatase 1B and aryl diketoacid derivatives. J Mol Graph Model. 2012, 38: 186-193. 10.1016/j.jmgm.2012.06.011.
Evans JL, Jallal B: Protein tyrosine phosphatases: their role in insulin action and potential as drug targets. Exp Opin Invest Drugs. 1999, 8: 139-160. 10.1517/13543784.8.2.139.
Cheng A, Dube N, Gu F, Tremblay ML: Coordinated action of protein tyrosine phosphatases in insulin signal transduction. Eur J Biochem. 2002, 269: 1050-1059. 10.1046/j.0014-2956.2002.02756.x.
Liu G, Xin Z, Liang H, Zapatero CA, Hajduk PJ, Janowick DA, Szczepankiewicz BG, Pei Z, Hutchins CW, Ballaron SJ, Stashko MA, Lubben TH, Berg CE, Rondinone CM, Trevillyan JM, Jirousek MR: Selective protein tyrosine phosphatase-1B inhibitors: targeting the second phosphotyrosine binding site with non-carboxylic acid-containing ligands. J Med Chem. 2003, 46: 3437-3440. 10.1021/jm034088d.
Wan ZK, Lee J, Xu W, Erbe DV, McCarthy DJ, Follows BC, Zhang Y: Monocyclic thiophenes as protein tyrosine phosphatase-1B inhibitors: capturing interactions with Asp48. Bioorg Med Chem Lett. 2006, 16: 4941-4945. 10.1016/j.bmcl.2006.06.051.
Liu G, Xin Z, Pei Z, Hajduk PJ, Zapatero CA, Hutchins CW, Hongyu ZH, Lubben TH, Ballaron SJ, Haasch DL, Kaszubska W, Rondinone CM, Trevillyan JM, Jirousek MR: Selective protein tyrosine phosphatase-1B inhibitors: targeting the second phosphotyrosine binding site with non-carboxylic acid-containing ligands. J Med Chem. 2003, 46: 4232-4235. 10.1021/jm034122o.
Shim YS, Kim KC, Chi DY, Lee KH, Cho H: Formylchromone derivatives as a novel class of protein tyrosine phosphatase-1B inhibitors. Bioorg Med Chem Lett. 2003, 13: 2561-2563. 10.1016/S0960-894X(03)00479-7.
Adams DR, Abraham A, Asano J, Breslin C, Dick CAJ, Lxkes U, Johnston BF, Johnston D, Kewnay J, Mackay SP, Mackenzie SJ, Mcfarlane M, Mitchell L, Takano Y: 2-Aryl-3,3,3-trifluoro-2-hydroxypropionic acids: a new class of protein tyrosine phosphatase-1B inhibitors. Bioorg Med Chem Lett. 2007, 17: 6579-6583. 10.1016/j.bmcl.2007.09.069.
Qingming W, Miaoli Z, Ruiting Z, Liping L, Caixia Y, Shu X, Xueqi F, Yuhua M, Qingwei H: Exploration of α-aminophosphonate N-derivatives as novel, potent and selective inhibitors of protein tyrosine phosphatases. Eur J Med Chem. 2012, 49: 354-364. 10.1016/j.ejmech.2012.01.038.
Zsuzsa I, Estelle W, Almudena L, Maria PP, Federica P, Eva S, Sandy B, John W, Fermin IM, Alfredo Martinez J, Philippe V, Christian C: Chronic benzylamine administration in the drinking water improves glucose tolerance, reduces body weight gain and circulating cholesterol in high-fat diet-fed mice. Pharmacol Res Off J Italian Pharmacol Soc. 2010, 61: 355-363. 10.1016/j.phrs.2009.12.014.
Stefan M, Robert W, Bernhard S, Susanne K, Roger A: Picolylamine-methylphosphonic acid esters as tridentate ligands for the labeling of alcohols with the fac-[M(CO)3]+ core (M = 99mTc, Re): synthesis and biodistribution of model compounds and of a 99mTc-labeled cobinamide. Nuclear Med Biol. 2005, 32: 473-484. 10.1016/j.nucmedbio.2005.04.007.
Simon TB, Karen RB, Teresa B, Ijen C, Mike C, Sarah D, Tim H, Allan MJ, Guy AK, Anthony RK, Burkhard K, Loic L, Angela M, Anil M, Sean L, Anthony P, Karine P, Mark R, Heather S, Melanie W, Ian AY: Discovery and optimization of potent and selective functional antagonists of the human adenosine A2B receptor. Bioorg Med Chem Lett. 2009, 19: 5945-5949. 10.1016/j.bmcl.2009.08.040.
Phillip JK, Thomas MB, Patrick RG, John TB, Alexander SK, Carrie KJ, Ashley EB, Jana KS, Jeffrey CP, Craig WL: Synthesis and structure–activity relationships of allosteric potentiators of the M4 muscarinic acetylcholine receptor. Chem Med Chem. 2009, 4: 1600-1607. 10.1002/cmdc.200900231.
Y Patrick YSL, Yu R, Prabhakar KJ, Paul EA, George VD, Charles JE, Chong HC, George E, Edward RH, Wayne FD, Liangzhu L, Pat NC, Robert JM, Qi H, Renhua L, Jay AM, Steven PS, Thomas RS, Lee TB, Marlene MR, Ronald MK, Linyee S, Dean LW, David MK, David AJ, Erickson-Viitanen S, Nicholas Hodge C: Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2¢ structure-activity relationship and molecular recognition of cyclic ureas. J Med Chem. 1996, 39: 3514-3525. 10.1021/jm9602571.
Jones BCNM, McGuigan C, O’Connor TJ, Jeffries DJ, Kinchington D: Synthesis and anti-HIV activity of some novel phosphoradiamidate derivatives of 3′-azido-3′-deoxythymidine (AZT). AntiVir Chem Chemother. 1991, 2: 35-39.
Serafinowska HT, Ashton RJ, Bailey S, Harnden MR, Jackson SM, Sutton D: Synthesis and in vivo evaluation of prodrugs of 9-[2-(phosphonomethoxy)ethoxy] adenine. J Med Chem. 1995, 38: 1372-1379. 10.1021/jm00008a015.
Keith KA, Hitchcock MJ, Lee WA, Holy A, Kern ER: Evaluation of nucleoside phosphonates and their analogs and prodrugs for inhibition of orthopoxvirus replication. Antimicrob Agents Chemother. 2003, 47: 2193-2198. 10.1128/AAC.47.7.2193-2198.2003.
Petr J, Ondrej B, Martin D, Ivan V, Zdenek Z, Gina B, George S, Tomas C, Richard M, Antonin H, Zlatko J: A novel and efficient one-pot synthesis of symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities. Eur J Med Chem. 2008, 46: 3748-3754.
Qun D, Rao KS, Tao J, Kevin F, Yan L, Frank T, William S, Daniel KC, Reddy KR, Paul DP, James MF, Scott CP, Mark DE: Discovery of phosphonicdiamideprodrugs and their use for the oral delivery of a series of fructose 1,6-bisphosphatase inhibitors. J Med Chem. 2008, 51: 4331-4339. 10.1021/jm8001235.
Claude G, Nicolas B, Chantal F, Raphael ED: Synthesis and antibacterial activity of novel enolphosphate derivatives. Bioorg Chem. 2010, 38: 218-223. 10.1016/j.bioorg.2010.06.006.
Asish KB, Florian S, Schmidt R: Design and synthesis of aryl/hetarylmethylphosphonate-UMP derivatives as potential glucosyltransferase inhibitors. Tetrahedron Lett. 2001, 42: 5393-5395. 10.1016/S0040-4039(01)00974-1.
Salkeeva LK, Nurmaganbetova MT, Minaeva EV, Kokzhalova BZ: Reactions of tert-Butyl N, N-Diethyl-N -(4-phenylthiazol-2-yl)-phosphorodiamidite with electrophilic reagents. Russ J Gen Chem. 2006, 76: 1397-1400. 10.1134/S1070363206090076.
Michael DC, Robert RS, Baihua H, James WJ, Robrt LM, David HK, Miller CP, Ullrich JW, Unwalla RJ, Wrobel JE, Quinet E, Nambai P, Bernotas RC, Elloso M: Anticholesterol Agents; Antidiabetic Agents; Cardiovascular Disorders; Antiarthritic Agents. 2009
Miyata K, Shoji Y, Tsuda Y, Tsutsumi K, Kamisaka E, Inoue Y: Inoue, JP-A-5043589. In ᅟ. ; 1993. http://www.wikipatents.com/JP-Patent-5043589/quinoline-derivative.
Nallusamy S, Samuel S: QSAR studies on HIV-1 protease inhibitors using Non-linearly transformed descriptors. Curr Comput Aided Drug Des. 2012, 8: 10-49. 10.2174/157340912799218534.
Valasani KR, Hu G, Chaney MO, Yan SS: Structure-based design and synthesis of benzothiazole phosphonate analogues with inhibitors of human ABAD-Aβ for treatment of Alzheimer’s disease. Chem Biol Drug Des. 2013, 81 (2): 238-249. 10.1111/cbdd.12068.
Ehsaneh T, Mahmoud D, Ahmad S, Ali MM, Djazayeri A, Qorbani M: The relationship between the activates of antioxidant enzymes in red blood cells and body mass index in Iranian type 2 diabetes and healthy subjects. J Diab Metabol Disord. 2012, 11: 1-5. 10.1186/2251-6581-11-1.
Roja R, Shekoufeh N, Bagher L, Mohammad A: A review on the role of antioxidants in the management of diabetes and its complications. Biomed Pharmacother. 2005, 59: 365-373. 10.1016/j.biopha.2005.07.002.
Suresh K, Sunil S, Vasudeva N, Varun R:In vivo anti-hyperglycemic and antioxidant potentials of ethanolic extract from Tecomella undulate. Diabetol Metabol Syndrome. 2012, 4: 1-7. 10.1186/1758-5996-4-1.
V Jan V, Marek J, Travnicek Z, Racanska E, Muselik J, Svajlenova O: Synthesis, structural characterization, antiradical and antidiabetic activities of copper(II) and Zinc(II) Schiff’s base complexes derived from salicylaldehyde and β-alanine. J Inorg Biochem. 2008, 102: 595-605. 10.1016/j.jinorgbio.2007.10.003.
Armarego WLF, Perrin DD: Purification of Laboratory Chemicals. 1997, Butterworth, Heinemann, Oxford, 4
Molecular Operating Environment (MOE): 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2011.10; Chemical Computing Group Inc; 2011.
Beresford AP, Selick HE, Tarbit MH: The emerging importance of predictive ADME simulation in drug discovery. Drug Discov Today. 2002, 7 (2): 109-116. 10.1016/S1359-6446(01)02100-6.
Marcocci L, Maguire JJ, Droy Lefaix MT, Packer L: The nitric oxide-scavenging properties of ginkgo biloba extract EGb 761. Biochem Biophys Res Commun. 1994, 201: 748-755. 10.1006/bbrc.1994.1764.
Ruch RJ, Cheng SJ: Klaunig: prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogenesis. 1989, 10: 1003-1008. 10.1093/carcin/10.6.1003.
Magid AFA, Carson KG, Harris BD, Maryanoff CA, Shah RD: Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. studies on direct and indirect reductive amination procedures. J Org Chem. 1996, 61: 3849-3862. 10.1021/jo960057x.
Kesari AN, Gupta RK, Singh SK, Diwakar S, Watal G: Hypoglycemic and anti-hyperglycemic activity of Aeglemarmelos seed extract in normal and diabetic rats. J Ethnopharmacol. 2006, 107: 374-379. 10.1016/j.jep.2006.03.042.
Thomas LCP: Interpretation of the Infrared Spectra of Organophosphorus Compounds. 1974, Hyden and Son Ltd, London
Rao VK, Michael OC, Victor WD, Yan SS: Acetylcholinesterase inhibitors: structure based design, synthesis, pharmacophore modeling, and virtual screening. J Chem Inf Model. 2013, 53 (8): 2033-2046. 10.1021/ci400196z.
Gulcin I, Bursal E, Hilal M, Ehitoglu S, Bilsel M, Goren AC: Polyphenol contents and antioxidant activity of lyophilized aqueous extract of propolis from Erzurum, Turkey. Food Chem Toxic. 2010, 48: 2227-2230. 10.1016/j.fct.2010.05.053.
Soares JR, Dins TCP, Cunha AP, Ameida LM: Antioxidant activity of some extracts of Thymus zygis. Free Rad Res. 1986, 26: 469-478. 10.3109/10715769709084484.
B Halliwell B, Gutteridge JMC: In Free Radicals, Ageing, and Disease, Free Radicals in Biology and Medicine. 2007, Clarendron Press, Oxford, 4
Halliwell B: Reactive oxygen species in living systems source, biochemistry, and role in human disease. Am J Med. 1991, 91: 14-22. 10.1016/0002-9343(91)90279-7.
Shridhar AH, Keshavayya J, Peethambar SK, Hoskeri JH: Synthesis and Biological activity of Bis alkyl 1,3,4-oxadiazole incorporated azo dye derivatives. Arab J Chem 2012. http://dx.doi.org/10.1016/j.arabjc.2012.04.018.
Osman H, Arshad A, Lam CK, Bagley MC: Microwave assisted synthesis and antioxidant properties of hydrazinylthiazolylcoumarin derivatives. Chem Centr J. 2012, 6: 32-41. 10.1186/1752-153X-6-32.
Safavi M, Foroumadi A, Abdollahi M: The importance of synthetic drugs for type 2 diabetes drug discovery. Expert Opin Drug Discov. 2013, 8 (11): 1339-1363. 10.1517/17460441.2013.837883.
Acknowledgements
The authors express their grateful thanks to BRNS (DAE) for sanctioning research project (2012/37C/21/BRNS/785), Mumbai, India for providing financial assistance through the project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CSK carried out synthesis and structural elucidation of title compounds, involved in screening of molecular modeling studies of designed compounds and drafted the manuscript. NKY had screen molecular modeling and ADMET studies of title compounds. JKMV was carried out Pharmacological studies of title compounds. ARC and NRC were provided the lab facilities and guidance in all aspects of this work. RS and MG were cooperated by their partial involvement in this work. All authors read and approved the final manuscript.
Electronic supplementary material
40199_2014_76_MOESM1_ESM.docx
Additional file 1:Construction of 3D-models and ligand database of the compounds. Lipinski Descriptors. Table S5. DPPH radical scavenging activity of the compounds 5a-f and 9a-f. Table S6. Nitric oxide (NO)scavenging activity of the compounds 5a-f and 9a-f. Table S7. Hydrogen peroxide (H2O2) scavenging activity of the compounds 5a-f and 9a-f. Figure S1: molecuar docking complex of 5a. Figure S2: molecuar docking complex of 5b. Figure S3: molecuar docking complex of 5c. Figure S4: molecuar docking complex of 5d. Figure S5: molecuar docking complex of 5e. Figure S6: molecuar docking complex of 5f. Figure S7: molecuar docking complex of 9a. Figure S8: molecuar docking complex of 9b. Figure S9: molecuar docking complex of 9c. Figure S10: molecuar docking complex of 9d. Figure S11: molecuar docking complex of 9e. Figure S12: molecuar docking complex of 9f. (DOCX 122 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Sekhar, K.C., Syed, R., Golla, M. et al. Novel heteroaryl phosphonicdiamides PTPs inhibitors as anti-hyperglycemic agents. DARU J Pharm Sci 22, 76 (2014). https://doi.org/10.1186/s40199-014-0076-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40199-014-0076-3