
Abstract
The skin is the exterior interface of the human body with the environment. Despite its harsh physical landscape, the skin is colonized by diverse commensal microbes. In this review, we discuss recent insights into skin microbial populations, including their composition and role in health and disease and their modulation by intrinsic and extrinsic factors, with a focus on the pathobiological basis of skin aging. We also describe the most recent tools for investigating the skin microbiota composition and microbe-skin relationships and perspectives regarding the challenges of skin microbiome manipulation.
Video abstract
Similar content being viewed by others

Introduction
The skin is the largest organ and the outermost interface between the human body and its environment. For decades, the skin surface was estimated to have an area as high as 2 m2, but recently, by taking into account the appendages, the estimate has increased to as high as 25 m2 [1]. Many regional differences overlap in the skin topography. For instance, temperature and humidity are higher at vaulted sites, such as the groin or armpit (approaching 37°C, the body’s core temperature) and lower at the body’s extremities (fingers and toes, approximately 30°C). Sebaceous gland density is an important variable factor involved in the secretion of many lipidic compounds, including fatty acids, that contribute to the acidification of the skin pH, which varies between 4.2 and 7.9 depending on the site of measurement [2].
These characteristics induce many possibilities for creating different ecological niches housing numerous commensal bacteria as well as fungi, viruses, archaea, and mites [3] in a network that varies in terms of its density and composition. Altogether, these microorganisms define the skin microbiota. According to Grice et al. [4], skin microbiota diversity may be higher than gut microbiota diversity. The vulnerability of this microbe network lies in the many intrinsic and extrinsic factors that affect it. The implications for wound healing and protection against potential pathogens or environmental conditions highlight a crucial role of skin homeostasis. Indeed, recent studies have identified associations between shifts in these commensal populations and physiological changes, such as aging and diverse dermatological diseases, not only in humans but in all vertebrates [5, 6].
With the recent advent of molecular biology and next-generation sequencing (NGS) as tools for microbiological identification, knowledge about the skin microbiota has grown exponentially. However, culture methods remain an essential tool for studying the characteristics of microorganisms in vivo. The manipulation of the skin microbiota represents a considerable challenge in clinical and cosmetic practice. This review discusses recent findings regarding the skin microbiome and its role in human health, aging, and disease.
Skin microbiota composition
Bacteria
Byrd et al. [3] reported the top ten bacterial species found on the skin through site area surveys in healthy volunteers based on high-throughput gene sequencing analysis. Human skin samples were found to be dominated by gram-positive bacteria belonging to the genera Staphylococcus spp., Corynebacterium spp., Enhydrobacter spp., Micrococcus spp., Cutibacterium spp., and Veillonella spp. A culture-based study by Myles et al. [7] that focused on the culturable fraction of gram-negative bacteria (GNB) from the human skin identified Roseomonas mucosa, Pseudomonas spp., Acinetobacter spp., Pantoea septica, and Moraxella osloensis as commensal residents. Other studies have verified that gram-negative bacteria (GNB), including Enterobacteriaceae, nonfermenting GNB, and anaerobes, are underestimated skin commensal organisms but are also part of the transient fraction of the skin microbiota [8, 9].
Candidate phyla
To date, few studies have reported the existence of TM7x (Saccharibacteria spp.) [10, 11] and TM6 sequences from skin samples [12]. These “candidate phyla” are considered to be not-yet-culturable bacteria and constitute a field of growing interest because such organisms are expected to be linked to pathologies such as periodontitis [13].
Archaea
As part of the Human Microbiome Project, Moissl-Eichinger et al. [14] focused their research on the detection of overlooked Archaea on human skin by analyzing 13 samples collected from healthy torso skin. Thaumarchaeota and Euryarchaeota were shown to be carried by all the human subjects analyzed [14, 15]. Phylogenetic analysis of Thaumarchaeota placed them close to ammonia-oxidizing archaea from the soil. Moreover, although it remains to be proven, the role of these archaea could be explained by chemolithotroph ammonia turnover, which could influence the pH regulation of the human skin and therefore the natural protective barrier of the body [15, 16].
Viruses
Within the viral fraction found on the skin, bacteriophages are predominant. The lytic activity of bacteriophages has been linked to the modulation of bacterial populations, and thus, bacteriophages participate in the homeostasis of the skin microbiota. Through culture-based approaches and genomic analysis of skin samples, Liu et al. [17] revealed an increased frequency of C. acnes phages isolated from healthy individuals compared to patients with Acne vulgaris and suggested that phages may play a role in modulating skin bacterial populations. Metagenomic shotgun sequencing analysis suggests that Cutibacterium and Staphylococcus phages are the most abundant skin phages, while other phages, such as Streptococcus and Corynebacterium phages, are also present but at lower relative abundances [18]. Byrd et al. [3] reported the top ten viruses found on the skin. Phages were identified as well as Acheta domestica; Densovirus; Alphapapillomavirus; Human papillomavirus (β), (γ) and (μ); Merkel cell polyomavirus; Molluscum contagiosum virus; Polyomavirus HPyV7; Polyomavirus; HpyV6 RD114 retrovirus; and Simian virus. Papillomaviruses and Molluscum contagiosum are known to cause dermatological lesions, such as warts. Merkel cell polyomavirus is implicated in the development of carcinoma. The question of the underappreciated abundance of phages was discussed recently by Hannigan et al. [19]. Whether the presence of these phages plays a role in skin microbiota dysbiosis or the expression of virulence or antibacterial genes needs to be further studied.
Eukaryota: fungi and demodex
To date, fungi, including Malassezia, Cryptococcus, Rhodotorula, and Candida species, have been identified as human skin commensal organisms. Culture-based studies have identified Malassezia spp. as the main genus of commensal skin fungi. The fungal community composition, unlike the bacterial fraction, was previously considered to be similar over all body areas [3]. However, recent studies showed that Malassezia spp. predominated at the central sites of the body and arms, while foot sites were colonized by a more diverse combination of fungi. Demodex are mites of the family Demodicidae and live in seborrheic areas of the skin, such as the face and hair [3, 18, 20]. Demodex are also widely found on the eyelids and the nasal ala. Two species have been identified from human samples: Demodex folliculorum and Demodex brevis [21], but such organisms remain difficult to breed. Notably, the microbiota of these organisms has been studied to increase our knowledge about how they are linked with cutaneous diseases, such as papulopustular rosacea [22].
Role of the skin microbiome
The maintenance of skin homeostasis plays a protective role against potential pathogens and environmental issues
The skin microbiome contributes to the barrier function of the skin and ensures skin homeostasis. The secretion of protease enzymes by skin microbes is involved in the desquamation process and stratum corneum renewal. Sebum and free fatty acid production are involved in pH regulation [23]. The secretion of lipase enzymes is involved in lipidic film surface breakdown. In addition, urease enzymes are implicated in urea degradation. Other roles of the microbiota include the production of biofilms, bacteriocins, and quorum sensing [24, 25]. Moreover, the skin microbiota plays an important role in protecting against potential pathogenic microorganisms by competition [26, 27] and antimicrobial peptide (AMP) production by commensal bacteria [28, 29] or Malassezia fungi, which produce a range of indoles that inhibit many other yeasts and molds [30].
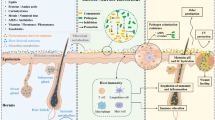
Training and communication with the immune system (Fig. 1)
Skin commensal bacteria have a close relationship with host immune cells from the beginning of their life, and skin resident T cells are thus trained to respond to potential transitory pathogenic bacteria [20, 31]. Meisel et al. [23] showed that the expression of 2820 genes was modulated in mice in response to microbial colonization. A notable proportion of these genes was related to the host immune response and showed roles in processes such as cytokine production, the complement cascade, and the signaling and homing of T cells. A specific strain of Staphylococcus epidermidis was shown to be able to produce 6-N-hydroxyaminopurine, which may confer protection against skin cancer [32].

Skin microbiota, its roles, and its relationship with the immune system. The skin microbiota is composed of bacteria, fungi, archaea, viruses, and mites (Demodex) that are related to the immune system through dialog with resident dendritic cells resulting from complement activation. a The immune system is enhanced by the quorum-sensing process between bacterial populations, which can limit the overgrowth of potential pathogens, or by the production of certain antibiotics, such as lugdunin (c). Microbiotic homeostasis is dependent on the production of antimicrobial peptides (AMPs) both by bacteria themselves and by host cells, such as keratinocytes and sebocytes (b and d).
Wound repair
As described above, skin commensal organisms are in constant crosstalk with the immune system and are thus also involved in wound healing. Leonel et al. [33] synthetized the current knowledge of this topic, which appears to be conflicting. For example, the absence of commensal skin microorganisms has been shown to have a positive effect on wound closure during healing [34]. On the other hand, in another study, the presence of Staphylococcus epidermidis was noted to be a positive factor related to unconventional repair mechanisms specific to commensal bacteria via the recruitment of regulatory CD8 T cells [35]. This finding is consistent with the beneficial skin microbiota effect noted by Lai et al. [36] and the negative effect of skin microbiota dysbiosis [37, 38]. Future investigations are needed to elucidate the influence of the skin microbiota in this process, given the complexity of its definition and its heterogeneity.
The skin microbiota composition depends on many factors (Fig. 2)
Intrinsic factors
Skin site, “biogeography” factor
Grice et al. [4] analyzed 20 different skin sites in 10 healthy humans. They found that Propionibacteria species and Staphylococci species predominated at sebaceous sites, and Corynebacteria species predominated at moist sites, although Staphylococci species were also represented. A mixed population of bacteria resided at dry sites, with a greater prevalence of β-Proteobacteria and Flavobacteriales.

Factors influencing the composition and function of the human skin microbiota and its function. The skin microbiota is shaped by numerous factors: extrinsic (such as lifestyle that embodies occupation, hygiene routine, use of drugs and cosmetics) and intrinsic (genetics, aging, sex, site of the body, etc.) These factors influence the roles of the skin microbiota, implying protection against potential pathogens or climate perturbation as well as the maintenance of skin integrity.
Intra- and interpersonal variability
Costello et al. showed that the phylogenetic diversity of different skin sites was greater than that of communities in the gut, the external auditory canal, or the oral cavity [39]. Interpersonal variation was found to be greater than intrapersonal variation over time. More recently, these observations have been supported by Perez et al. [40], who showed that the arms present significantly less intragroup variation than the axilla or the scalp, and the axilla exhibits the greatest intragroup variation.
Ethnicity
Ethnicity has been shown to contribute to skin microbiome variation and is partly linked to lifestyle. Indeed, Harker et al. [41] reported the differences in the axillary microbiota, such as significantly lower abundances of Staphylococcus species and greater abundances of Corynebacterium species, linked to different genotypes of the gene ABCC11. Between A/A and A/G or G/G individuals, dimorphism was observed between East Asian people and European or African people. Leung et al. [42] indicated that the microbial composition of Chinese people was different from that of other ethnic groups through analysis of metagenomics data from different studies analyzing the palms of hands. Perez et al. [40] showed that the arm microbiota of African American men was relatively homogenous and significantly different from those of all other groups, including the African-continental group. Similarly, the axillary microbiota of East Asian men was highly homogenous and significantly differed from that of other groups. Li et al. [43] found that a unique microbial composition was associated with East Asians compared to Caucasians and Hispanics. East Asians presented higher levels of total bacteria and proteobacteria than the other groups. The Corynebacterium species distribution was analyzed, and Corynebacterium variabile was found to be present exclusively in Hispanics, while Corynebacterium kroppenstedtii was only detected in the East Asian group. However, the definition of ethnicity in the era of globalization with permanent migratory crossings remains elusive.
Gender
Physiological differences between male and female skin environments, such as differences in hormone metabolism, the perspiration rate, and skin surface pH, have been described. Fierer et al. [44] found significant differences between men and women in terms of alpha diversity, but these differences seemed to be related to specific sites and to affect specific age groups. The hand microbiota of women was characterized by greater alpha diversity than that of men, but no specific taxa were found. In terms of relative abundance, they showed that Cutibacterium and Corynebacterium were more abundant in men than in women. In women, Enterobacteriales, Moraxellaceae, Lactobacillaceae, and Pseudomonadaceae were more abundant than in men. Oh et al. [45] observed no sex-associated differences through the study of males and females between the ages of 2 and 40 years regardless of their age based on swabs of the antecubital and popliteal fossae, the volar forearm, and the nares. By studying the axillary vault, Callewaert et al. [46] found that two main groups could be distinguished by characterizing whether the predominantly colonizing genus was Staphylococcus or Corynebacterium. Females predominantly clustered within the Staphylococcus cluster, whereas males clustered more frequently with the Corynebacterium cluster. Prohic et al. [47] did not find a significant influence of sex by studying the distribution of Malassezia species, whereas a significant impact on archaeal diversity and the archaeal community composition was observed [14]. Leung et al. [48] showed that males were characterized by higher abundances of Cutibacterium, Staphylococcus, and Enhydrobacter, whereas Streptococcus was more abundant in the female population. Jo et al. [49] suggested that sex may affect the mycobiome structure during sexual maturation. The Epicoccum and Cryptococcus genera were found at sebaceous sites in males, whereas Malassezia was enriched in females. Zhai et al. [50] showed that males presented greater species richness than females, but the sex differences in the community structure were only present in certain age groups and at particular skin sites. These differences could be found on the upper back of adolescents and elderly people and the cheeks of young adults. Li et al. [43] observed that males presented greater amounts of Corynebacteria than females, though this difference was not significant. Moreover, males exclusively hosted Corynebacterium amycolatum and Corynebacterium kroppenstedtii, in contrast to females, who hosted only Corynebacterium urealyticum and Corynebacterium variabile. By conducting a study based on cultured bacteria, Shami et al. [51] found no significant effect of sex on the number of bacteria isolated from four groups (young people, elderly people, males, and females).
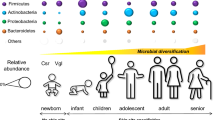
Aging
Although it is known that the skin microbiome is relatively stable over time in the medium term [18], aging is known to be one of the main factors influencing the skin microbiota composition. In 2019, Dimitriu et al. [52] sampled 495 people of various origins at four skin sites and the mouth and considered aging to be the fourth most important factor affecting skin microbiome variation following lifestyle, physiology, demographic proprieties, and pigmentation.
Indeed, aging is associated with many shifts in skin features, such as spot and wrinkle appearance, modified sebaceous gland activity, and dermal compound production [53] (Fig. 3), which impact the previously established ecological conditions for cutaneous microorganisms.

Skin features associated with aging
Several studies have described higher bacterial alpha diversity in skin samples in elderly people. Somboonna et al. [54] studied the skin bacterial composition of 30 healthy Thai females aged 19 to 57 years at the same sites and found that Planctomycetes and Nitrospirae were more prevalent in the teenage group. A Japanese cohort was analyzed in 2017 by Shibagaki et al. [55], who revealed 38 different bacterial species, including many oral bacteria, which significantly differed between the two age groups with skin site dependency. They showed a reduction in the relative abundance of the dominant skin genus Cutibacterium in the cheek, forearm, and forehead microbiota of older adults; an increased proportion of Corynebacterium in the older group on the cheek and forehead; and an increased proportion of Acinetobacter on the scalp in the older group. In another study, Jugé et al. [56] analyzed the forehead skin microbiota of 34 healthy Western European women. They observed an increase in Proteobacteria and a decrease in Actinobacteria populations on older skin. Within the latter phylum, there was a significant increase in the relative abundance of Corynebacterium and a decrease in the relative abundance of Cutibacterium. These data are consistent with Shibagaki et al. [55], who described the previously identified phenomenon of decreased production of sebum associated with aging, which could induce a loss of nutrients available for such bacteria and induce a spread of opportunists. Dimitriu et al. [52] also demonstrated that two mutually exclusive Corynebacterium OTUs could be correlated with skin aging. Indeed, a Corynebacterium OTU with a similar trend to Corynebacterium kroppenstedti displaced another Corynebacterium OTU. In 2014, Prohic et al. [47] collected samples from the trunk and scalp of 100 people by scraping and observed variation in Malassezia species with age. Malassezia furfur was characteristic of the trunk skin of children, whereas Malassezia restricta was predominant on the scalp of 21- to 35-year-old individuals. The Malasseiza population was more prominent in the 36- to 50-year-old group, and M. sympodialis was the predominant species on the trunk skin in older subjects.
Among less-studied skin commensal microorganisms, such as Demodex, Jacob et al. [21] showed that the prevalence of Demodex mites on the skin was positively correlated with aging and reached up to 95% in people over 71 years old. For Archaea, Moissl-Eichinger et al. [14] showed a greater abundance in human subjects older than 60 years compared to middle-aged human subjects.
Extrinsic factors
Mode of delivery
In newborns, bacterial skin colonization has been shown to be influenced by the mode of delivery, the postpartum environment, and the influence of medical staff [57,58,59]. This primary microbiota is transitory and largely influenced by environmental factors and subsequently evolves towards a microbiota that is close to that of adult skin, particularly to the microbiota of hydrated zones. From the age of 3 months, regionalization of the skin microbiota of the child is observed.
Lifestyle, hygiene routine, and cosmetics
Differences are observed in the skin microbiome of people living in different environments (rural or urban) [60,61,62], which are also correlated with the presence of domestic animals [63]. In contrast, the microbiota of people who live together tends to converge, even if they are not genetically related or intimate with each other [64]. Staudinger et al. [65] showed that the use of makeup, including foundation and powder, significantly increased community diversity on the forehead skin. The beneficial effects of some cosmetic compounds, such as preservatives, against the growth and biofilm formation of cutaneous S. aureus or pathogenic C. acnes have been described [66]. These chemical compounds are also involved in the inhibition of commensal bacteria survival [67]. Emulsifiers have been shown to favor the growth of potential pathogens, such as S. aureus [68]. Other studies have revealed the modulation of the diversity of archaeal and bacterial populations, and chemical skin compounds are correlated with changes in the hygiene/cosmetic routine (use of deodorant, moisturizer, or “historical” soap formulated with potash). Such shifts could result in an increased nutrient supply from these products [69,70,71,72]. These results led to the filing of numerous patents aimed at stabilizing or enriching the skin microbiota with beneficial bacteria (e.g., Jessica Wilson, “Personal cleansing compositions and methods of stabilizing the microbiome” 2016 Patent US20190053993A1; Greg Hillebrand, “Method and topical composition for modification of a skin microbiome” 2013 Patent EP3049533A1).
Antibiotics
Zhang et al. [37] demonstrated that the oral intake of vancomycin decreases the bacterial density and alters the bacterial composition in skin wounds, which may contribute to delayed wound repair in mice. In accordance with these data, SanMiguel et al. [73] showed that topical antibiotics can alter the resident skin bacteria for several days and implicated a decrease in the commensal Staphylococcus spp. population, which is known to compete for colonization with pathogenic Staphylococcus aureus. Recently, Park and Lee [74] found that oral administration of doxycycline was linked to a decrease in Cutibacterium acnes relative abundance, an increase in Cutibacterium granulosum, and thus an improvement in the clinical signs in 20 acneic patients, highlighting the implications of antibiotic use for the modulation of the skin microbiota.
Geographical location, climate, and seasonality
A recent study showed a greater benefit of an alpine climate compared to a maritime climate, which differ in pollution and UV radiation levels, for the treatment of atopic dermatitis in children [75]. This observation needs to be confirmed in subjects with healthy skin [76]. Other authors showed that after seawater exposure, exogenous bacteria were present on the skin for at least 24 h after swimming and that ocean water exposure removed normal resident bacteria from the human skin. Likewise, elevation, which is related to extreme environmental conditions, has been shown to disturb skin microbiome stability [77]. Moreover, airborne pollution [78] has been shown to degrade skin microbial population diversity.
Diseases
Table 1 synthetizes the identified associations with commensal community dysregulation related to dermatological pathologies, which encompass widespread conditions, such as acne vulgaris and seborrheic or atopic dermatitis, and less common conditions, such as vitiligo or lupus erythematosus.
How to investigate the skin microbiota
Sampling method
The uniqueness of skin characteristics makes it necessary to standardize and validate the methods used in microbiome research, which would allow comparisons between different studies [27]. The first parameter to keep in mind is the study design, which comprises the strict screening of the subjects involved in the study and the collection of all information that could influence the microbiome variation. The second parameter is sample processing. Samples obtained from swabbing, scraping, and tape strips provide information on the superficial microbiota composition, whereas biopsies offer the opportunity to study microorganisms that could inhabit the deepest layers of the skin [95]. Recently, Ogai et al. [96] completed a comparison between these sampling methods and showed no difference in the results of studying the microbiota by using swabs or tape strips for NGS analysis, but tape strip sampling was shown to be superior when the results were obtained by culture. Verbanic et al. [97] noted the necessity of improving the preparation of samples obtained via the swabbing method so that a sufficient DNA yield is obtained for 100% of samples (the percentage is 25% with traditional preparation). Pedrosa et al. [98] noted that direct plate contact with the skin for the recovery of Malassezia species was more convenient than the tape stripping method.
Transport and storage conditions
Kong et al. [27] noted that immediate freezing at −80°C after sampling was generally preferred and that freeze-thaw cycles must be avoided. The optimization of sample collection and processing through the use of a protectant medium (transport medium that conserves viability and prevents the growth of microorganisms) and the control of storage temperature and time have a considerable effect on the results obtained by analyzing stool samples [99, 100]. Such conclusions remain to be assessed specifically in relation to the improvement of skin microbiota analysis.
Microorganism identification methods: culture vs. nonculture tools
The culture of microorganisms is a historical method for studying their characteristics and properties. With recent advances in molecular biology, this fundamental tool has been shelved in favor of next-generation sequencing methods, which are more sensitive and faster than culture. However, next-generation sequencing does not provide all the information needed to understand the habits of microorganisms in vivo; for example, it provides no information about the viability of the detected organisms [98]. A goal that is as important as the improvement of sampling and storage methods is the improvement of culture parameters in efforts aimed at isolating the viable and culturable fraction of the skin microbiome, which presents its own particularities and shows certain consistent traits [64]. For example, Myles et al. [7] showed that when using a low-nutrient culture medium (R2A), inhibition of the gram-positive fraction by treating the sample with vancomycin and a reduced incubation temperature led to the isolation of the gram-negative fraction of the skin microbiota. Moreover, other parameters of the protocol could be adjusted to obtain more efficient culture media for the growth of diverse skin microorganisms and to improve the methods of colony identification [101, 102]. In these efforts, the culturomics method was improved by Lagier et al. [103], which allowed the discovery of multiple unknown bacteria. By using these methods (i.e., the combination of multiple culture media and conditions), Timm et al. [104] collected more than 800 strains, including more than 30 bacterial genera and 14 fungal genera. However, because this technique requires fastidious and time-consuming work, an increasing number of scientific teams have reinstated this method uniquely or with the use of complementary metagenomic tools [30, 105, 106].
The democratization of metagenomic technologies has induced a shift in interest related to human-associated microorganisms. The skin microbiota has been largely underestimated in terms of diversity, which has persisted because of culture techniques that induce bias due to the growth of microbes in artificial settings [106]. To apply this kind of method for skin microbiome analyses, particular attention is needed at each step of the protocol, including the DNA extraction method, library construction, sequencing step (e.g., primer selection, the chosen platform [88], and the use of blanks and controls), and subsequent analysis (e.g., the selected database and software) [27, 106,107,108]. Furthermore, advanced methods to isolate and cultivate difficult strains by reverse genomics have been recently proposed [109].
Future insights
Study of the microbe-skin relationship: the development of 3D skin models
Many biological models have been produced in an attempt to reconstitute the skin-microbiome interaction with different complexity levels. The first studies consisted of culturing human skin cells, mainly keratinocytes or sebocytes, with bacteria or their metabolites. The main goal of these studies was to understand the pathways involved in pathogen infections or commensal benefits for the skin. Keratinocytes incubated with sterile filtered Staphylococcus aureus medium showed increased production of proteolytic enzymes, followed by the degradation of skin barrier proteins, such as desmoglein-1 and filaggrin [110]. In contrast, some metabolites produced by S. epidermidis could increase the keratinocyte production of antimicrobial peptides via Toll-like receptor 2 activation [36].
Using living bacteria, several studies showed that immediately after inoculation, different Staphylococcus strains showed an increase in epidermal tight junctions (TJs) [111, 112]. However, after several hours of colonization, S. aureus decreased the number of TJs and, subsequently, that of two other types of epidermal junctions, adherent junctions (AJs) and desmosomes, whereas under the same conditions, S. epidermidis showed a minor effect. Other studies focused on cell viability and inflammation revealed that pathogenic strains such as S. aureus or C. acnes or their metabolites induced cell cytotoxicity and increased the production of pro-inflammatory cytokines in skin cells [113, 114].
Skin commensal organisms have also been incubated with skin metabolites to mimic the crosstalk between skin cells and the skin microbiota. As the largest neuroendocrine organ of the human body, the skin produces neurotransmitters, especially stress mediators, including catecholamines or substance P. S. epidermidis, S. aureus, and C. acnes can detect these molecules via specific receptors and respond with increased biofilm formation or production of toxins, resulting in a more virulent phenotype that causes more skin cell damage [115, 116].
Polymicrobial biofilms formed by a mixture of commensal strains (Staphylococcus epidermidis and Micrococcus luteus) and pathogens (Staphylococcus aureus and Pseudomonas aeruginosa) were also used to study the interactions among commensal organisms, pathogens, and human keratinocytes. The authors observed that the commensal organisms reduced the damage caused to the keratinocyte monolayer by pathogens, reduced biofilm thickness, and formed a layer between the keratinocytes and pathogens [117].
Due to the faster growth rate of bacteria than human cells, 2D models cannot be maintained for more than 24 h. Moreover, cells cultivated in monolayers do not reflect the skin surface. These cells are more reflective of the conditions in wounds or defective skin barriers. These limitations have led to the development of more complex models that better reproduce the skin barrier with its dry environment to study long-term interactions between the skin and its microbiota.
Thus, 3D skin models have been colonized with bacteria. These models are now widely used for dermatological and cosmeceutical studies. The simplest model, the reconstructed human epidermis (RHE), is composed of primary human keratinocytes grown on a decellularized dermis or a porous membrane. Air-liquid culture allows the formation of a fully differentiated epidermis and the formation of a functional barrier. When a living dermis is also present, the model is referred to as full-thickness skin (FT-skin).
Many studies have used these models to study the bacterial, fungal, or yeast infection process. Using an RHE model, Lerebour et al. [118] described differential adhesion properties between S. aureus, a hydrophilic strain, and S. epidermidis, a relatively hydrophobic strain, suggesting that rendering the skin surface more hydrophobic would restrict microbial adhesion. When the epidermis is locally removed, either by punching or thermal injury, these models can reproduce a skin wound environment by making the dermis accessible to bacteria. These models have been widely used to study the infection process by following microbial growth and tissue damage [119,120,121,122,123,124,125,126]. These infection models have mostly been used to identify potential antibacterial treatments. For example, when a biofilm-forming S. aureus was inoculated at high density (i.e., >107 CFU) onto a 3-mm-punched FT-skin model, plasma treatment reduced the number of adherent colonies after 24 h [119]. In another study, wound infection with methicillin-resistant S. aureus (MRSA) after thermal injury in an FT-skin model revealed significant growth of MRSA after 24–48 h. Skin exposure to MRSA increased the expression of inflammatory mediators, such as TLR2, IL-6, and IL-8, and the antimicrobial proteins human β-defensin-2, human β-defensin-3, and RNAse7. Moreover, locally applied mupirocin effectively reduced MRSA counts in a thermal wounded skin model by more than 99.9% within 24 h [120].
More recently, with the emerging role of the skin microbiome in skin health, 3D models have been colonized with commensal bacteria in the absence of any stress or injury. Unique commensal bacterial strains, among which S. epidermidis and C. acnes are the most represented, have been inoculated and cultivated for up to 4 days on the skin surface [127, 128]. The main endpoint in these studies is bacterial growth or bacterial competition. For example, C. acnes seemed to better colonize immature skin, under the differentiated epidermis. Competition was observed when S. epidermidis and C. acnes were inoculated concomitantly, with S. epidermidis decreasing C. acnes growth, while the inverse was not true [128].
Most of the published research that has used 3D models to investigate host-microbiota interactions has thus focused on the impact of individual species. Collecting bacteria from the skin, which is a very poor environment, and culturing them in conventional cell culture media probably affect their metabolism and may not reflect the natural crosstalk occurring at the skin surface.
There is a clear need to develop and improve experimental strategies for the colonization of 3D skin models with complete microbiota communities, including those directly isolated from individual humans, to more closely mimic the in vivo skin-microbiota interplay.
In 2019, Cenizo et al. [129] sampled the skin microbiota of the inner forearm of a woman and immediately inoculated the sample onto the surface of a 3D skin model. The model was followed for 7 days in culture and showed the stabilization of the number of living bacteria at a density similar to what was found on the collected skin. The microbiota-colonized model showed higher proliferation of the epidermis basal layer and increases in epidermal junctions and desquamation. This model also showed higher time stability than the same model colonized with a unique strain in which the rapid growth of S. epidermidis destroyed the tissue. These results suggested that the interactions occurring within diverse microbiota could prevent the outgrowth of single strains. Such models offer ways to study the impact of external factors on the composition of the skin microbiota as well as its implications for the skin response to these factors.
Even more complex models may help to establish 3D skin models as a replacement for animal models in the future. 3D skin models can include immune, nervous, pigmentation, and endothelial cells [124, 130, 131]. These models can be bioprinted to reduce their production time and improve reproducibility [132, 133] and can be inoculated with swabs collected from pathological skin lesions in which the microbiota is known to play a role (e.g., atopic dermatitis, psoriasis, or acne) as well as offer new drug evaluations.
Transplantation of skin microbiota
By analogy with fecal transplantation, which is a powerful therapeutic tool for digestive disorders [134, 135], skin microbiota transplantation studies are moving forward [136,137,138] and could provide a promising approach for the treatment of diseases, such as atopic dermatitis. Transplantation as a tool for correcting unsuitable armpit odor is currently a possibility under consideration. One process is focused on the removal of the malodor-causing-microbiome by the means of antibiotics, which is then replaced by a healthy nonodorous axillary microbiome. A second process consists of the application of probiotics, such as lactic bacteria or skin bacilli, but their incapacity to durably colonize the niches induced the need to find other nonodor-causing commensals that could inhibit unsuitable ones, notably Staphylococcus hominis, Corynebacterium tuberculostearicum, and Anaerococcus spp. [139]. This phenomenon is probably governed by more subtle principles, and it is reasonable to think that modifications may involve the microorganisms at the subtype level. The transplantation method depends on the triangle donor microbiome composition (A), the recipient microbiome composition (B), and the load of the transplant (C). Indeed, a recipient microbiome composed of C. acnes subtype H1 and Leifsonia spp. allows a better engraftment of donor strains [137]. By assessing the viability of skin microbial communities unidirectionally (from the forearm to the back of the same volunteers), Perin et al. [138] described the partial efficiency of unenriched skin microbial community transfer, but more information is needed regarding the viability of the microbes transplanted. Myles et al. pointed to Roseomonas mucosa, a gram-negative commensal bacterium associated with decreasing atopic dermatitis severity [136]. Nakatsuji et al. [28] successfully tested the transfer of bacteria selected for their ability to inhibit S. aureus in atopic dermatitis patients, which highlighted the protective effect of commensal coagulase-negative Staphylococcus (CoNS).
Prebiotics and probiotics
At a time when microbiomes and their fluctuations are known to be associated with several dysfunctions, the skin microbiome is of growing interest in the field of cosmetics, focusing on the exploitation of these proprieties to improve human well-being through formulations that contain prebiotics, probiotics, or skin microbiome-friendly ingredients known as cosmeceutics.
Different firms have developed formulations containing a Hylocereus undatus fruit extract that can reduce the perception of skin imperfections. This extract may positively influence the skin microbiota balance, and several patents highlight this finding (Liki Von Oppen-Bezalel et al. patent n° WO2016147189A1; Korean patent n° KR20150118078A, etc.). Indeed, this plant was shown by Som et al. to be a major source of antioxidant substances, which are key in the skin aging mechanism [140]. Similarly, Banerjee et al. [141] controlled the formulation of an emulsion cream for topical application and considered its impact on the commensal skin flora. Bacterial extracts were also tested, such as an extract of Shingomonas hydrophobicum [142] and lactobionic acid from Pseudomonas taetrolens, for antiaging activity [143]. Other formulations containing bacterial derivative compounds (e.g., the Lactobacillus extract filtrate in Skinolance®), prebiotic peptides (ACTIBIOME™, FENSEBIOME™), or vitamins, such as niacinamide (Univerler patent WO2019086327), are provided by raw material sellers and are indicated to show prebiotic activity. Globally, this kind of information remains elusive, and firm scientific conclusions are rarely available [144].
Lactobacillus and Bifidobacterium species, which are implicated in health, were analyzed for their probiotic properties related to skin homeostasis. The tested species included Lactobacillus reuteri [145], Lactobacillus acidophilus [146], Lactobacillus plantarum [147,148,149], a formulation based on a patented Lactobacillus mixture (CN110121353A), Lactobacillus helveticus [150], Lactobacillus rhamnosus applied synergistically with the plant Agastache rugosa [151], and Bifidobacterium breve [152].
Recently, Nitrosomonas eutropha, an ammonia-oxidizing agent, was targeted for its antiaging properties [153, 154] and included in the Mother Dirt AO+ Mist Skin Probiotic Spray patent (JP2017519486A).
Many patents are being produced that focus on diverse bacterial strains that could improve skin well-being and the antiaging properties of cosmetics (Streptococcus pneumoniae and Streptococcus thermophilus in patents KR20180121269 and KR20180121268; the newly discovered strain Epidermidibacterium keratini in patent WO201804224; Pseudoalteromomonas antartica in patent JP2018500279A, etc.). Nevertheless, scientific data supporting their efficacy are rarely available.
Key points and conclusions
-
The skin microbiome is composed of a variety of organisms, including bacteria, archaea, fungi, and even small arthropods, which interact with each other and could be implicated in the host health status.
-
The skin microbiome composition depends on many factors. These factors form an intricate network that novel sequencing technologies allow us to better understand. However, standardization of studies is required to reach strong conclusions on which innovation process could be best.
-
Optimized evaluation tools, such as 3D skin models, offer ways to study the impact of modulation factors on the composition of the skin microbiota as well as its implications for the skin response.
-
Presently, understanding the skin microbiome is at a turning point. The beneficial and protective role of bacterial communities in close relationship with their host is understood to be clinically manipulated (illustrated by “transplantation-like” technology) or to be an important industrial concern through the investigation of microbial-derivated products with bioactive activities.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AJs:
-
Adherent junctions
- AMP:
-
Antimicrobial peptides
- CoNS:
-
Coagulase-negative Staphylococcus
- GNB:
-
Gram-negative bacteria
- HPV:
-
Human papillomavirus
- NGS:
-
Next-generation sequencing
- TJs:
-
Tight junctions
References
Gallo RL. Human skin is the largest epithelial surface for interaction with microbes. J Investigative Dermatol. 2017;137:1213–4.
Schmid-Wendtner M-H, Korting HC. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol Physiol. 2006;19:296–302.
Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16:143–55.
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2.
Ross AA, Rodrigues Hoffmann A, Neufeld JD. The skin microbiome of vertebrates. Microbiome. 2019;7:79.
Sfriso R, Egert M, Gempeler M, Voegeli R, Campiche R. Revealing the secret life of skin - with the microbiome you never walk alone. Int J Cosmet Sci. 2020;42:116–26.
Myles IA, Reckhow JD, Williams KW, Sastalla I, Frank KM, Datta SK. A method for culturing Gram-negative skin microbiota. BMC Microbiol. 2016;16:60.
Cassir N, Thomas G, Hraiech S, Brunet J, Fournier P-E, La Scola B, et al. Chlorhexidine daily bathing: impact on health care–associated infections caused by gram-negative bacteria. American Journal of Infection Control. 2015;43:640–3.
Cassir N, Papazian L, Fournier P-E, Raoult D, La Scola B. Insights into bacterial colonization of intensive care patients’ skin: the effect of chlorhexidine daily bathing. Eur J Clin Microbiol Infect Dis. 2015;34:999–1004.
Dinis JM, Barton DE, Ghadiri J, Surendar D, Reddy K, Velasquez F, et al. In search of an uncultured human-associated TM7 bacterium in the environment. Yang C-H, editor. PLoS ONE. 2011;6:e21280.
Ferrari B, Winsley T, Ji M, Neilan B. Insights into the distribution and abundance of the ubiquitous Candidatus Saccharibacteria phylum following tag pyrosequencing. Sci Rep. 2015;4:3957.
McLean JS, Lombardo M-J, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, et al. Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proceed Nat Acad Sci. 2013;110:E2390–9.
Bor B, Bedree JK, Shi W, McLean JS, He X. Saccharibacteria (TM7) in the human oral microbiome. J Dent Res. 2019;98:500–9.
Moissl-Eichinger C, Probst AJ, Birarda G, Auerbach A, Koskinen K, Wolf P, et al. Human age and skin physiology shape diversity and abundance of Archaea on skin. Scientific Rep. 2017;7 Available from: http://www.nature.com/articles/s41598-017-04197-4. [cited 2019 Jan 8].
Koskinen K, Pausan MR, Perras AK, Beck M, Bang C, Mora M, et al. First insights into the diverse human archaeome: specific detection of Archaea in the gastrointestinal tract, lung, and nose and on skin. Schleper CM, Jansson JK, editors. mBio. 2017;8:e00824–17 /mbio/8/6/mBio.00824-17.atom.
Pausan MR, Csorba C, Singer G, Till H, Schöpf V, Santigli E, et al. Exploring the archaeome: detection of archaeal signatures in the human body. Front Microbiol. 2019;10:2796.
Liu J, Yan R, Zhong Q, Ngo S, Bangayan NJ, Nguyen L, et al. The diversity and host interactions of Propionibacterium acnes bacteriophages on human skin. ISME J. 2015;9:2078–93.
Oh J, Byrd AL, Park M. NISC Comparative Sequencing Program, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell. 2016;165:854–66.
Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, SanMiguel AJ, et al. The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. Heitman J, editor. mBio. 2015;6:e01578–15.
Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9:244–53.
Jacob S, VanDaele MA, Brown JN. Treatment of Demodex -associated inflammatory skin conditions: a systematic review. Dermatologic Therap. 2019;32. [cited 2020 Feb 6]. https://doi.org/10.1111/dth.13103.
Murillo N, Aubert J, Raoult D. Microbiota of Demodex mites from rosacea patients and controls. Microbial Pathogenesis. 2014;71–72:37–40.
Meisel JS, Sfyroera G, Bartow-McKenney C, Gimblet C, Bugayev J, Horwinski J, et al. Commensal microbiota modulate gene expression in the skin. Microbiome. 2018;6:20.
Baldwin HE, Bhatia ND, Friedman A, Eng RM, Seite S. The role of cutaneous microbiota harmony in maintaining a functional skin barrier. J Drugs Dermatol. 2017;16:12–8.
Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med. 2019;11:eaat8329.
Grice EA, Kong HH, Renaud G, Young AC, Program NISCCS, Bouffard GG, et al. A diversity profile of the human skin microbiota. Genome Research. 2008;18:1043–50.
Kong HH, Andersson B, Clavel T, Common JE, Jackson SA, Olson ND, et al. Performing skin microbiome research: a method to the madness. J Investigative Dermatol. 2017;137:561–8.
Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 2017;9:eaah4680.
O’Sullivan JN, Rea MC, O’Connor PM, Hill C, Ross RP. Human skin microbiota is a rich source of bacteriocin-producing staphylococci that kill human pathogens. FEMS Microbiol Ecol. 2019:95. [cited 2020 Feb 6]. https://doi.org/10.1093/femsec/fiy241/5259109.
Gaitanis G, Tsiouri G, Spyridonos P, Stefos Τ, Stamatas GN, Velegraki A, et al. Variation of cultured skin microbiota in mothers and their infants during the first year postpartum. Pediatric Dermatol. 2019. https://doi.org/10.1111/pde.13829.
Coates M, Lee MJ, Norton D, MacLeod AS. The skin and intestinal microbiota and their specific innate immune systems. Front Immunol. 2019;10:2950.
Nakatsuji T, Chen TH, Butcher AM, Trzoss LL, Nam S-J, Shirakawa KT, et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci Adv. 2018;4:eaao4502.
Leonel C, Sena IFG, Silva WN, Prazeres PHDM, Fernandes GR, Mancha Agresti P, et al. Staphylococcus epidermidis role in the skin microenvironment. J Cell Mol Med. 2019;23:5949–55.
Canesso MCC, Vieira AT, Castro TBR, Schirmer BGA, Cisalpino D, Martins FS, et al. Skin wound healing is accelerated and scarless in the absence of commensal microbiota. JI. 2014;193:5171–80.
Linehan JL, Harrison OJ, Han S-J, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell. 2018;172:784–96 e18.
Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, et al. Commensal bacteria regulate Toll-like receptor 3–dependent inflammation after skin injury. Nat Med. 2009;15:1377–82.
Zhang M, Jiang Z, Li D, Jiang D, Wu Y, Ren H, et al. Oral antibiotic treatment induces skin microbiota dysbiosis and influences wound healing. Microbial Ecology. 2015;69:415–21.
Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, et al. Temporal stability in chronic wound microbiota is associated with poor healing. J Investigative Dermatol. 2017;137:237–44.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7.
Perez Perez GI, Gao Z, Jourdain R, Ramirez J, Gany F, Clavaud C, et al. Body site is a more determinant factor than human population diversity in the healthy skin microbiome. McDowell A, editor. PLOS ONE. 2016;11:e0151990.
Harker M, Carvell A-M, Marti VPJ, Riazanskaia S, Kelso H, Taylor D, et al. Functional characterisation of a SNP in the ABCC11 allele - effects on axillary skin metabolism, odour generation and associated behaviours. J Dermatol Sci. 2014;73:23–30.
Leung MHY, Tong X, Wilkins D, Cheung HHL, Lee PKH. Individual and household attributes influence the dynamics of the personal skin microbiota and its association network. Microbiome. 2018;6:26.
Li M, Budding AE, van der Lugt-Degen M, Du-Thumm L, Vandeven M, Fan A. The influence of age, gender and race/ethnicity on the composition of the human axillary microbiome. Int J Cosmetic Sci. 2019;41(4):371–7. https://doi.org/10.1111/ics.12549.
Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proceed Nat Acad Sci. 2008;105:17994–9.
Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genom Med. 2012;4:77.
Callewaert C, Kerckhof F-M, Granitsiotis MS, Van Gele M, Van de Wiele T, Boon N. Characterization of Staphylococcus and Corynebacterium clusters in the human axillary region. van Schaik W, editor. PLoS ONE. 2013;8:e70538.
Prohic A, Simic D, Sadikovic TJ, Krupalija-Fazlic M. Distribution of Malassezia species on healthy human skin in Bosnia and Herzegovina: correlation with body part, age and gender. Iran J Microbiol. 2014;6:253–62.
Leung MHY, Wilkins D, Lee PKH. Insights into the pan-microbiome: skin microbial communities of Chinese individuals differ from other racial groups. Sci Rep. 2015;5:11845.
Jo J-H, Deming C, Kennedy EA, Conlan S, Polley EC, Ng W-I, et al. Diverse human skin fungal communities in children converge in adulthood. J Investigative Dermatol. 2016;136:2356–63.
Zhai W, Huang Y, Zhang X, Fei W, Chang Y, Cheng S, et al. Profile of the skin microbiota in a healthy Chinese population. J Dermatol. 2018;45:1289–300.
Shami A, Al-Mijalli S, Pongchaikul P, Al-Barrag A, AbduRahim S. The prevalence of the culturable human skin aerobic bacteria in Riyadh, Saudi Arabia. BMC Microbiol. 2019;19:189.
Dimitriu PA, Iker B, Malik K, Leung H, Mohn WW, Hillebrand GG. New insights into the intrinsic and extrinsic factors that shape the human skin microbiome. mBio. 2019;10(4):e00839–19. https://doi.org/10.1128/mBio.00839-19.
Bonté F, Girard D, Archambault J-C, Desmoulière A. Skin changes during ageing. In: Harris JR, Korolchuk VI, editors. Biochemistry and cell biology of ageing: Part II Clinical science [Internet]. Singapore: Springer Singapore; 2019. p. 249–80. [cited 2020 Feb 6]. https://doi.org/10.1007/978-981-13-3681-2_10.
Somboonna N, Wilantho A, Srisuttiyakorn C, Assawamakin A, Tongsima S. Bacterial communities on facial skin of teenage and elderly Thai females. Arch Microbiol. 2017;199:1035–42.
Shibagaki N, Suda W, Clavaud C, Bastien P, Takayasu L, Iioka E, et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Scientific Rep. 2017;7 Available from: http://www.nature.com/articles/s41598-017-10834-9. [cited 2019 Jan 8].
Jugé R, Rouaud-Tinguely P, Breugnot J, Servaes K, Grimaldi C, Roth M-P, et al. Shift in skin microbiota of Western European women across aging. J Appl Microbiol. 2018;125:907–16.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceed Nat Acad Sci. 2010;107:11971–5.
Nagata R, Nagano H, Ogishima D, Nakamura Y, Hiruma M, Sugita T. Transmission of the major skin microbiota, Malassezia, from mother to neonate: transmission of Malassezia. Pediatr Int. 2012;54:350–5.
Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. 2017;23:314–26.
Ying S, Zeng D-N, Chi L, Tan Y, Galzote C, Cardona C, et al. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. Badger JH, editor. PLOS ONE. 2015;10:e0141842.
Kirjavainen PV, Karvonen AM, Adams RI, Täubel M, Roponen M, Tuoresmäki P, et al. Farm-like indoor microbiota in non-farm homes protects children from asthma development. Nat Med. 2019;25:1089–95.
McCall L-I, Callewaert C, Zhu Q, Song SJ, Bouslimani A, Minich JJ, et al. Home chemical and microbial transitions across urbanization. Nat Microbiol. 2020;5:108–15.
Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife. 2013;2:e00458.
Bewick S, Gurarie E, Weissman JL, Beattie J, Davati C, Flint R, et al. Trait-based analysis of the human skin microbiome. Microbiome. 2019;7:101.
Staudinger T, Pipal A, Redl B. Molecular analysis of the prevalent microbiota of human male and female forehead skin compared to forearm skin and the influence of make-up. J Appl Microbiol. 2011;110:1381–9.
Gannesen AV, Borrel V, Lefeuvre L, Netrusov AI, Plakunov VK, Feuilloley MGJ. Effect of two cosmetic compounds on the growth, biofilm formation activity, and surface properties of acneic strains of Cutibacterium acnes and Staphylococcus aureus. MicrobiologyOpen. 2019;8:e00659.
Wang Q, Cui S, Zhou L, He K, Song L, Liang H, et al. Effect of cosmetic chemical preservatives on resident flora isolated from healthy facial skin. J Cosmet Dermatol. 2019;18:652–8.
Nielsen CK, Kjems J, Mygind T, Snabe T, Meyer RL, et al. Front Microbiol. 2016;7. [cited 2020 Apr 7]. https://doi.org/10.3389/fmicb.2016.01878/full.
Callewaert C, Hutapea P, Van de Wiele T, Boon N. Deodorants and antiperspirants affect the axillary bacterial community. Arch Dermatol Res. 2014;306:701–10.
Blaak J, Staib P. The relation of pH and skin cleansing. In: Surber C, Abels C, Maibach H, editors. Current problems in dermatology [Internet]. S. Karger AG; 2018. p. 132–42. Available from: https://www.karger.com/Article/FullText/489527. [cited 2020 Apr 8].
Lee HJ, Jeong SE, Lee S, Kim S, Han H, Jeon CO. Effects of cosmetics on the skin microbiome of facial cheeks with different hydration levels. MicrobiologyOpen. 2018;7:e00557.
Bouslimani A, da Silva R, Kosciolek T, Janssen S, Callewaert C, Amir A, et al. The impact of skin care products on skin chemistry and microbiome dynamics. BMC Biol. 2019;17:47.
SanMiguel AJ, Meisel JS, Horwinski J, Zheng Q, Grice EA. Topical antimicrobial treatments can elicit shifts to resident skin bacterial communities and reduce colonization by Staphylococcus aureus competitors. Antimicrobial Agents and Chemotherap. 2017;61:e00774–17 /aac/61/9/e00774-17.atom.
Park YJ, Lee HK. The role of skin and orogenital microbiota in protective immunity and chronic immune-mediated inflammatory disease. Front Immunol. 2018;8:1955.
Mierlo MMF, Totté JEE, Fieten KB, den Broek TJ, Schuren FHJ, Pardo LM, et al. The influence of treatment in alpine and moderate maritime climate on the composition of the skin microbiome in patients with difficult to treat atopic dermatitis. Clin Experimental Allergy. 2019;49:1437–45.
Nielsen MC, Jiang SC. Alterations of the human skin microbiome after ocean water exposure. Marine Pollution Bull. 2019;145:595–603.
Li H, Wang Y, Yu Q, Feng T, Zhou R, Shao L, et al. Elevation is associated with human skin microbiomes. Microorganisms. 2019;7:611.
Araviiskaia E, Berardesca E, Bieber T, Gontijo G, Sanchez Viera M, Marrot L, et al. The impact of airborne pollution on skin. J Eur Acad Dermatol Venereol. 2019;33(8):1496–505. https://doi.org/10.1111/jdv.15583. Epub 2019 Apr 26.
Platsidaki E, Dessinioti C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Res. 2018;7:1953.
O’Neill AM, Gallo RL. Host-microbiome interactions and recent progress into understanding the biology of acne vulgaris. Microbiome. 2018;6:177.
Hiruma M, Cho O, Hiruma M, Kurakado S, Sugita T, Ikeda S. Genotype analyses of human commensal scalp fungi, Malassezia globosa, and Malassezia restricta on the scalps of patients with dandruff and healthy subjects. Mycopathologia. 2014;177:263–9.
Francuzik W, Franke K, Schumann R, Heine G, Worm M. Propionibacterium acnes abundance correlates inversely with Staphylococcus aureus: data from atopic dermatitis skin microbiome. Acta Derm Venerol. 2018;98:490–5.
Tanaka A, Cho O, Saito C, Saito M, Tsuboi R, Sugita T. Comprehensive pyrosequencing analysis of the bacterial microbiota of the skin of patients with seborrheic dermatitis: skin microbiota. Microbiol Immunol. 2016;60:521–6.
Moallaei H, Namazi MJ, Bouchara JP, Pourhammed S. Malassezia species in students from universities of Sabzevar, Northeastern Iran. J de Mycologie Médicale. 2018;28:70–5.
Tarkowski W, Moneta-Wielgoś J, Młocicki D. Do Demodex mites play a role in pterygium development? Medical Hypotheses. 2017;98:6–10.
Visser MJE, Kell DB, Pretorius E. Bacterial dysbiosis and translocation in psoriasis vulgaris. Front Cell Infect Microbiol. 2019;9:7.
Chang H-W, Yan D, Singh R, Liu J, Lu X, Ucmak D, et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome. 2018;6:154.
Stehlikova Z, Kostovcik M, Kostovcikova K, Kverka M, Juzlova K, Rob F, et al. Dysbiosis of skin microbiota in psoriatic patients: co-occurrence of fungal and bacterial communities. Frontiers in Microbiology. 2019;10:438.
Ganju P, Nagpal S, Mohammed M, Nishal Kumar P, Pandey R, Natarajan VT, et al. Microbial community profiling shows dysbiosis in the lesional skin of Vitiligo subjects. Sci Rep. 2016;6:18761.
Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. The Malassezia genus in skin and systemic diseases. Clin Microbiol Rev. 2012;25:106–41.
Natsuga K, Cipolat S, Watt FM. Increased bacterial load and expression of antimicrobial peptides in skin of barrier-deficient mice with reduced cancer susceptibility. J Investigative Dermatol. 2016;136:99–106.
Wood DLA, Lachner N, Tan J-M, Tang S, Angel N, Laino A, et al. A natural history of actinic keratosis and cutaneous squamous cell carcinoma microbiomes. Fraser CM, editor. mBio. 2018;9:e01432–18 /mbio/9/5/mBio.01432-18.atom.
Redel H, Gao Z, Li H, Alekseyenko AV, Zhou Y, Perez-Perez GI, et al. Quantitation and composition of cutaneous microbiota in diabetic and nondiabetic men. J Infect Dis. 2013;207:1105–14.
Huang C, Yi X, Long H, Zhang G, Wu H, Zhao M, et al. Disordered cutaneous microbiota in systemic lupus erythematosus. J Autoimmunity. 2020;108:102391.
Prast-Nielsen S, Tobin A-M, Adamzik K, Powles A, Hugerth LW, Sweeney C, et al. Investigation of the skin microbiome: swabs vs. biopsies. Bri J Dermatol. 2019;181:572–9.
Ogai K, Nagase S, Mukai K, Iuchi T, Mori Y, Matsue M, et al. A comparison of techniques for collecting skin microbiome samples: swabbing versus tape-stripping. Front Microbiol. 2018;9:2362.
Verbanic S, Kim CY, Deacon JM, Chen IA. Improved single-swab sample preparation for recovering bacterial and phage DNA from human skin and wound microbiomes. BMC Microbiology. 2019;19:214.
Pedrosa AF, Lisboa C, Faria-Ramos I, Silva RM, Miranda IM, Rodrigues AG. Malassezia species retrieved from skin with pityriasis versicolor, seborrhoeic dermatitis and skin free of lesions: a comparison of two sampling methods. Bri J Dermatol. 2018; [cited 2020 Jan 17]. https://doi.org/10.1111/bjd.16521.
Bellali S, Lagier J-C, Raoult D, Bou KJ. Among live and dead bacteria, the optimization of sample collection and processing remains essential in recovering gut microbiota components. Front Microbiol. 2019;10:1606.
Nagata N, Tohya M, Takeuchi F, Suda W, Nishijima S, Ohsugi M, et al. Effects of storage temperature, storage time, and Cary-Blair transport medium on the stability of the gut microbiota. Drug Discov Ther. 2019;13:256–60.
Chang Y, Hou F, Pan Z, Huang Z, Han N, Bin L, et al. Optimization of culturomics strategy in human fecal samples. Front Microbiol. 2019;10:2891.
Bonnet M, Lagier JC, Raoult D, Khelaifia S. Bacterial culture through selective and non-selective conditions: the evolution of culture media in clinical microbiology. New Microbes and New Infections. 2020;34:100622.
Lagier J-C, Dubourg G, Million M, Cadoret F, Bilen M, Fenollar F, et al. Culturing the human microbiota and culturomics. Nat Rev Microbiol. 2018;16:540–50.
Timm CM, Loomis K, Stone W, Mehoke T, Brensinger B, Pellicore M, et al. Isolation and characterization of diverse microbial representatives from the human skin microbiome. Microbiome. 2020;8:58.
Zapka C, Leff J, Henley J, Tittl J, De Nardo E, Butler M, et al. Comparison of standard culture-based method to culture-independent method for evaluation of hygiene effects on the hand microbiome. Clemente JC, editor. mBio. 2017;8:e00093–17 /mbio/8/2/e00093-17.atom.
Sandhu SS, Pourang A, Sivamani RK. A review of next generation sequencing technologies used in the evaluation of the skin microbiome: what a time to be alive. Dermatol Online J. 2019;25(7):13030/qt3hv5z3q3.
Meisel JS, Hannigan GD, Tyldsley AS, SanMiguel AJ, Hodkinson BP, Zheng Q, et al. Skin microbiome surveys are strongly influenced by experimental design. J Investigative Dermatol. 2016;136:947–56.
Castelino M, Eyre S, Moat J, Fox G, Martin P, Ho P, et al. Optimisation of methods for bacterial skin microbiome investigation: primer selection and comparison of the 454 versus MiSeq platform. BMC Microbiol. 2017;17:23.
Cross KL, Campbell JH, Balachandran M, Campbell AG, Cooper SJ, Griffen A, et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat Biotechnol. 2019;37:1314–21.
Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus induces increased serine protease activity in keratinocytes. J Investigative Dermatol. 2017;137:377–84.
Ohnemus U, Kohrmeyer K, Houdek P, Rohde H, Wladykowski E, Vidal S, et al. Regulation of epidermal tight-junctions (TJ) during infection with exfoliative toxin-negative Staphylococcus strains. J Investigative Dermatol. 2008;128:906–16.
Bäsler K, Galliano M-F, Bergmann S, Rohde H, Wladykowski E, Vidal-y-Sy S, et al. Biphasic influence of Staphylococcus aureus on human epidermal tight junctions: S. aureus and tight junctions. Ann NY Acad Sci. 2017;1405:53–70.
Kwon HI, Jeong NH, Jun SH, Son JH, Kim S, Jeon H, et al. Thymol attenuates the worsening of atopic dermatitis induced by Staphylococcus aureus membrane vesicles. Int Immunopharmacol. 2018;59:301–9.
Sanford JA, O’Neill AM, Zouboulis CC, Gallo RL. Short-chain fatty acids from Cutibacterium acnes activate both a canonical and epigenetic inflammatory response in human sebocytes. JI. 2019;202:1767–76.
N’Diaye A, Mijouin L, Hillion M, Diaz S, Konto-Ghiorghi Y, Percoco G, et al. Effect of substance P in Staphylococcus aureus and Staphylococcus epidermidis virulence: implication for skin homeostasis. Front Microbiol. 2016;7. [cited 2020 Apr 1]. https://doi.org/10.3389/fmicb.2016.00506/abstract.
Borrel V, Thomas P, Catovic C, Racine P-J, Konto-Ghiorghi Y, Lefeuvre L, et al. Acne and stress: impact of catecholamines on Cutibacterium acnes. Front Med. 2019;6:155.
Jordana-Lluch E, Garcia V, Kingdon ADH, Singh N, Alexander C, Williams P, et al. A simple polymicrobial biofilm keratinocyte colonization model for exploring interactions between commensals, pathogens and antimicrobials. Front Microbiol. 2020;11:291.
Lerebour G, Cupferman S, Bellon-Fontaine MN. Adhesion of Staphylococcus aureus and Staphylococcus epidermidis to the EpiskinR reconstructed epidermis model and to an inert 304 stainless steel substrate. J Appl Microbiol. 2004;97:7–16.
Kim PY, Kim Y-S, Koo IG, Jung JC, Kim GJ, Choi MY, et al. Bacterial inactivation of wound infection in a human skin model by liquid-phase discharge plasma. Neyrolles O, editor. PLoS ONE. 2011;6:e24104.
Haisma EM, Rietveld MH, de Breij A, van Dissel JT, El Ghalbzouri A, Nibbering PH. Inflammatory and antimicrobial responses to methicillin-resistant Staphylococcus aureus in an in vitro wound infection model. Cole AM, editor. PLoS ONE. 2013;8:e82800.
Popov L, Kovalski J, Grandi G, Bagnoli F, Amieva MR. Three-dimensional human skin models to understand Staphylococcus aureus skin colonization and infection. Front Immunol. 2014;5. https://doi.org/10.3389/fimmu.2014.00041/abstract [cited 2020 Apr 1].
den Reijer PM, Haisma EM, Lemmens-den Toom NA, Willemse J, Koning RA, Demmers JAA, et al. Detection of Alpha-Toxin and Other Virulence Factors in Biofilms of Staphylococcus aureus on Polystyrene and a Human Epidermal Model.. PLoS ONE. 2016;11(1):e0145722. https://doi.org/10.1371/journal.pone.0145722.
Liang P-P, Huang X-Z, Yi J-L, Chen Z-R, Ma H, Ye C-X, et al. A Trichophyton rubrum infection model based on the reconstructed human epidermis - Episkin®. Chinese Med J. 2016;129:54–8.
Rademacher F, Simanski M, Gläser R, Harder J. Skin microbiota and human 3D skin models. Experimental Dermatol. 2018;27:489–94.
Pedrosa AF, Lisboa C, Branco J, Pellevoisin C, Miranda IM, Rodrigues AG. Malassezia interaction with a reconstructed human epidermis: keratinocyte immune response. Mycoses. 2019;62:932–6. https://doi.org/10.1111/myc.12965.
Jahanshahi M, Hamdi D, Godau B, Samiei E, Sanchez-Lafuente C, Neale K, et al. An engineered infected epidermis model for in vitro study of the skin’s pro-inflammatory response. Micromachines. 2020;11:227.
Bojar RA. Studying the human skin microbiome using 3D in vitro skin models. Appl Vitro Toxicol. 2015;1:165–71.
Cadau S, Valla-Dury L, Cenizo V, Le-Beux C, Rival D, Gault M, et al. Studying microbiote competition and skin interaction using organotypic 3D skin models. ATROA. 2017;2 Available from: https://medcraveonline.com/ATROA/studying-microbiote-competition-and-skin-interaction-using-organotypic-3d-skin-models.html. [cited 2020 Apr 1].
Landemaine L, Cenizo V, Lemaire G, Portes P. Colonization of a 3d skin model with a complete microbiota is more beneficial to the skin barrier than with staphylococcus epidermidis alone. Int Federation Soc Cosmetic Chemists. 2018. https://www.labskin.co.uk/loccitane-colonization-of-a-3d-skin-model/.
Dezutter-Dambuyant C, Black A, Bechetoille N, Bouez C, Maréchal S, Auxenfans C, et al. Evolutive skin reconstructions: from the dermal collagen-glycosaminoglycan-chitosane substrate to an immunocompetent reconstructed skin. Biomed Mater Eng. 2006;16:S85–94.
Auxenfans C, Fradette J, Lequeux C, Germain L, Kinikoglu B, Bechetoille N, et al. Evolution of three dimensional skin equivalent models reconstructed in vitro by tissue engineering. Eur J Dermatol. 2009;19:107–13.
Randall MJ, Jüngel A, Rimann M, Wuertz-Kozak K. Advances in the biofabrication of 3D skin in vitro: healthy and pathological models. Front Bioeng Biotechnol. 2018;6:154.
Tarassoli SP, Jessop ZM, Al-Sabah A, Gao N, Whitaker S, Doak S, et al. Skin tissue engineering using 3D bioprinting: an evolving research field. J Plastic, Reconstructive Aesthetic Surg. 2018;71:615–23.
Hocquart M, Lagier J-C, Cassir N, Saidani N, Eldin C, Kerbaj J, et al. Early fecal microbiota transplantation improves survival in severe Clostridium difficile infections. Clin Infect Dis. 2018;66:645–50.
Hocquart M, Pham T, Kuete E, Tomei E, Lagier JC, Raoult D. Successful fecal microbiota transplantation in a patient suffering from irritable bowel syndrome and recurrent urinary tract infections. Open Forum Infect Dis. 2019;6:ofz398.
Myles IA, Earland NJ, Anderson ED, Moore IN, Kieh MD, Williams KW, et al. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight. 2018;3:e120608.
Paetzold B, Willis JR, Pereira de Lima J, Knödlseder N, Brüggemann H, Quist SR, et al. Skin microbiome modulation induced by probiotic solutions. Microbiome. 2019;7:95.
Perin B, Addetia A, Qin X. Transfer of skin microbiota between two dissimilar autologous microenvironments: a pilot study. Blumenberg M, editor. PLoS ONE. 2019;14:e0226857.
Callewaert C, Lambert J, Van de Wiele T. Towards a bacterial treatment for armpit malodour. Exp Dermatol. 2017;26:388–91.
Som AM, Ahmat N, Abdul Hamid HA, Azizuddin N. A comparative study on foliage and peels of Hylocereus undatus (white dragon fruit) regarding their antioxidant activity and phenolic content. Heliyon. 2019;5:e01244.
Banerjee K, Thiagarajan N, Thiagarajan P. Formulation and characterization of a Helianthus annuus - alkyl polyglucoside emulsion cream for topical applications. J Cosmet Dermatol. 2019;18:628–37.
Gervason S, Napoli M, Dreux-Zhiga A, Lazzarelli C, Garcier S, Alexandre B, et al. Attenuation of negative effects of senescence in human skin using an extract from Shingomonas hydrophobicum: development of new skin care solution. Int J Cosmet Sci. 2019;41(4):391–7. https://doi.org/10.1111/ics.12534. Epub 2019 Jun 17.
Goderska K. The antioxidant and prebiotic properties of lactobionic acid. Appl Microbiol Biotechnol. 2019;103:3737–51.
Michalek IM, Lelen-Kaminska K, Caetano dos Santos FL. Peptides stimulating synthesis of extracellular matrix used in anti-ageing cosmetics: are they clinically tested? A systematic review of the literature. Australas J Dermatol. 2019;60. [cited 2020 Feb 6]. https://doi.org/10.1111/ajd.13036.
Khmaladze I, Butler É, Fabre S, Gillbro JM. Lactobacillus reuteri DSM 17938—a comparative study on the effect of probiotics and lysates on human skin. Exp Dermatol. 2019;28:822–8.
Im A, Lee B, Kang D, Chae S. Protective effects of tyndallized Lactobacillus acidophilus IDCC 3302 against UVB-induced photodamage to epidermal keratinocytes cells. Int J Mol Med. 2019; [cited 2020 Feb 6]. https://doi.org/10.3892/ijmm.2019.4161.
Lee DE, Huh C-S, Ra J, Choi I-D, Jeong J-W, Kim S-H, et al. Clinical evidence of effects of Lactobacillus plantarum HY7714 on skin aging: a randomized, double blind, placebo-controlled study. J Microbiol Biotechnol. 2015;25:2160–8.
Nam B, Kim SA, Nam W, Jeung WH, Park S-D, Lee J-L, et al. Lactobacillus plantarum HY7714 restores TNF-α induced defects on tight junctions. Prev Nutr Food Sci. 2019;24:64–9.
Ong JS, Taylor TD, Yong CC, Khoo BY, Sasidharan S, Choi SB, et al. Lactobacillus plantarum USM8613 aids in wound healing and suppresses Staphylococcus aureus infection at wound sites. Probiotics & Antimicro Prot. 2019 [cited 2020 Feb 6]. https://doi.org/10.1007/s12602-018-9505-9.
Rong J, Liu S, Hu C, Jin F, Wang L. Oral intake of Lactobacillus helveticus NS8 alleviates ovalbumin-induced atopic dermatitis in SKH-1 hairless mice. Indian J Microbiol. 2018;58:312–8.
Shin D, Lee Y, Huang Y-H, Lim H-W, Jang K, Kim D-D, et al. Probiotic fermentation augments the skin anti-photoaging properties of Agastache rugosa through up-regulating antioxidant components in UV-B-irradiated HaCaT keratinocytes. BMC Complement Altern Med. 2018;18:196.
Satoh T, Murata M, Iwabuchi N, Odamaki T, Wakabayashi H, Yamauchi K, et al. Effect of Bifidobacterium breve B-3 on skin photoaging induced by chronic UV irradiation in mice. Beneficial Microbes. 2015;6:497–504.
Lee NY, Ibrahim O, Khetarpal S, Gaber M, Jamas S, Gryllos I, et al. Dermal microflora restoration with ammonia-oxidizing bacteria Nitrosomonas eutropha in the treatment of keratosis pilaris: a randomized clinical trial. J Drugs Dermatol. 2018;17:285–8.
Notay M, Saric-Bosanac S, Vaughn AR, Dhaliwal S, Trivedi M, Reiter PN, et al. The use of topical Nitrosomonas eutropha for cosmetic improvement of facial wrinkles. J Cosmet Dermatol. 2020;19(3):689–93. https://doi.org/10.1111/jocd.13060. Epub 2019 Jul 1.
Acknowledgements
Not applicable.
Funding
This work was supported by the AMU and M&L Laboratories collaboration contract N° PVM: 2018-200, by the Agence Nationale de la Recherche (ANR), including the “Programme d’investissement d’avenir” under the reference Méditerranée Infection 10-1AHU-03 and European funding FEDER PRIMI.
Author information
Authors and Affiliations
Contributions
MB and VC analyzed the bibliography and wrote and edited the original manuscript. NC wrote, supervised, and edited the manuscript. BLS conceptualized, supervised, and edited the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
M Boxberger received a PhD grant supported by the L’Occitane Society, V Cenizo is employed by L’Occitane Society. All other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Boxberger, M., Cenizo, V., Cassir, N. et al. Challenges in exploring and manipulating the human skin microbiome. Microbiome 9, 125 (2021). https://doi.org/10.1186/s40168-021-01062-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01062-5