
Abstract
Small cell lung cancer (SCLC) is a recalcitrant cancer characterized by early metastasis, rapid tumor growth and poor prognosis. In recent decades, the epidemiology, initiation and mutation characteristics of SCLC, as well as abnormal signaling pathways contributing to its progression, have been widely studied. Despite extensive investigation, fewer drugs have been approved for SCLC. Recent advancements in multi-omics studies have revealed diverse classifications of SCLC that are featured by distinct characteristics and therapeutic vulnerabilities. With the accumulation of SCLC samples, different subtypes of SCLC and specific treatments for these subtypes were further explored. The identification of different molecular subtypes has opened up novel avenues for the treatment of SCLC; however, the inconsistent and uncertain classification of SCLC has hindered the translation from basic research to clinical applications. Therefore, a comprehensives review is essential to conclude these emerging subtypes and related drugs targeting specific therapeutic vulnerabilities within abnormal signaling pathways. In this current review, we summarized the epidemiology, risk factors, mutation characteristics of and classification, related molecular pathways and treatments for SCLC. We hope that this review will facilitate the translation of molecular subtyping of SCLC from theory to clinical application.
Similar content being viewed by others
Introduction
Small cell lung cancer (SCLC) accounts for approximately 11% of all lung cancer cases [1]. Tobacco exposure is strongly associated with the incidence of SCLC. SCLC is considered as a recalcitrant tumor because of its early metastasis, rapid tumor growth and poor prognosis. Approximately 70% of patients have distant metastasis when they are first diagnosed with SCLC. Since the 1980s, a minority of very early-stage patients have received surgery as initial treatment and adjuvant platinum-based chemotherapy as follow-up treatment; most patients with advanced stage SCLC have received concurrent radiation and platinum-based chemotherapy [2]. Regardless of whether the patients had metastasis, the initial response rate to concurrent chemoradiotherapy was satisfactory. However, the response is short-lived, and the median survival time is less than one year [3]. Over the past decade, numerous clinical trials of combination immunotherapies have been conducted. Some of them have shown potential antitumor activity [4], and atezolizumab was recommended as a first-line treatment in 2019 [5]; however, the efficacy of immunotherapy still needs to be improved.
In the past few years, the prognosis of patients with non-small cell lung cancer (NSCLC) has improved significantly, mainly because of genotype-directed targeted therapies against tumor-specific somatic mutant forms [6]. In contrast, the identification of targetable driver mutations in SCLC lags behind that of other tumors, and SCLC has always been treated as a single disease entity. There have been no revolutionary new treatments for decades. SCLC has frequent copy number alterations (CNAs) and high tumor mutation burdens (TMBs). Genome sequencing and animal models have revealed more mutation hallmarks and related molecular pathways of SCLC. Some research groups have attempted to characterize the biologically distinct subtypes of SCLC based on the differential expression of transcriptional regulators. The molecular subtype classification is continuously being updated for the study of disease progression. A recognized subtype classification is urgently needed to develop more effective and personalized approaches for treatment.
In this review, we present the current trends regarding the epidemiology of, risk factors for, mutation characteristics of and treatments for SCLC. To explore the new directions of therapeutic research, we also highlight the latest discoveries in subtypes and related molecular pathways.
Epidemiology
Prevalence and prognosis
According to a recent population-based study, the annual number of new lung cancer cases worldwide is approximately 2.2 million, with SCLC accounting for 11% (232,000 cases) of these cases [1]. Previous research estimated that SCLC leads to 250,000 deaths worldwide each year [7]. The incidence of SCLC exhibits notable regional and sex disparities. In terms of regional prevalence trends, the incidence of SCLC exhibits a declining pattern in developed areas, while it exhibits a gradually increasing trend in underdeveloped areas [8]. In 2012, the highest incidence of SCLC among males was observed in Central and Eastern Europe as well as East Asia; over the same period, the highest incidence among females was observed in North America and Northern Europe [9]. A higher rate of smoking leads to a higher incidence of SCLC among males. This disparity is gradually diminishing toward equality, reflecting the declining rate of smoking among males and additional exposure to nonsmoking factors in females [10]. Variances in lung cancer incidence by country and sex are consistent with smoking trends, but there is a lag of 3–4 decades [9]. In recent decades, the implementation of smoking cessation initiatives has led to a gradual reduction in the relative SCLC incidence rate worldwide [11]. However, the disease burden caused by SCLC is expected to increase concomitantly with the increasing incidence of lung cancer in the future [12].
Owing to the rapid tumor growth and early metastasis of SCLC, the prognosis of SCLC is more unfavorable than that of other subtypes of lung cancer, with a median survival period of less than one year [3]. Thus, investigation of the orienting metastatic features of SCLC will be valuable. The most common sites of SCLC metastasis include brain, bone, adrenal glands, liver and contralateral lung. Recently, Chan et al. found that PLCG2 was significantly overexpressed in the metastatic sites. Among the metastatic sties, the liver and lymph node metastasis, the most common of metastatic site, had the highest level of PLCG2 [13]. Na et al. discovered that KMT2C was the frequently mutated gene in both primary tumor and metastatic samples. Subsequent in vivo and in vitro experiments further revealed that KMT2C-DNMT3A-MEIS/HOX axis was responsible for the metastasis in liver, lymph node, and other organs [14]. Collectively, these results offer a novel insight of SCLC metastasis and may be useful for the medical development.
Risk factors
The occurrence of SCLC is closely linked to tobacco exposure both biologically and epidemiologically. Tobacco smoke contains more than 70 confirmed carcinogens, including more than 20 directly linked to the development of lung cancer [15]. The comprehensive genomic profiles of SCLC highlighted the significance of tobacco carcinogens in the initiation of SCLC [16]. The prevalence of ever-smokers among SCLC patients is as high as 94%, surpassing that of all subtypes of lung cancer [17]. Smoking is associated with a worse prognosis in patients diagnosed with SCLC [18, 19]. A significant dose‒response relationship has been observed between smoking intensity and the risk of developing SCLC, while cessation of smoking has been shown to be correlated with a reduction in SCLC incidence [20]. Moreover, secondhand smoke is a strong risk factor for SCLC. Individuals exposed to secondhand smoke are more susceptible to developing SCLC than nonsmokers [21]. Previous studies have shown that secondhand smoke is more strongly associated with SCLC than with other histological types of lung cancer [22].
The proportion of nonsmokers among SCLC patients ranges from approximately 2% to 2.5%, and a majority of them are female [23, 24]. In addition to tobacco exposure, risk factors for SCLC include residential radon, air pollution, occupational carcinogens, and hormonal and dietary factors. Among nonsmokers, residential radon is considered the leading cause of lung cancer [25]. Air pollutants that have been found to be closely associated with the incidence of SCLC mainly include nitrogen dioxide (NO2) and particulate matter 10 (PM10). Annual residential exposure to NO2 and PM10 is positively correlated with SCLC morbidity [26, 27]. Exposure to carcinogens involved in chemical industrial production processes, such as chloromethyl methyl ether, diesel engine exhaust, polycyclic aromatic hydrocarbons (PAHs), arsenic, silica asbestos, some heavy metals and their compounds, has been reported to contribute to lung cancer [28,29,30]. The intake of hormones and dietary habits also contribute to the prevention of lung cancer [31,32,33,34]. However, few studies have examined the effects of nonsmoking-related factors on SCLC alone, and the underlying mechanisms have yet to be fully elucidated. Furthermore, respiratory comorbidities such as COPD have been identified as independent risk factors for SCLC [20]. Genetic susceptibility also plays a significant role in the onset of SCLC; the details of this relationship will be further described in subsequent sections (Fig. 1).

Risk factors associated with SCLC. The initiation of SCLC was associated with different factors, especially heavy smoking
Origins of SCLC
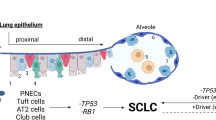
The rapid growth of SCLC often leads to advanced stage at the time of first diagnosis; therefore, few studies have exampled the cellular origins of SCLC [35]. Due to technological advancements in genome and transcriptome sequencing and the widespread access to cell-specific Cre recombinase and clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein (Cas) 9 in SCLC cell lines and genetically engineered mouse models (GEMMs), insight into the origin of SCLC has gradually increased (Fig. 2).

The cellular origin of SCLC. SCLC may originate from AT2 cells, NE cells, club cells and basal cells and transform from NSCLC cells
Given the universal inactivation of TP53 and RB1 detected in humans, these genes are considered necessary for the tumorigenesis of SCLC [36, 37]. Initial mouse models revealed that the inactivation of Rb1 and Trp53 in lung epithelial cells via Adeno-Cre virus could lead to the neuroendocrine (NE) SCLC [38]. Subsequent triple-knockout GEMMs (Rb1/Trp53/Pten, Rb1/Trp53/P130) or Rb1/Trp53 knockout GEMMs with Myc, Nfib or Fgfr1 amplification could accelerate the initiation and progression of SCLC in lung epithelial cells [38,39,40,41,42,43,44]. Studies using the cell type-restricted Adeno-Cre virus in GEMMs with Rb1 and Trp53 inactivation have shown that NE cells and alveolar type 2 (AT2) cells expressing surfactant protein C (SPC) contribute to the formation of SCLC. In contrast to the study by Mollaoglu et al. [39], Chen et al. recently reported that AT2, club and NE cells can transform into SCLC cells via the combination of Myc amplification and the inactivation of Rb1 and Trp53 in a mouse model [44]. In the context of Rb1fl/flTrp53fl/fl GEMMs with Fgfr1 amplification, SCLC can originate from tracheobronchial-basal cells expressing K14 [38,39,40,41,42,43]. Furthermore, the inhibition of NOTCH, RB1 and TP53 in human embryonic stem cells (hESCs) enables the generation of pulmonary NE cells (PNECs) and the formation of SCLC-like cells [45].
Distinct from the NE cell lineage, a variant form of SCLC with low NE features harbors markers of the tuft cell lineage and may originate from chemosensory epithelial cells of the primary and secondary bronchi [46]. Recent studies confirmed that non-NE SCLC cells could shift from the NE fate originating from PNECs via MYC/NOTCH or Yap/TAZ/NOTCH signaling [47, 48]. Additionally, some SCLCs can be derived from EGFR-, ALK-, ROS1- or RET-driven lung adenocarcinoma (LUAD) upon the acquisition of tyrosine kinase inhibitor (TKI) resistance [49,50,51,52]. Moreover, NSCLC patients with EGFR/TP53/RB1 mutations or with apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC)–induced hypermutation may be at increased risk of transformation [53, 54]. Notably, transformation to SCLC may also occur in patients with NSCLC who receive immunotherapies [55,56,57]. For combined SCLC (CSCLC), including SCLC and NSCLC components, few studies have assessed its origin. Our previous study and research of Zhao et al. revealed that the same pluripotent clone with subsequent divergent oncogenic changes may be responsible for the different components of CSCLC [58, 59].
Characterization of mutations in SCLC
Recalcitrant SCLC has been shown to exhibit extensive chromosomal aberrations and genomic alterations, which could contribute to the development and progression of SCLC with a high TMB. In a SCLC mouse model with Trp53 and Rb1 inactivation, DNA CNAs were detected on chromosomes 4, 8, 12, 14, 16 and 19 [60]. In human SCLC samples, amplification of chromosomes, including 1p, 1q, 3q, 5p, 8q, 14p, 14q, 18p and 18q, and deletions in 3p, 4p, 4q, 5q, 13q, 15q, 16q, 17p and 17q were detected in parallel to previous studies [16, 37]. Further multi-omics analysis revealed trans-associations on chromosome 5q, which are involved in DNA repair, replication and cell cycle progression [61].
Through comprehensive genomic analyses, SCLC was characterized by bi-allelic inactivation of Tp53 and Rb1 [16, 38]. Whole-genome sequencing (WGS) of 110 treatment-naive SCLC tumors revealed that the prevalence rates of genomic mutation in TP53 and RB1 were 94% and 78%, respectively [16]. Similarly, of the 3600 “real-world” SCLC patients, 92 and 74% had genomic mutations in TP53 and RB1, respectively [62]. However, our multi-omics analysis revealed lower rates of TP53 and RB1 mutations in some SCLC patients (Table 1). The low rates of TP53 and RB1 mutations may be associated with different ancestry [62]. Moreover, other mechanisms may be responsible for TP53 and RB1 inactivation: (1) higher DNA deletions in TP53 and RB1 [61]; (2) chromothripsis, which is a catastrophic event that could lead to the inactivation of RB1 [16]; (3) epigenetic alterations that may result in the inactivation of TP53 and RB1 [63, 64]; and (4) human viruses (such as human papillomaviruses, HPV) that can functionally inactivate TP53 and RB1 [62].
In addition to the TP53 and RB1 mutations, alterations in other genes, including MYC, ZFHX3, PTEN, and NOTCH family genes as well as KMT2D, CREBBP and EP300, were detected. MYC family genes, including MYC, MYCL and MYCN, are mutually exclusively expressed and exhibit approximately 20% amplification in SCLC [48]. In the lungs of GEMMs with inactivation of Trp53 and Rb1, MYC amplification has been shown to promote the formation of SCLC, which is characterized by high aggression and metastasis and poor survival [39], thus leading to the dynamic evolution of SCLC [39, 48].
ZFHX3, a transcription factor (TF) with four homeodomains, 23 zinc finger domains and other motifs, has been suggested to be a tumor suppressor for different cancers [65]. In metastatic tumors, frequent mutations in ZFHX3 can be detected [66, 67]. In our recent study, we found that the rate of ZFHX3 mutation was 19% in Chinese patients, which is higher than that in a previous study, and that ZFHX3 mutation could serve as a biomarker for immunotherapy response in SCLC patients [61].
NOTCH family genes were recurrently mutated with a pattern of frequent inactivation. Mutation of the NOTCH signaling pathway occurs in approximately 25% of human SCLC cases [16]. Activation of the NOTCH signaling pathway inhibited tumor growth and increased survival. Moreover, the active NOTCH signaling pathway can increase the expression of antigen processing and presentation machinery (APM) genes in SCLC [68]. PTEN is an oncogenic phosphatidylinositol 3 kinase (PI3K) inhibitor that is lost in SCLC. A significant reduction in tumor latency and overall survival (OS) can be observed in Trp53/Rb1/PTEN triple-knockout (TKO) GEMMs [60]. Approximately 8% of SCLC have KMT2D mutations, including truncating nonsense/frameshift/splice site mutations [69]. KMT2D deletion has been reported to lead to significant defects in cell type-specific gene expression and cell differentiation [70]. Additionally, deletions and truncating mutations of CREBBP and EP300 in the histone acetyltransferase (HAT) domain are frequently found in SCLC [71].
Evolutionary dynamics of SCLC subtypes
From a clinical perspective, SCLC is considered as a single disease entity, which may explain the failure of different drugs that have been examined for its treatment. By integrating data on human tumors, cell lines and different mouse models, distinct subtypes of SCLC have been characterized, and precise subtype-specific treatments have been proposed [72]. In the current section, investigations of SCLC classification will be reviewed.
Histologically, SCLC is first dichotomized into classic and variant subtypes on the basis of morphology and growth characteristics [73]. Further investigation has revealed the heterogeneity of SCLC based on NE features, which is similar to the above findings. The low-NE subtype has morphological features of the variant subgroup and grows in a loose aggregated or single form, unlike the high-NE subtype [74].
From a genomic perspective, comprehensive whole-exome or whole-genome sequencing of SCLC has demonstrated only universal mutations in TP53 and RB1. Moreover, unlike LUAD, no genetic subtypes of SCLC and no revolutionized specific therapeutic vulnerabilities have been identified [16, 36, 37, 75]. With the accumulation of human SCLC tumors, a “real-world” study identified three potential genetic subtypes: a cohort without TP53/RB1 alteration, a cohort with STK11 mutation, and a cohort that may transform from NSCLC with typical oncogenic driver mutations [62].
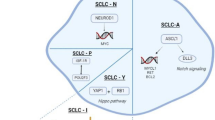
Given the integrated analysis of human and mouse model data, unexpected molecular classification of SCLC has been proposed. The initial exploration of the selective susceptibility of Seneca Valley virus (SVV-001) to various subtypes of SCLC revealed that two TFs, Achaete-scute homolog 1 (ASCL1) and neurogenic differentiation factor 1 (NEUROD1), play key roles in NE differentiation [76]. Subsequent in vivo and in vitro experiments further confirmed that ASCL1 and NEUROD1 drive SCLC subtypes [39, 48, 77]. Unsupervised clustering analysis of a large panel of SCLC cell lines revealed that insulinoma-associated protein 1 (INSM1), an NE TF, and yes-associated protein 1 (YAP1), a vital mediator activated in the Hippo signaling pathway, may define two subtypes of SCLC [78]. Subtypes with high YAP1 expression displayed low levels of ASCL1 and NEUROD1. Conversely, variable levels of ASCL1 and NEUROD1 can be detected in subtypes with higher INSM1 expression [78]. CRISPR screening of SCLC cell lines revealed that POU class 2 homeobox 3 (POU2F3), a powerful TF, was expressed exclusively and necessary in the variant subtype of SCLC lacking NE marker expression [46]. Thereafter, synthesized analysis of both SCLC cell lines and tumor RNA data further suggested that SCLC can be definitively distinguished by the TFs ASCL1, NEUROD1, YAP1 and POU2F3 [72]. The activation of NOTCH signaling pathways by MYC in ASCL1 subtypes can drive the activation of the NEUROD1 and YAP1 subtypes in an orderly manner, indicating that no distinct subtypes instead of different stages of dynamic evolution may exist in SCLC [48]. When exploring the expression of the four TFs at the protein level, their expression profiles tended to be more heterogeneous [79]. POU2F3, which was uniquely expressed in 7% of SCLC patients, showed mutually exclusive associations with ASCL1 and NEUROD1. However, low levels of YAP1 coexisted with other subtypes. For the NE subtype of SCLC, the coexpression ratio of ASCL1 and NEUROD1 was more prevalent than that of ASCL1-positive or NEUROD1-positive SCLC [79]. To better refine the subtypes of SCLC, RNA sequencing data from surgically resected SCLC (n = 81) and the IMpower 133 clinical trial (n = 276) revealed four SCLC subtypes: the ASCL1, NEUROD1, POU2F3 and Inflamed subtypes. Notably, the Inflamed subtype had low levels of the three TFs and can strikingly benefit from immune checkpoint inhibitors (ICIs) [80]. A subsequent study demonstrated that these subtypes can be identified by tumor- and circulation-free DNA methylation [81]. In contrast to the above classification, four different subtypes were discerned by de novo non-negative matrix factorization (NMF) using the IMpower 133 data [82]. Robustly high levels of NEUROD1 and ASCL1 can be detected in the NMF1 and NMF2 subtypes, respectively. However, inflamed features can be found in both NMF3 and NMF4. In contrast to NMF3, NMF4 had unique POU2F3 expression and non-NE features. In addition, patients with the NMF3 subtypes with NE features and low levels of T-effector-high/tumor-associated macrophages (TAMs) can benefit from immunotherapy [82] (Fig. 3).

Therapeutic vulnerabilities and emerging subtypes of SCLC. A schematic summarizing the proposed potential therapeutic targets and emerging molecular subtypes of SCLC is shown. On the left side of the diagram, potential therapeutic targets, including those involved in the cell cycle and DNA damage and repair pathway, epigenetics pathway, metabolism pathway, NOTCH pathway, apoptosis pathway and tumor immunity, are displayed. (The activation effect is denoted by an arrow, while the inhibition effect is represented by a vertical bar). On the right side, the evolution of SCLC subtypes is shown in chronological order [46, 61, 72, 73, 76, 78, 80, 82]. Abbreviations are shown below. Achaete-scute homolog 1, ASCL1; neurogenic differentiation factor 1, NEUROD1; POU class 2 homeobox 3, POU2F3; yes-associated protein 1, YAP1; insulinoma-associated protein 1, INSM1; neuroendocrine, NE
Emerging evidence substantiates that multi-omics analysis of tumors can offer a better understanding of disease and may provide a more specific therapeutic regimen [83,84,85]. Recently, our team has demonstrated four novel subtypes of SCLC by synthesizing multi-omics data, including mRNA, protein and phosphorylation data [61]. The nmf1 subtype is characterized by cell cycle progression and DNA damage, suggesting that this subtype has a high proliferation rate. The nmf2 subtype, which has a lower amount of multi-omics data, exhibits the highest TMB and highest expression of delta-like ligand-3 (delta-like ligand 3, DLL3). The nmf3 subtype, which is enriched in the extracellular matrix and focal adhesion pathway at the protein level, exhibits robust receptor tyrosine kinase (RTK) signaling pathway activity according to the phosphorylation data. Uniquely, the nmf4 subtype is mainly associated with RNA metabolism pathways and a high number of MYC targets.
To explore the association between multi-omics subtypes and previous subtypes, the TU-SCLC cohort was grouped into four subtypes using the NMF-defined gene list reported by Gay et al. [80]. The correlation between multi-omics and the established subtypes in the TU cohort was significant (P = 1.71E-20, Fisher’s exact test). Additionally, the nmf1 subtypes with half ASCL1-driven and half NEUROD1-driven tumors had higher levels of ASCL1 and NEUROD1; the nmf2 subtype had the highest number of ASCL1-driven tumors coupled with higher levels of DLL3; and the nmf3 subtype with a non-NE phenotype had mostly inflamed tumors. The nmf4 subtype with lower ASCL1 and NEUROD1 expression included all POU2F3-driven tumors except for one with high YAP1 expression.
Apart from the above subtypes of SCLC, other classifications have also been proposed, which may be useful for identifying potential distinct therapeutic vulnerabilities. Relying on the DNA methylation and RNA-seq data of primary SCLC, three distinct subtypes (M1, M2 and SQ-P for the methylation cluster and E1, E2 and SQ-P for the gene expression cluster) were identified by Poirier et al. [86]. High levels of NEUROD1 and low levels of ASCL1 were observed in the E1 cluster. In contrast, the opposite results were detected in the E2 cluster. For the SQ-P cluster, similar gene expression profiles were discerned compared with those of lung squamous cell carcinomas, which lacked NE markers. Intriguingly, unsupervised hierarchical clustering of RNA-seq data revealed two groups [16]. Group 2 represented the majority of SCLC patients and exhibited high levels of CHGA, GRP, ASCL1 and DLK1. Furthermore, Wooten et al. defined four subtypes of SCLC via systems-level analyses of RNA-seq data from SCLC cell lines, human tumors and patient-derived tumor xenograft (PDX)/cell line-derived xenograft (CDX) mice [87]. Under these conditions, a canonical NE subtype referred to as the ASCL1 group, an ASCL1 + NE variant assigned as the NEv2 group, or SCLC-A2, an NE variant subtype termed the NEv1 group aligning with the NEUROD1 group, and a non-NE variant subtype termed the YAP1 group, were identified. Unbiased hierarchical clustering of SCLC CDX RNA-seq data revealed four subtypes, namely, ASCL1, NEUROD1, POU2F3 and the TF ATOH1, which are important for neuronal differentiation [88]. Recently, another four clusters of SCLC were classified by Chen et al. [89]. Cluster 1 characterized by low levels of ASCL1 and NEUROD1 and high levels of POU2F3 and NOTCH2, was associated with immune-related features termed the immune subtype. Clusters 2 and 3 were parallel to the ASCL1 and NEUROD1 groups, respectively. Cluster 4, which was characterized by the expression of Clara cell secretory protein (CCSP), may originate from club cells and was therefore defined as the SCLC-C group.
Different from bulk RNA sequencing used for classification, single-cell RNA sequencing (scRNA-seq) can better address the heterogeneity of SCLC. In contrast to LUAD, the inter-patient and intra-tumor heterogeneity of SCLC malignant cells was much higher. Based on the distinct expression patterns of canonical TFs, Chan et al. discovered the most likely classifications of each cell, and identified the major subclone of each tissue as ASCL1, NEUROD1, POU2F3, except YAP1 [13]. Similarly, our study concerning the analysis of our scRNA-seq in metastatic SCLC also identified the different ASCL1 and NEUROD1 expression patterns, which further uncovered the diversity of inter-patient and intra-tumor heterogeneity [90].
Taken together, similarities have been detected among these classifications of SCLC. However, uniform and rigorous consensus clustering of SCLC patients, which may overcome the lack of therapeutic vulnerabilities, is still under debate, and further analysis is needed.
Precise therapeutic vulnerabilities of SCLC subtypes
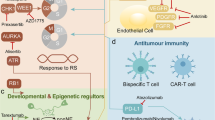
Subtype-specific molecular characterization and alterations in key signaling pathways provide a basis for exploring specific therapeutic strategies (Fig. 4). Three potential genetic subtypes identified in the “real-world” study showed that the cohort without TP53/RB1 alteration may benefit from targeting the virus or regaining the function of TP53; the cohort with STK11 mutation may exclude the efficacy of ICIs, and the cohort transformed from NSCLC with typical oncogenic driver mutations may avoid transformation with better treatment [62]. Apart from the above genetic subtypes, our recent study revealed that patients with the ZFHX3 mutation subtype of SCLC may benefit from ICIs [61]. Unexpectedly, Aurora kinase A (AURKA) inhibitors have been shown to have a durable effect on SCLC with RB1 loss of function [91].

Therapeutic vulnerabilities of specific SCLC clusters. The SCLC clusters from the perspectives of genomics, transcriptomics and multi-omics are shown, and their potential available therapeutic vulnerabilities are discussed
The heterogeneity of SCLC subtypes driven by TFs has strengthened the exploration of precise therapeutic options. Chromatin immunoprecipitation sequencing (ChIP-seq) revealed the unique downstream targets of ASCL1 and NEUROD1, providing the initial insights into the specific treatment involved [77]. The functional oncogenes targeted by ASCL1 in SCLC include BCL2, NFIB, SOX2, RET, MYCL1, and DLL3. In vivo and in vitro experiments confirmed that a BCL2 inhibitor can induce the apoptosis of SCLC cell lines with high BCL2 levels, indicating that a BCL2 inhibitor may be effective for treating the ASCL1 subtype of SCLC [92]. Similarly, DLL3, an inhibitory ligand of the NOTCH pathway, may be a prospective target for the ASCL1 subtype, despite failure of the DLL3-targeted drug (rovalpituzumab tesirine, Rova-T) in SCLC, which may be closely associated with the undistinguished population [93, 94]. Additionally, CREBBP, which encodes a histone acetyltransferase, was found to have frequent genetic mutations in SCLC [16, 61]. The loss of CREBBP in the Rb1/Trp53 GEMM (mainly belonging to the ASCL1 subtype) drastically contributed to the progression of SCLC, which can show an exceptional response to the histone deacetylase (HDAC) inhibitor pracinostat [71]. Suppression of the histone demethylase lysine-specific demethylase 1 (LSD1) with the selective inhibitor ORY-1001 can reactivate the NOTCH pathway and attenuate the level of ASCL1, suggesting the potential therapeutic potential of LSD inhibitors in the ASCL1 subtype [95]. Within the ASCL1 subtype, a bimodal distribution of Schlafen family member 11 (SLFN11) was detected [95]. The critical response to cisplatin and the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib can be confirmed in the ASCL1 subtype, which has a high level of SLFN11 [77, 80]. Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), another highly expressed target gene in the ASCL1 subtype of SCLC, can be targeted by labetuzumab govitecan [80].
The level of MYC, a transcriptional target of NEUROD1, was amplified or overexpressed in ASCL1-low SCLC subtypes and correlated with poor prognosis and treatment resistance [39]. A targeted drug screening experiment demonstrated that SCLC patients with high MYC levels were more vulnerable to treatment with an Aurora kinase A/B (AURKA/AURKB) inhibitor, which indicated that patients with the NEUROD1 subtype may benefit from Aurora kinase inhibitors [39, 96]. In addition, inosine monophosphate dehydrogenase-1 and -2 (IMPDH1 and IMPDH2), which are selectively expressed in ASCL1-low SCLC, are downstream targets of MYC. The use of an IMPDH inhibitor can better suppress SCLC growth [97]. Similarly, MYC-driven SCLC is vulnerable to arginine deprivation and mTOR inhibition in combination with a checkpoint kinase 1 (CHK1) inhibitor [98, 99]. Taken together, these findings indicate that these targeted inhibitors may be potential therapeutic candidates for treating NEUROD1 and other ASCL1-low subtypes in which MYC is overexpressed. High levels of somatostatin receptor 2 (SSTR2), which is a well-established target of somatostatin analogs, were detected in the NEUROD1 subtype.
The POU2F3 and Inflamed subtypes, which are rare subsets of SCLC, have unique therapeutic vulnerabilities. CRISPR screening revealed the essential role of activating the insulin-like growth factor 1 receptor (IGF1R) signaling pathway in the POU2F3 subtype. The IGF1R inhibitor linsitinib may be a promising targeted drug for the POU2F3 subtype. A recent study revealed that compared with other subtypes, the POU2F3 subtype is more vulnerable to PARP inhibitors, antimetabolites containing nucleoside analogs, antifolates and mammalian switch/sucrose non-fermentable (mSWI/SNF) ATPase degrader [80, 100]. MICA, which encodes MCH class I polypeptide–related sequence A, is highly expressed in the POU2F3 and Inflamed subtypes and can be targeted by IPH43 in these two subtypes [80]. The Inflamed subtype is a novel group that has the highest immune infiltration, cytolytic activity score, highest expression of Bruton’s tyrosine kinase (BTK) and mesenchymal status in SCLC. The available evidence has shown that the Inflamed subtype may benefit from ICIs, BTK inhibitors, and HDAC inhibitors, which can restore epithelial markers [80].
In terms of the unique therapeutic vulnerabilities of multi-omics, external data were integrated with functional experiments. The nmf1 subtype associated with exceptionally high NE scores, a high cell proliferation rate, high E2F activity and replication stress may be better targeted by drugs that exacerbate genome instability. Thus, we hypothesized that E/P-based chemotherapy may be the proper treatment option for these nmf1-subtype tumors [61]. Emerging evidence has shown that concurrent inhibition of Ataxia telangiectasia and Rad3-related protein (ATR) and DNA topoisomerase I (TOP1) can result in durable tumor regression in SCLC characterized by high replication stress [101]. In contrast, DLL3-targeted drugs may be the best choice for the nmf2 subtype with the highest DLL3 level, which was confirmed in the George et al. dataset [16, 61]. The nmf3 subtype, characterized by elevated RTK signaling pathway activity, was shown to benefit from RTK inhibitors in PDX models. In contrast to the findings of a previous study, the nmf4 subtype was confirmed to have the highest MYC and POU2F3 levels via multi-omics and immunohistochemical assays. Subsequent experiments demonstrated that the nmf4 subtype may be suitable for AURKA/B inhibitors.
In conclusion, the OS rate of SCLC is low, and better treatment options are lacking. The exploration of SCLC subtypes and their unique therapeutic vulnerabilities may provide better insights, but further investigation is needed.
Targeted therapy
Systematic therapy of platinum-based chemoradiotherapy has occupied the backbone role in the management of SCLC for decades. Currently, extensive research has been conducted in the field of targeted therapy for SCLC, focusing on the abnormal signaling pathways involved in cell cycle and DNA damage and repair (DDR), epigenetic regulation, cell metabolism, and tumor immunity. Despite the exploration of numerous drugs targeting various pathways for treating SCLC, only ICIs have demonstrated satisfied efficacy. Subsequently, we will present a comprehensive review for these studies. Due to the profound significance and unique characteristics of immunotherapy, it will be discussed in detail as a separate topic. Key completed and ongoing trials targeting various pathways, including the cell cycle and DDR pathway, epigenetics, metabolism, as well as NOTCH pathway and apoptosis, are shown in Tables 2 and 3, categorized by target and agent.
Cell cycle and DNA damage and repair
The frequent loss of RB1 and TP53 in SCLC renders this tumor more vulnerable to DNA damage, thus leading to the upregulation of mediators involved in cell cycle control and the DDR pathway to maintain genomic instability and evade cell death [102]. Inhibition or loss of DDR proteins exacerbates the accumulation of DNA damage and increases the susceptibility of SCLC to various agents that cause DNA damage [103]. Therefore, DDR proteins such as PARP, ATR, CHK1 and WEE1 have been identified as potential targets for SCLC treatment.
Poly (ADP-ribose) polymerase, PARP
PARP is a prominent drug target among DDR proteins [104]. PARP inhibitors impede DNA repair and synergize with drugs that induce DNA damage. Additionally, PARP inhibition significantly upregulates PD-L1 expression and augments the antitumor effect of ICIs through the STING-mediated immune pathway [105, 106]. Thus, extensive investigations have been conducted to evaluate the addition of PARP inhibitors to standard chemoradiotherapy, immunotherapy, or other DNA damage agents for the treatment of SCLC [107]. Currently, PARP inhibitors that have been utilized in clinical trials for SCLC mainly include olaparib, talazoparib, veliparib, niraparib, fluzoparib, rucaparib, AZD5305 and RP12146.
PARP inhibitors are primarily used as subsequent therapeutic options after the initial treatment of extensive-stage SCLC (ES-SCLC). The efficacy of PARP inhibitor monotherapy as maintenance therapy is limited, resulting in relatively few studies [108,109,110,111]. The combination of temozolomide (TMZ) with the PARP inhibitors veliparib or olaparib was investigated in relapsed SCLC patients in two phase 2 trials. The results revealed that the addition of PARP inhibitors enhances the antitumor effect of TMZ, but no significant OS benefit has been observed (OS 8.2 months vs. 7.0 months, P = 0.50) [112, 113]. The efficacy of olaparib combined with durvalumab was tested in two single-arm trials involving relapsed ES-SCLC patients, but these trials did not meet their primary efficacy endpoints [114, 115]. PARP inhibitors have also been investigated as first-line treatment regimens for SCLC. In two phase 2 trials, the addition of veliparib to platinum-based frontline chemotherapy improved progression-free survival (PFS) in treatment-naïve patients with ES-SCLC [116, 117].
The combination of chemotherapy and immunotherapy has emerged as the recommended first-line treatment for ES-SCLC [4, 118]. In this setting, the addition of PARP inhibitors is currently being investigated in several ongoing clinical trials (NCT05245994, NCT04728230, and NCT04624204). Given the diverse responses observed in finished trials, it is important to identify patients who may benefit from PARP inhibitors. Focusing on patients who are sensitive to chemotherapy seems to be a promising approach (NCT03923270, NCT05162196, NCT03830918, NCT04782089, and NCT03958045). In addition, biomarkers play a significant role in predicting the response to PARP inhibitors. Increased sensitivity to PARP inhibitors is associated with increased SLFN11 expression [112, 119,120,121], decreased EMT scores and E-cadherin levels [122], decreased DNA-PKcs expression [123], increased E2F1 expression [102], decreased ATM expression [122] and the use of fluorinated [18F]-radiolabeled PARPis [124]. However, among these biomarkers, only SLFN11 has been applied for patient selection in ongoing trials (NCT05718323, NCT04334941).
In general, as valuable drugs targeting the DDR pathway, PARP inhibitors exacerbate tumor susceptibility by inhibiting DNA repair, aggravating DNA damage and enhancing tumor immunity, thereby achieving promising efficacy in first-line treatment of SCLC. In the future, guided by subtype-specific therapy, ASCL1-subtype with high levels of SLFN11 may represent a prioritized population for PARP inhibitors.
Ataxia telangiectasia and Rad3-related protein, ATR
As a crucial component of DDR proteins, ATR plays a pivotal role in sensing DNA damage and preserving genomic instability [125, 126]. Upon activation by DNA damage, ATR stops cell progression to the G2 phase through the G2/S checkpoint, thereby preventing cell apoptosis [127]. Three ATR inhibitors, berzosertib (M6620), ceralasertib (AZD6738) and elimusertib (BAY1895344), have been investigated in SCLC. The combination of ATR inhibitors with DNA TOP1 inhibitors was found to augment their antitumor efficacy and potentially improve the response to immunotherapy in SCLC characterized by low expression of the STING pathway [101, 128]. In single-arm trials, berzosertib combined with topotecan achieved good tolerance and critical clinical benefit in relapsed platinum-resistant SCLC patients [101, 129]. Recently, a two-arm phase 2 trial demonstrated that berzosertib plus topotecan did not improve PFS (HR = 0.80 [95% CI 0.46–1.41]; P = 0.44) in relapsed SCLC patients compared to topotecan alone, but it significantly prolonged OS (HR = 0.53 [95% CI 0.29–0.96]; P = 0.03) [130]. Two trials (NCT03428607, NCT02937818) investigated the combination of AZD6738 and olaparib in relapsed or refractory SCLC patients; however, neither achieved the predetermined therapeutic endpoint [108].
Ongoing trials are currently investigating the safety and efficacy of berzosertib combined with topotecan (NCT04768296), irinotecan (NCT02595931), or sacituzumab govitecan (NCT04826341) in relapsed SCLC patients. A previous study identified an ATR inhibitor as the most effective agent for potentiating lurbinectedin in SCLC [131]. Further clinical trials are underway to confirm its safety and efficacy in relapsed SCLC patients (NCT04802174). The addition of AZD6738 to immunotherapy as a second/third-line treatment (NCT04361825) or chemoimmunotherapy as a first-line treatment (NCT04699838) is under investigation in single-arm trials. The results of an epigenome-wide DNA methylation analysis suggested that sensitivity to ATR inhibitors may be correlated with genomic methylation levels and TREX1 expression [132]. To date, no biomarker-guided trial has been identified.
Overall, ATR inhibitors exert anti-tumor effects by promoting apoptosis induced by DNA damage and enhancing tumor immunity. The combination of ATR inhibitors with topotecan, lurbinectedin or immunotherapy has potential advantages in relapsed SCLC. However, further evidence is needed to better understand the role of ATR inhibitors in subtype-specific therapy.
Checkpoint kinase 1, CHK1
CHK1 is a serine/threonine protein kinase involved in DNA damage-induced cell cycle arrest and is considered as a potential therapeutic target for SCLC [133,134,135]. Currently, the CHK1 inhibitors utilized in the treatment of SCLC patients include prexasertib (LY2606368) and SRA-737. In both SCLC cells and mouse models, promising antitumor efficacy has been achieved by CHK1 inhibitor monotherapy or in combination with chemotherapy or PARP inhibition. These findings highlight the potential of CHK1 inhibitors to overcome resistance to chemotherapy or PARP inhibitors [99, 136,137,138]. In addition, CHK1 inhibition activates the function of cytotoxic T lymphocytes via the innate immune STING pathway and enhances the antitumor effect of ICIs [105].
The recommended dose of LY2606368 monotherapy was established in a phase 1 trial, but a subsequent phase 2 trial failed to achieve the anticipated efficacy in platinum-resistant ES-SCLC patients [139, 140]. Another oral CHK1 inhibitor, i.e., SRA-737, was tested in a phase 1/2 trial. The combination of SRA-737 and low-dose gemcitabine resulted in a partial response rate of 11.1% (1/9) in SCLC patients [141]. Furthermore, the addition of SRA737 and low-dose gemcitabine enhances the antitumor efficacy of PD-L1 blockade, highlighting a potential triple combination therapy [142]. The inhibition of WEE1 reversed LY2606368 resistance in SCLC cell lines, thereby providing evidence for the synergistic potential of CHK1 and WEE1 inhibitors [143].
To sum up, CHK1 inhibitors impede DNA damage repair, resulting in the formation of replication barriers and induction of apoptosis in cancer cells. However, the efficacy of CHK1 inhibitors for SCLC remains unsatisfactory. The overexpression of MYC has been identified as a candidate biomarker for CHK1 inhibitors [99, 144]. Thus, despite suboptimal performance, CHK1 inhibitors may be applicable to NEUROD1 and other ASCL1-low subtypes characterized by MYC overexpression.
WEE1
WEE1 is a protein tyrosine kinase that inactivates cyclin-dependent kinase (CDK) 1/2 in the cell cycle and protects against DNA replication through the regulation of histone synthesis and epigenetic modification [145,146,147]. The combination of a WEE1 inhibitor and a PARP inhibitor exhibits promising antitumor efficacy within circulating tumor cell (CTC)-derived explant SCLC models [148, 149]. Inhibition of WEE1 promotes the immune response via the STING-TBK1-IRF3 pathway, enhances the antitumor effect of PD-L1 antibodies through the STAT1 pathway, and significantly suppresses tumor progression in SCLC models (including MYC-stabilized SCLC) [150].
Adavosertib (AZD1775) is an oral WEE1 inhibitor that has been tested in several advanced solid tumors. In a three-arm trial (NCT02937818), the efficacy of AZD1775 in combination with carboplatin was evaluated in platinum refractory ES-SCLC patients, with a median OS of 4.67 months, indicating the potential efficacy in SCLC. An ongoing trial is investigating the safety and efficacy of the novel WEE1 inhibitor Debio-0123 in combination with etoposide and carboplatin in patients with relapsed SCLC (NCT05815160).
In summary, WEE1 does not appear to be a worthy target in the treatment of SCLC, as the efficacy of WEE1 inhibitors is quite limited and its potential has yet to be fully explored in subtype-specific therapy.
Aurora kinase A/B, AURKA/B
The aurora kinase family is classified as serine/threonine kinases that play crucial roles in regulating the G2/M transition and spindle assembly checkpoint during the cell cycle [151]. RB1 gene mutations and MYC overexpression or amplification frequently occur in SCLC, leading to high sensitivity to AURKA/B inhibitors [39, 91, 144, 152,153,154]. A recent study demonstrated the potential of AURKA/B inhibitors to augment the antitumor efficacy of PD-L1 blockade via the restoration of inflammatory gene expression [155]. Currently, three AURKA inhibitors, namely, alisertib (MLN8237), JAB-2485, and erbumine (LY3295668), are used to treat SCLC.
A multicenter phase 1/2 trial investigated the efficacy of the oral AURKA inhibitor alisertib as monotherapy in advanced tumors. Among 48 relapsed SCLC patients, 10 (20.8%) achieved an objective response, and a median FPS of 2.1 months was observed [156]. In a separate phase 1 trial, alisertib was combined with nab-paclitaxel, resulting in a partial response in 1 out of 5 refractory SCLC patients [157]. In a phase 2 trial, the combination of alisertib and paclitaxel as a second-line treatment demonstrated superior PFS benefits for patients with relapsed SCLC expressing MYC compared to monotherapy with paclitaxel (PFS 4.6 months vs. 2.3 months; HR = 0.29, 95% CI 0.12–0.72) [158]. An ongoing phase 2 trial (NCT06095505) is evaluating the safety and efficacy of alisertib in progressed ES-SCLC patients who are receiving or have completed first-line treatment with chemotherapy combined with anti-PD-L1 immunotherapy. Further ongoing trial will determine the role of AURKA inhibitor.
In conclusion, AURKA inhibitors present promising antitumor efficacy through the inhibition of MYC. AURKA may be considered as a potential therapeutic vulnerability for NEUROD1, POU2F3 and Inflamed subtypes based on the transcriptional classification, and nmf4 subtype based on the multi-omics classification that exhibit overexpression of MYC in subtype-specific therapy.
Cyclin-dependent kinase 4/6, CDK4/6
The aberrant activation of CDK4/6 results in excessive phosphorylation of the Rb protein, leading to dysregulation of the G1/S transition and promoting tumorigenesis [159,160,161]. The antitumor effect of CDK4/6 inhibitors has not been confirmed in SCLC, but they have shown strong protective effects against chemotherapy-induced myelosuppression (CIM) [162]. The transient and reversible arrest of hematopoietic stem and progenitor cells (HSPCs) in the G1 phase is achieved through the inhibition of CDK4/6, thereby providing protection against cytotoxic injury induced by chemotherapy [163]. Several clinical trials have demonstrated the protective effects of the CDK4/6 inhibitor trilaciclib in CIM [164,165,166]. Thus, the Food and Drug Administration (FDA) has granted approval to trilaciclib for alleviating CIM in ES-SCLC patients [167]. Additionally, the incorporation of trilaciclib into chemotherapy plus ICIs effectively preserved immune system function and augmented the antitumor response in preclinical models [168].
An ongoing phase 4 trial is evaluating progression and survival in ES-SCLC patients when trilaciclib is added to topotecan-containing chemotherapy (NCT05874401). The effect of trilaciclib in combination with lurbinectedin is currently being investigated in a phase 2 trial (NCT05578326). The efficacy and safety of abemaciclib, a novel CDK4/6 inhibitor, are under evaluation in Rb wild-type refractory ES-SCLC (NCT04010357). All in all, although CDK4/6 inhibitors do not specifically target any subtypes, they hold significant and extensive implications as protective agents to alleviate the adverse effects of chemotherapy.
Epigenetics
The accumulation of genomic structural and functional changes is widely recognized as a primary force driving cancer development. Epigenetic modification represents one such mechanism. Epigenetic modifications are characterized by heritable changes in gene activity that occur without altering the DNA sequence. Abnormal modifications regulate gene expression patterns that promote tumorigenesis and facilitate the acquisition of hallmark tumor capabilities [169]. The two primary types of epigenetic modifications are DNA methylation and histone modifications. Clinicians are leveraging these events as adjunctive tools in clinical decision-making. Furthermore, the reversibility of epigenetic modifications has led to the emergence of epigenetic therapy as a promising strategy for treating SCLC [170, 171].
Lysine-specific demethylase 1, LSD1
As a form of histone modification, histone methylation represents one of the critical epigenetic hallmarks of SCLC. Encoded by KDM1A, LSD1 functions as a histone demethylase that selectively removes monomethylated and dimethylated groups from histone H3K4 and H3K9 sites, thereby influencing gene transcription [172]. Through interaction with SNAG domain-containing proteins, namely, INSM1 and GFI1B, LSD1 facilitates the transcriptional activation of genes associated with NE phenotypes and augments the proliferation of SCLC cells. Perturbation of this interaction attenuates the expression of pivotal genes, such as ASCL1, and impedes tumor proliferation [173]. Similarly, ZFP36L1 has been identified as a target gene of LSD1, which binds to and destabilizes SOX2 and INSM1, thus regulating the NE differentiation of SCLC cells [174]. A recent study demonstrated that LSD1 inhibition activates NOTCH signaling, leading to a subsequent reduction in ASCL1 expression in SCLC [95]. Additionally, the selective LSD1 inhibitor GSK2879552 was found to induce growth inhibition in SCLC cell lines [175]. These findings underscore the potential of LSD1 as a therapeutic target for SCLC.
At present, a range of drugs targeting LSD1 have been identified, including GSK2879552, CC-90011 and bomedemstat (IMG-7289). Currently, another reversible LSD1 inhibitor, CC-90011, is being evaluated in combination with cisplatin/etoposide with or without nivolumab for untreated ES-SCLC patients (NCT03850067). The combination of LSD1 inhibitors with immunotherapy represents an innovative therapeutic strategy, as several studies have suggested that the inhibition of LSD1 enhances the antitumor efficacy of immune checkpoint blockade [176,177,178,179]. A concomitant trial is being conducted to assess the effect of bomedemstat and maintenance immunotherapy with atezolizumab in newly diagnosed ES-SCLC patients (NCT05191797). Collectively, the clinical efficacy of LSD1 remains unproven with no successful clinical trials. Two early-stage clinical trials are ongoing to verify its effectiveness. If successful, these combinations could offer new hope for improving outcomes in SCLC patients.
Histone deacetylases, HDACs
Apart from histone methylation, histone acetylation is another crucial epigenetic hallmark in SCLC. Histone acetylation/deacetylation modulates the transcriptional regulation of genes implicated in the initiation, progression, and metastasis of SCLC by modifying chromatin accessibility [170]. Overexpression of HDACs in cancer cells results in increased deacetylation, which adversely affects the expression of tumor suppressor genes [180]. Administration of HDACs inhibitors in murine models was observed to upregulate YAP through attenuation of the activity of the RE1-silencing transcription factor-corepressor-HDAC complex, hence suppressing metastasis and improving survival in SCLC [181]. Furthermore, a series of studies revealed that HDAC inhibitors potentiate the efficacy of conventional chemotherapeutic regimens [182, 183]. This synergistic interaction might be mediated via the induction of S-phase arrest and decreased base excision repair induced by HDAC inhibition [184].
Currently, four HDAC inhibitors (vorinostat, belinostat, panobinostat, and romidepsin) have received FDA approval for use in certain cancers. Related research is undertaken in SCLC as well. For instance, a multicenter, nonrandomized phase 2 trial evaluated the antitumor activity of panobinostat in patients with previously treated SCLC [185]. At the first assessment, more than 30% tumor shrinkage was observed in 2 of the 19 patients [185]. Another phase 1 trial assessing the combination of belinostat with cisplatin and etoposide demonstrated promising results in SCLC patients [186]. An objective response was observed in 7 (47%) of 15 patients with NE tumors (including SCLC) [186].
Overall, the existing FDA approvals for HDAC inhibitors in other cancers and the positive outcomes in early trials highlight their potential as a viable therapeutic option for SCLC. Further clinical trials are essential to establish the efficacy and safety of HDAC inhibitors in SCLC.
Enhancer of Zeste homolog 2, EZH2
EZH2 is a pivotal oncogene linked to methylation processes in SCLC. Specifically, EZH2 is the core component of polycomb repressive complex 2 (PRC2), a histone methyltransferase responsible for methylating lysine at position 27 on histone H3 (H3K27me3), which is crucial for maintaining epigenetic gene silencing [187]. EZH2 regulation is mediated by the pRB-E2F axis, and its expression is frequently augmented in SCLC as a consequence of RB1 alterations [86, 188]. Through modulation of apoptosis and cell cycle regulation, EZH2 promotes E2F-driven SCLC tumorigenesis [189]. EZH2-mediated epigenetic modifications also lead to the upregulation of TWIST1 and the suppression of SLFN11 in SCLC, contributing to resistance to chemotherapy [190]. Moreover, the growth and chemoresistance of SCLC cells have been proven to be mediated by TUG1, which regulates LIMK2b via EZH2 [191].
Hence, therapeutic interventions targeting EZH2 might be beneficial for SCLC patients. Currently, the EZH2 inhibitor PF-06821497 is under clinical investigation as a monotherapy in patients with relapsed or refractory SCLC (NCT03460977). Based on the correlations between EZH2 and chemoresistance, a phase 1/2 trial (NCT03879798) is assessing the safety and efficacy of the EZH1/2 inhibitor DS-3201b in combination with irinotecan for patients with recurrent SCLC. Moreover, the overexpression of EZH2 may contribute to radioresistance [192], suggesting that patients with radioresistant SCLC could also benefit from EZH2 inhibition. Furthermore, a correlation between EZH2 expression and the response to immunotherapy has been identified in various types of cancer [193], indicating that combining EZH2 inhibitors with immunotherapy may enhance treatment efficacy. The phase 1b/2 KEYNOTE F19 trial (NCT06022757) is investigating the role of XNW5004 in combination with pembrolizumab for relapsed SCLC patients.
Totally, albeit the clinical application of EZH2 inhibitors in SCLC is still being explored. The diverse mechanisms by which EZH2 inhibition may enhance treatment efficacy through overcoming chemoresistance, radioresistance, and improving immunotherapy response, suggest that EZH2 is a promising target for SCLC therapy.
Other pathways
Metabolism
The relentless pursuit of effective treatments for SCLC has led to a focus on the metabolic underpinnings of this disease. The rapid growth and progression of cancer are sustained by altered metabolic pathways, including the way cells process glucose and amino acids such as arginine. Such metabolic alterations not only fuel cancer growth but also offer potential targets for therapy. These vulnerabilities are being exploited through various pathways, aiming to cut off the cancer's energy supply and building blocks necessary for its growth.
Glucose metabolism pathway
Glycolysis, oxidative phosphorylation, and the pentose phosphate pathway are three primary branches of glucose metabolism [194]. Glycolysis, which is prominent in the Warburg effect, is the major source of energy for cancer cells. This metabolic reprogramming allows cancer cells to produce high energy levels even under anaerobic or hypoxic conditions at the expense of high glucose intake. Hence, elevated glucose uptake is often observed in most SCLC patients and is associated with a poor prognosis [195, 196]. Elevated glycolysis, aligning with the Warburg effect, has been detected in cell lines overexpressing MYC [197]. Administration of the glycolysis inhibitor PFK158 in xenograft models led to delayed tumor progression and a reduction in the expression of genes associated with glycolysis [197]. Currently, both preclinical and clinical trials are being performed to test drugs that interfere with glucose metabolic pathways or downstream molecules. For instance, the antidiabetic drug metformin was found to improve both the OS and PFS of diabetic CSCLC patients (OS 19.0 vs 11.5 months, p < 0.001; DFS 10.5 vs 7.0 months, p < 0.001) [198]. Additionally, metformin may reverse acquired resistance to PD-1 inhibitors in SCLC [199]. A phase 2 trial (NCT03994744) is evaluating the safety and efficacy of combining metformin with sindilizumab, a PD-1 inhibitor, in pretreated ES-SCLC patients. Despite their promising therapeutic effects, metabolism-based therapies may encounter challenges such as nonspecific toxicity [200, 201]. Many obstacles still need to be overcome in this field.
Arginine
As the precursor for polyamine biosynthesis, NO generation and mTOR pathway activation, arginine plays a vital role in multiple cellular physiological processes. A key enzyme in the synthesis of arginine is ASS1, the expression of which is often reduced in SCLC. Loss of ASS1 causes notable bioenergetic alterations in SCLC, resulting in arginine dependence, which is correlated with chemoresistance and poor clinical outcomes [98, 202,203,204]. Treatment with pegylated arginine deiminase (ADI-PEG 20) to induce arginine depletion markedly impeded tumor growth and enhanced the survival of mice bearing MYC-driven tumors [98]. Hence, arginine deprivation may serve as a subtype-specific therapeutic vulnerability for patients with SCLC.
Currently, a therapeutic regimen comprising ADI-PEG 20 in combination with gemcitabine and docetaxel is under active clinical investigation for SCLC patients who progressed after frontline therapy (NCT05616624). Concurrently, the potential synergistic effect of ADI-PEG 20 administered in combination with pembrolizumab (NCT03371979) has also been explored in a phase 1 trial. However, the outcomes of these studies have not yet been reported. These efforts suggest a promising direction, with two ongoing clinical trials exploring ADI-PEG20 combinations, but it is clear that arginine-targeted therapies still have a considerable journey ahead in proving their efficacy.
NOTCH pathway
Comprising NOTCH receptors, DSL family ligands, and numerous signal transduction molecules, the NOTCH signaling pathway orchestrates several cellular functions: cell proliferation, stem cell maintenance, differentiation, and apoptosis [205]. Alterations that result in the loss of NOTCH signaling function have been frequently observed among SCLC patients [16, 206]. Previous studies have also revealed that NOTCH signaling is implicated in chemoresistance and modulation of the immune microenvironment, thus underscoring its potential as an antitumor target in SCLC [16, 206].
DLL3 is a single-pass type I transmembrane protein and a member of the inhibitory ligands of the NOTCH pathway. DLL3 interacts with NOTCH receptors, exerting inhibitory effects on the NOTCH pathway in SCLC. DLL3 is overexpressed in SCLC and certain neuroendocrine tumors, whereas its expression is minimal in healthy individuals [207]. This expression pattern has sparked significant interest in developing DLL3 as a novel therapeutic target for SCLC and other malignancies [207].
Rova-T is an antibody‒drug conjugate that targets DLL3 with a specialized humanized monoclonal antibody. The initial human study by Rova-T reported an objective response rate (ORR) of 18% in previously treated SCLC patients [93]. However, the phase 2 TRINITY study reported that grade 3 to 5 adverse events (AEs) were seen in 213 (63%) patients in the third-line and beyond settings [94]. The phase 3 TAHOE and MERU trials, which evaluated Rova-T with topotecan as second-line therapy and Rova-T as maintenance therapy after first-line treatment, were both halted early due to failure of predetermined PFS and OS [208, 209].
Tarlatamab (AMG 757) is a bispecific T-cell engager that targets dual DLL3 and CD3 [210]. A promising response durability was observed in SCLC patients treated with tarlatamab monotherapy, with a reported ORR of 23.4% (95% CI 15.7–32.5) [211]. Recently, the phase 2 DeLLphi-301 trial (NCT05060016) demonstrated persistent antitumor activity of tarlatamab in patients with relapsed/refractory SCLC. Compared to patients in the 100 mg dose group, patients in the 10 mg dose group had superior outcomes, with an objective remission rate of up to 40% (97.5% CI 29–52), as opposed to 32% (97.5% CI 21–44) [212]. To compensate for the absence of a standard treatment control group, the phase 3 DeLLphi-304 study (NCT05740566) will compare the efficacy of tarlatamab with that of the standard care in relapsed SCLC patients. Additionally, the efficacy of tarlatamab after chemoradiotherapy in patients with limited-stage SCLC (LS-SCLC) is being evaluated in the phase III DeLLphi-306 study (NCT06117774). Additionally, the combined effects of tarlatamab and chemoimmunotherapy are being explored in multiple clinical trials. An ongoing phase 1b study (NCT04885998) is evaluating the safety and efficacy of tarlatamab in combination with AMG 404 in SCLC patients. In the phase 3 DeLLphi-305 trial (NCT06211036), tarlatamab and durvalumab versus durvalumab alone is being compared in first-line ES-SCLC following platinum, etoposide and durvalumab treatment. Its combined effect with carboplatin, etoposide, and PD-L1 inhibitors in ES-SCLC is also being investigated in a phase 1b trial (NCT05361395). Similar to tarlatamab, BI 764532 redirects T cells to eradicate tumor cells and serves as a DLL3-targeted treatment for SCLC. Two trials are testing the effects of different doses of BI 764532 monotherapy in SCLC (NCT04429087 and NCT05882058). The combination of BI 764532 with the PD-1 inhibitor ezabenlimab is being explored in SCLC patients positive for DLL3 (NCT05879978). The efficacy of different doses of BI 764532 in addition to standard treatment or topotecan are also being tested in SCLC (NCT05990738, NCT06077500). Additionally, trials are recruiting volunteers for other drugs, such as HPN328, PT217 and 89Zr-DFO-SC16.56 (NCT04199741, NCT04471727, and NCT05652686).
The efficacy of CAR-T-cell therapy in blood cancers has prompted investigations into its potential application for solid tumors. In a phase 1 trial of AMG 119 involving five relapsed/refractory SCLC patients, one patient achieved a partial response (PR) with 43% shrinkage in lesion size, and the other patients exhibited a 16% reduction in lesion size along with the resolution of several liver metastases (NCT03392064). These initial data support the continued development of DLL3 CAR-T-cell therapy for SCLC. An ongoing phase 1 trial is recruiting volunteers for further validation in patients with ES-SCLC (NCT05680922). Similarly, CAR-NK-cell therapy has also demonstrated short-term effects, with noticeable shrinkage of the remaining metastatic lesions in SCLC patients [213]. A related trial using DLL3-CAR-NK cell therapy for treating ES-SCLC is underway (NCT05507593).
Other molecules, such as NOTCH2 and NOTCH3, also represent alternative targets for modulating NOTCH signaling. Tarextumab (anti-NOTCH2/3, OMP-59R5) is a human monoclonal antibody that targets NOTCH2 and NOTCH3 receptors. A study utilizing PDX tumors preliminarily demonstrated the antitumor effect of tarextumab [214]. Subsequently, a phase 1 dose-escalation study indicated good tolerability of tarextumab in patients with advanced solid tumors [215]. However, a combined phase 1b/2 PINNACLE trial investigating tarextumab and chemotherapy in SCLC patients was terminated due to a lack of improvement in PFS (NCT01859741).
In addition to serving as a therapeutic target, NOTCH signaling mutations are associated with improved clinical benefits in SCLC patients undergoing immunotherapy. In a cohort of 662 patients receiving ICIs, the NOTCH4 mutation group had better objective remission rates, clinical benefit rates, and longer PFS and OS, indicating that NOTCH signaling is a determinant of the response to ICIs in SCLC patients [68].
Beyond doubt, the inhibitor of NOTCH signaling pathway, particularly DLL3, has shown more promising results for SCLC. As a potential marker in the multi-omics model of the nmf2 subtype, DLL3 emerges as an attractive therapeutic target. Despite challenges with Rova-T, promising results from drugs like tarlatamab and BI 764532 highlight the therapeutic promise of targeting NOTCH signaling. Further study and combination therapies may better improve prognosis of SCLC patients.
Apoptosis
BCL-2
Through the modulation of the mitochondrial outer membrane, BCL2 family proteins control the cellular decision between survival and apoptosis [216]. The overexpression of BCL2, an important member of the BCL2 family, is frequently observed in SCLC and is linked to the development of drug resistance and poor prognosis [217]. An FDA approved BCL2 inhibitor, venetoclax, was proven to block tumor growth and induce tumor regression in mice bearing high BCL2 expressing SCLC [217]. Moreover, high BCL2 levels have been demonstrated to suppress DNA damage and apoptosis induced by the AURKB inhibitor AZD2811. Resistant models could be significantly sensitized by the combination of AZD2811 with the BCL2 inhibitor venetoclax [218]. Additionally, the synergistic effect of receptor tyrosine kinase-like orphan receptor 1 inhibition with BCL2 inhibition was observed in SCLC models [219].
The combination of oblimersen (G3139), an antisense BCL2 oligonucleotide, with carboplatin and etoposide or paclitaxel was shown to be well tolerated in phase 1 trials [220, 221]. However, drugs such as obatoclax (a first-generation BCL2 inhibitor), AT-101 (a small-molecule BCL2 inhibitor), ABT-263, and isotretinoin failed to obtain clinical benefits in phase 2 trials [222,223,224,225]. The addition of oblimersen to a standard regimen also failed to improve any clinical outcomes in the phase 2 CALGB 30103 trial [226].
Of note, BCL2 may be a potential molecular target for SCLC. Despite promising preclinical results, BCL-2 inhibitors have failed in phase 2 trials. Additional clinical trials are needed to fully elucidate its therapeutic potential.
Immunotherapy
Extensive-stage SCLC, ES-SCLC
SCLC is considered a good candidate for ICIs owing to its increased TMB and the presence of autoimmune paraneoplastic phenomena. Currently, several ICIs, such as atezolizumab, durvalumab, serplulimab and adebrelimab, which target the PD-1/PD-L1 pathway, have been approved for the treatment of ES-SCLC by the National Comprehensive Cancer Network (NCCN) or Chinese Society of Clinical Oncology (CSCO) guidelines. However, the process is filled with thistles and thorns.
Ipilimumab, a monoclonal antibody against CTLA4, was first used following an early paclitaxel/carboplatin-induced regimen in patients with ES-SCLC, and the immune-related PFS improved significantly compared with that in the control group [227]. CheckMate 032, a multicenter, phase 1/2 trial executed later, explored the role of nivolumab, an anti-PD-1 antibody, as well as the role of nivolumab combined with ipilimumab in previously treated SCLC patients. The final results confirmed the durable antitumor activity and manageable safety of ICIs [228]. Similar promising antitumor activity and safety of pembrolizumab, another anti-PD-1 antibody, was achieved in previously treated SCLC patients with recurrence or metastasis, as shown by KEYNOTE-028 and KEYNOTE-158 [229, 230]. However, in CheckMate 331, a randomized phase 3 trial, the advantage of nivolumab as a second-line treatment in patients with relapsed SCLC could not be verified [231]. Taken together, these clinical trials indicate that the role of ICIs in SCLC as a posterior treatment is contradictory, and the consolidated role of ICIs in different phases needs further investigation.
To explore the role of ICIs in maintenance therapy for ES-SCLC, several studies have been performed. A phase II study revealed that maintenance of pembrolizumab, a PD-1 inhibitor, in ES-SCLC patients receiving standard chemotherapy failed to improve patient prognosis compared with historical data [232]. In the CheckMate 451 study, compared with placebo, nivolumab plus ipilimumab or nivolumab monotherapy maintenance did not achieve desirable outcomes in patients with ES-SCLC following first-line chemotherapy [233]. Thus, further regimens should be administered.
Owing to the potential promising efficacy and contradictory role of ICIs, different immune-monoclonal antibodies combined with first-line chemotherapy followed by ICIs as maintenance therapy were tested in patients with ES-SCLC. The regimen of ipilimumab plus etoposide and platinum was first applied in ES-SCLC via a phase 3 randomized trial, and the outcomes showed that immunotherapy did not lead to a better OS [234]. A similar failure to improve OS was observed in the KEYNOTE-604 study, which used pembrolizumab as a first-line therapy in patients with ES-SCLC [235]. Undoubtedly, the above disappointing conclusions cast a shadow on the exploration of immunotherapy. With persistent effort, the IMpower133 randomized double-blind phase III trial revealed that the addition of atezolizumab, an anti-PD-L1 antibody, to standard chemotherapy could significantly prolong the median OS in patients with ES-SCLC for 2 months [4]. Thereafter, CASPIAN randomized phase 3 trials also confirmed that the anti-PD-L1 antibody durvalumab in combination with first-line treatment could yield positive results, with an extended median OS of 3 months in patients with ES-SCLC [236]. Accordingly, atezolizumab and durvalumab are approved as first-line immunotherapies combined with standard chemotherapy for treating ES-SCLC by the US FDA. The CAPSTONE-1 and ASTRUM-005 phase 3 trials also yielded positive results using different PD‐L1 (adebrelimab) and PD-1 (serplulimab) inhibitors, respectively, and these two drugs were approved by the National Medical Products Administration (NMPA) for first-line treatment of ES-SCLC [237, 238]. In a recent RATIONALE-312 study, another PD-1 inhibitor, tislelizumab, demonstrated superior OS and PFS when combined with chemotherapy as a first-line treatment for ES-SCLC [239].
Unsurprisingly, ICIs, as a first-line treatment, have yielded promising benefits and represent a novel treatment approach for ES-SCLC. Nonetheless, the role of ICIs combined with different regimens remains elusive. Radiotherapy combined with ICIs can exhibit synergistic effects by remodeling the tumor microenvironment (TME) [240]. However, this regimen needs further investigation via evidence-based data, such as the RAPTOR/NRG LU007 trial (NCT04402788) and LEAD trial (NCT05092412), although current guidelines suggest that thoracic radiotherapy could be used with immunotherapy [241]. The combination of ICIs with targeted therapies may have promising applications. The phase II PASSION study evaluated the PD-1 inhibitor camrelizumab plus the antiangiogenic drug apatinib in patients with ES-SCLC. The results showed that the above treatment significantly enhanced PFS in ES-SCLC patients who did not respond to first-line platinum-based chemotherapy, with a favorable safety profile [242]. Moreover, the phase 3 ETER701 trial demonstrated that the addition of the PD-L1 inhibitor TQB2450 and anlotinib to standard chemotherapy significantly improved the OS and PFS of patients with ES-SCLC [243]. For the role of two ICIs in ES-SCLC, different regimens should be used. However, the SKYSCRAPER-02 trial revealed that atezolizumab combined with extra tiragolumab (an anti-TIGIT monoclonal antibody) did not significantly improve PFS or OS compared to that of the control group [244].
Limited-stage SCLC, LS-SCLC
Although immunotherapy has achieved remarkable success in patients with ES-SCLC, its efficacy in patients with LS-SCLC remains unconfirmed. Two single-arm phase 1 trials exploring the efficacy of chemoradiotherapy (CRT) combined with duvarizumab [245] or pembrolizumab [246] in treating LS-SCLC achieved 2-year survival rates of 67.8 and 65.8%, respectively. Despite the promising clinical efficacy and tolerable toxicity observed in single-arm trials, consolidation therapy with ICIs following concurrent CRT (CCRT) failed to improve the survival of patients with LS-SCLC in the STIMULI trial [247]. The phase 2 STIMULI trial was designed to explore the efficacy and safety of nivolumab in combination with ipilimumab as maintenance therapy for patients with LS-SCLC who have not progressed after receiving CRT and prophylactic cranial irradiation (PCI) [247]. No significant improvement in PFS was observed, potentially due to drug toxicity reactions that limit the number of patients able to receive maintenance therapy [247].
Further investigations are currently underway to investigate the efficacy of combining ICIs with CRT. The phase 3 ADRIATIC trial (NCT03703297) demonstrated that durvalumab with or without tremelimumab after concurrent CRT significantly improved the PFS and OS of patients with LS-SCLC [248, 249]. The safety run-in results of a phase 3 study (NCT04691063) revealed that SHR-1316 combined with concurrent chemoradiotherapy achieved promising clinical efficacy and tolerable safety [250]. A subsequent randomized, double-blind and placebo-controlled study is currently ongoing. Similarly, two phase 2 trials (NCT03540420 and NCT03811002) of atezolizumab have also completed recruitment, potentially confirming the role of immunotherapy in LS-SCLC.
Emerging targets for immunotherapy
CD47 is highly expressed on the surface of SCLC cells and interacts with signal-regulatory protein alpha (SIRPα) receptors on macrophages, thereby inhibiting phagocytic activity and facilitating immune evasion [251, 252]. Thus, antibodies targeting the CD47/SIRPα axis can activate macrophages and enhance antitumor immunity [252,253,254]. Recent studies have demonstrated that CD47 inhibitors augment the therapeutic efficacy of local radiotherapy and exert distant effects by suppressing the growth of nonirradiated tumors [255]. PT217, a bispecific antibody targeting DLL3 and CD47, is under investigation among patients with SCLC and other NE cancers in the phase 1 SKYBRIDGE study (NCT05652686).
The immune checkpoint B- and T-lymphocyte attenuator (BTLA), which is detected at high levels on T and B lymphocytes, dendritic cells and macrophages, can interact with herpesvirus entry mediator (HVEM) expressed on tumor cells and T and B lymphocytes, NK cells and myeloid cells [256]. The BTLA/HVEM signaling pathway is negatively associated with the immune response via the recruitment of phosphatases 1 and 2 [257]. An emerging study demonstrated that treatment with an anti-BTLA antibody (tifcemalimab) combined with toripalimab and chemotherapy has tolerable side effects in patients with ES-SCLC, with an 86.5% ORR and a 100% disease control rate (NCT05000684) [258]. The role of tifcemalimab in LS-SCLC is also being investigated (NCT06095583).
As an immune checkpoint regulator, B7 homolog 3 protein (B7-H3) modulates T-cell activation through its costimulatory and coinhibitory roles, making it a promising target for SCLC treatment [259, 260]. B7-H3 is overexpressed in SCLC and has been linked to unfavorable outcomes [261, 262]. Ifinatamab deruxtecan (I-DXd) is an antibody‒drug conjugate that targets B7-H3 and delivers the topoisomerase I inhibitor deruxtecan. This drug is being evaluated in several ongoing trials. DS7300-A-J101 (NCT04145622) is a phase I/II clinical trial that enrolled patients with advanced, unresectable or metastatic solid tumors. In a subgroup of 21 SCLC patients, the study reported an ORR of 52.4%, with a complete response (CR) rate of 4.8% and a median OS of 12.2 months. In the phase 2 IDeate-Lung01 trial (NCT05280470), the efficacy, safety and pharmacokinetics of I-DXd were investigated in pretreated ES-SCLC patients. The recently initiated phase 1b/2 IDeate-Lung03 trial (NCT06362252) aimed to assess the efficacy of I-DXd plus atezolizumab, with or without chemotherapy, as first-line induction or maintenance therapy in patients with ES-SCLC.
Natural Killer Group 2A (NKG2A) is an inhibitory receptor found on the surface of both T and NK cells [263]. The inhibition of NKG2A unleashes the function of T and NK cells and promotes antitumor immunity [264]. The efficacy of the NKG2A inhibitor monalizumab combined with durvalumab plus platinum-based chemotherapy has been evaluated in a single-arm phase II MOZART trial (NCT05903092).
In addition to ICIs, the initiation of innovative immunotherapy based on the infusion of immune cells, such as dendritic cells (DCs) and cytokine-induced killer (CIK) cells, has gained considerable attention. In a phase 2 trial, a vaccine (Ad.p53-DC) containing dendritic cells transfected with wild-type TP53 failed to improve the response to chemotherapy in recurrent ES-SCLC patients, but its safety and therapeutic immune potential remain encouraging [265]. However, a subsequent trial (NCT03406715) of the Ad.p53-DC vaccine combined with ipilimumab and nivolumab was terminated, and the results were limited. Additionally, the combination of CIK cell transfusion and chemotherapy has shown promise with a 4-month PFS [266]. Maintenance therapy with the PD-1 inhibitor sintilimab after first-line CIK cell therapy plus chemotherapy also presented satisfactory safety and antitumor efficacy [267]. Thus, standard immunochemotherapy combined with immune cell therapy seems to be a promising strategy.
Virotherapy is an emerging field in the treatment of lung cancer. Oncolytic viruses are a class of viral agents capable that selectively target neoplastic cells and augment the antitumor immune response [268]. Two types of viruses, namely, Seneca Valley virus (NTX-010) [269] and a modified oncolytic myxoma virus (MYXV) [270], have been used in SCLC trials. Although a phase 2 trial revealed that ES-SCLC patients did not benefit from NTX-010 treatment following platinum-based chemotherapy, the potential of viroimmunotherapy remains promising [269]. Another oncolytic virus, RT-01, is currently being evaluated in a single-arm phase 1 trial (NCT05205421) among ES-SCLC patients.
Conclusion and future directions
SCLC, closely correlated with heavy smoking, is considered as a recalcitrant cancer. Despite the unambiguous analysis of genomic alterations, no valuable targeted therapy analogous to adenocarcinoma with genomic alterations has been confirmed for SCLC. Due to treatment limitations and the intrinsic aggressive features of SCLC, patients still have poor outcomes. Systematic exploration of SCLC subtypes and signaling pathways may provide novel insight for SCLC treatment. Based on diverse transcriptional data, different SCLC subtypes and specific treatments for these subtypes were identified. Integrated multi-omics analysis revealed four novel subtypes of SCLC, which may provide insight into therapeutic regimens. In addition to the classification of SCLC, investigations of abnormal signaling pathways (mainly those involved in the cell cycle and DNA damage and repair, epigenetics, metabolism and others) can also lead to progress in treatment. However, many problems still exist, and further attention should be devoted to these issues.
Based on different data and method used for classification, diverse subtypes were shown. Yet, uniform and rigorous consensus clustering of SCLC patients is still under debate, not to mention the biomarker for each subtype. For classic ASCL1, NEUROD1, YAP1 and POU2F3 subtypes, immunohistochemistry can be used. However, low levels of YAP1 coexisted with other subtypes, and high rate of coexpression of ASCL1 and NEUROD1 can be detected. In terms of ASCL1, NEUROD1, POU2F3 and Inflamed subtypes, they can largely be defined by differential level of ASCL1, NEUROD1, POU2F3, and low expression of these three factors. With regard to multi-omics subtypes, nmf4 and nmf2 can be mostly defined by POU2F3 and DLL3 expression, the rest of them are still lack of specific biomarker. And further efforts are still needed to solve the clustering and biomarkers.
The low number of tumor samples has blocked the exploration of SCLC. The high quality of samples from clinical trials can better illustrate the treatment response at the molecular level. For instance, transcriptional data from IMpower133 revealed that patients with the Inflamed subtypes can benefit greatly from immunotherapy. Thus, clinical trials with the support of samples could promote the progression of SCLC.
The bank of the PDX/CDX model can not only serve as a tool for basic study and treatment, but also provide adequate tissues for multi-omics, which can offer additional information for SCLC. However, the complicated process, high prices and low rate of model development limit progress, and further efforts should be made.
Fewer drugs have been approved for SCLC in recent decades. With the development of pharmaceutical technology, including the use of ADCs and bispecific antibodies, emerging treatments may be effective for treating SCLC, and further clinical trials should be carried out.
Currently, SCLC has always been treated as a single disease entity. The classification of SCLC is diverse and uncertain. More accurate and rigorous subtypes of SCLC should be determined. Moreover, biomarkers and precise treatments for different subtypes should also be elucidated.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- SCLC:
-
Small cell lung cancer
- NSCLC:
-
Non-small cell lung cancer
- CNAs:
-
Copy number alterations
- TMBs:
-
Tumor mutation burdens
- NO2 :
-
Nitrogen dioxide
- PM10 :
-
Particulate matter 10
- PAHs:
-
Polycyclic aromatic hydrocarbons
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- GEMMs:
-
Genetically engineered mouse models
- NE:
-
Neuroendocrine
- AT2:
-
Alveolar type 2
- SPC:
-
Surfactant protein C
- hESCs:
-
Human embryonic stem cells
- PNECs:
-
Pulmonary NE cells
- EGFR:
-
Epidermal growth factor receptor
- LUAD:
-
Lung adenocarcinoma
- TKI:
-
Tyrosine kinase inhibitor
- APOBEC:
-
Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like
- CSCLC:
-
Combined SCLC
- WGS:
-
Whole genome sequencing
- HPV:
-
Human papillomavirus
- REST:
-
RE1-silencing transcription factor
- APM:
-
Antigen processing and presentation machinery
- PI3K:
-
Phosphatidylinositol 3 kinase
- TKO:
-
Triple-knockout
- H3K4:
-
Histone H3 and lysine 4
- CREBBP:
-
CREB binding protein
- EP300:
-
E1A binding protein p300
- HAT:
-
Histone acetyltransferase
- SVV-001:
-
Seneca valley virus
- TFs:
-
Transcriptional factors
- NEUROD1:
-
Neurogenic differentiation factor 1
- INSM1:
-
Insulinoma-associated protein 1
- YAP1:
-
Yes-associated protein 1
- ASCL1:
-
Achaete-scute homolog 1
- POU2F3:
-
POU class 2 homeobox 3
- ICIs:
-
Immune checkpoint inhibitors
- NMF:
-
Non-negative matrix factorization
- TAMs:
-
Tumor-associated macrophages
- DLL3:
-
Delta-like ligand-3 (δ-like ligand 3)
- RTKs:
-
Receptor tyrosine kinases
- CCSP:
-
Clara cell secretory protein,
- scRNA-seq:
-
Single-cell RNA sequencing
- AURKA/B:
-
Aurora kinase A/B
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- Rova-T:
-
Rovalpituzumab teserine
- HDACs:
-
Histone deacetylases
- LSD1:
-
Lysine-specific demethylase 1
- SLFN11:
-
Schlafen family member 11
- PARP:
-
Poly (ADP-ribose) polymerase
- CEACAM5:
-
Carcinoembryonic antigen-related cell adhesion molecule 5
- IMPDH1 and IMPDH2:
-
Inosine monophosphate dehydrogenase-1 and -2
- CHK1:
-
Checkpoint kinase 1
- SSTR2:
-
Somatostatin receptor 2
- IGF1R:
-
Insulin-like growth factor 1 receptor
- mSWI/SNF:
-
Mammalian switch/sucrose non-fermentable
- BTK:
-
Bruton’s tyrosine kinase
- ATR:
-
Ataxia telangiectasia and Rad3-related protein
- TOP1:
-
Topoisomerase I
- PDX:
-
Patient-derived tumor xenograft
- CDX:
-
Cell-line derived xenograft
- OS:
-
Overall survival
- LS-SCLC:
-
Limited-stage SCLC
- PCI:
-
Prophylactic cranial irradiation
- NCCN:
-
National comprehensive cancer network
- TOP2:
-
Topoisomerase II
- FDA:
-
Food and drug administration
- CRT:
-
Chemoradiotherapy
- CCRT:
-
Concurrent CRT
- PFS:
-
Progression-free survival
- ES-SCLC:
-
Extensive-stage SCLC
- CSCO:
-
Chinese society of clinical oncology
- NMPA:
-
National medical products administration
- TME:
-
Tumor microenvironment
- SIRPα:
-
Signal-regulatory protein alpha
- B7-H3:
-
B7 homolog 3 protein
- ORR:
-
Objective response rate
- CR:
-
Complete response
- PR:
-
Partial response
- NKG2A:
-
Natural killer group 2A
- DC:
-
Dendritic cell
- CIK:
-
Cytokine-induced killer
- TRT:
-
Thoracic radiation therapy
- WBRT:
-
Whole brain radiation therapy
- DDR:
-
DNA damage repair
- AEs:
-
Adverse events
- TMZ:
-
Temozolomide
- H3K27me3:
-
Methylating lysine at position 27 on histone H3
- ADC:
-
Antibody–drug conjugate
- CDK:
-
Cyclin-dependent kinase
- CTC:
-
Circulating tumor cell
- CIM:
-
Chemotherapy-induced myelosuppression
- HSPCs:
-
Hematopoietic stem and progenitor cells
- EZH2:
-
Enhancer of Zeste Homolog 2
- PRC2:
-
Polycomb repressive complex 2
- H3K27me3:
-
Methylating lysine at position 27 on histone H3
- BTLA:
-
B- and T-lymphocyte attenuator
- HVEM:
-
Herpesvirus entry mediator
References
Zhang Y, et al. Global variations in lung cancer incidence by histological subtype in 2020: a population-based study. Lancet Oncol. 2023;24:1206–18. https://doi.org/10.1016/S1470-2045(23)00444-8.
Pignon JP, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med. 1992;327:1618–24. https://doi.org/10.1056/NEJM199212033272302.
Wang S, et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci Rep. 2017;7:1339. https://doi.org/10.1038/s41598-017-01571-0.
Horn L, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9. https://doi.org/10.1056/NEJMoa1809064.
Ragavan M, Das M. Systemic therapy of extensive stage small cell lung cancer in the era of immunotherapy. Curr Treat Options Oncol. 2020;21:64. https://doi.org/10.1007/s11864-020-00762-8.
Jordan EJ, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. https://doi.org/10.1158/2159-8290.CD-16-1337.
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17:725–37. https://doi.org/10.1038/nrc.2017.87.
Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. https://doi.org/10.1007/978-3-319-24223-1_1.
Islami F, Torre LA, Jemal A. Global trends of lung cancer mortality and smoking prevalence. Transl Lung Cancer Res. 2015;4:327–38. https://doi.org/10.3978/j.issn.2218-6751.2015.08.04.
Ragavan M, Patel MI. The evolving landscape of sex-based differences in lung cancer: a distinct disease in women. Eur Respir Rev. 2022. https://doi.org/10.1183/16000617.0100-2021.
Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7:3. https://doi.org/10.1038/s41572-020-00235-0.
Luo G, et al. Projections of lung cancer incidence by 2035 in 40 countries worldwide: population-based study. JMIR Public Health Surveill. 2023;9:e43651. https://doi.org/10.2196/43651.
Chan JM, et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell. 2021;39:1479–96. https://doi.org/10.1016/j.ccell.2021.09.008.
Na F, et al. KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming. Nat Cancer. 2022;3:753–67. https://doi.org/10.1038/s43018-022-00361-6.
Hecht SS. Lung carcinogenesis by tobacco smoke. Int J Cancer. 2012;131:2724–32. https://doi.org/10.1002/ijc.27816.
George J, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. https://doi.org/10.1038/nature14664.
Siegel DA, Fedewa SA, Henley SJ, Pollack LA, Jemal A. Proportion of never smokers among men and women with lung cancer in 7 US States. JAMA Oncol. 2021;7:302–4. https://doi.org/10.1001/jamaoncol.2020.6362.
Liu X, et al. Characterization of never-smoking and its association with clinical outcomes in Chinese patients with small-cell lung cancer. Lung Cancer. 2018;115:109–15. https://doi.org/10.1016/j.lungcan.2017.11.022.
Sun JM, et al. Small-cell lung cancer detection in never-smokers: clinical characteristics and multigene mutation profiling using targeted next-generation sequencing. Ann Oncol. 2015;26:161–6. https://doi.org/10.1093/annonc/mdu504.
Huang R, et al. Associated links among smoking, chronic obstructive pulmonary disease, and small cell lung cancer: a pooled analysis in the international lung cancer consortium. EBioMedicine. 2015;2:1677–85. https://doi.org/10.1016/j.ebiom.2015.09.031.
Abdel-Rahman O. Incidence and mortality of lung cancer among never smokers in relationship to secondhand smoking: findings from the PLCO trial. Clin Lung Cancer. 2020;21:415–20. https://doi.org/10.1016/j.cllc.2020.04.009.
Kim CH, et al. Exposure to secondhand tobacco smoke and lung cancer by histological type: a pooled analysis of the international lung cancer consortium (ILCCO). Int J Cancer. 2014;135:1918–30. https://doi.org/10.1002/ijc.28835.
Varghese AM, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol. 2014;9:892–6. https://doi.org/10.1097/JTO.0000000000000142.
Ou SH, Ziogas A, Zell JA. Prognostic factors for survival in extensive stage small cell lung cancer (ED-SCLC): the importance of smoking history, socioeconomic and marital statuses, and ethnicity. J Thorac Oncol. 2009;4:37–43. https://doi.org/10.1097/JTO.0b013e31819140fb.
Rodriguez-Martinez A, Torres-Duran M, Barros-Dios JM, Ruano-Ravina A. Residential radon and small cell lung cancer. A systematic review. Cancer Lett. 2018;426:57–62. https://doi.org/10.1016/j.canlet.2018.04.003.
Hystad P, Demers PA, Johnson KC, Carpiano RM, Brauer M. Long-term residential exposure to air pollution and lung cancer risk. Epidemiology. 2013;24:762–72. https://doi.org/10.1097/EDE.0b013e3182949ae7.
Lamichhane DK, et al. Lung cancer risk and residential exposure to air pollution: a Korean population-based case-control study. Yonsei Med J. 2017;58:1111–8. https://doi.org/10.3349/ymj.2017.58.6.1111.
Field RW, Withers BL. Occupational and environmental causes of lung cancer. Clin Chest Med. 2012;33:681–703. https://doi.org/10.1016/j.ccm.2012.07.001.
Driscoll T, et al. The global burden of disease due to occupational carcinogens. Am J Ind Med. 2005;48:419–31. https://doi.org/10.1002/ajim.20209.
Weiss W, Boucot KR. The respiratory effects of chloromethyl methyl ether. JAMA. 1975;234:1139–42.
Wang Q, et al. Dietary quality using four dietary indices and lung cancer risk: the golestan cohort study (GCS). Cancer Causes Control. 2021;32:493–503. https://doi.org/10.1007/s10552-021-01400-w.
Wang Q, Ru M, Zhang Y, Kurbanova T, Boffetta P. Dietary phytoestrogen intake and lung cancer risk: an analysis of the prostate, lung, colorectal and ovarian (PLCO) cancer screening trial. Carcinogenesis. 2021;42:1250–9. https://doi.org/10.1093/carcin/bgab072.
Baik CS, Strauss GM, Speizer FE, Feskanich D. Reproductive factors, hormone use, and risk for lung cancer in postmenopausal women, the nurses’ health study. Cancer Epidemiol Biomarkers Prev. 2010;19:2525–33. https://doi.org/10.1158/1055-9965.EPI-10-0450.
Pesatori AC, et al. Hormone use and risk for lung cancer: a pooled analysis from the international lung cancer consortium (ILCCO). Br J Cancer. 2013;109:1954–64. https://doi.org/10.1038/bjc.2013.506.
Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366:1385–96. https://doi.org/10.1016/S0140-6736(05)67569-1.
Peifer M, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. https://doi.org/10.1038/ng.2396.
Rudin CM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–6. https://doi.org/10.1038/ng.2405.
Meuwissen R, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9. https://doi.org/10.1016/s1535-6108(03)00220-4.
Mollaoglu G, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell. 2017;31:270–85. https://doi.org/10.1016/j.ccell.2016.12.005.
Schaffer BE, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010;70:3877–83. https://doi.org/10.1158/0008-5472.CAN-09-4228.
Cui M, et al. PTEN is a potent suppressor of small cell lung cancer. Mol Cancer Res. 2014;12:654–9. https://doi.org/10.1158/1541-7786.MCR-13-0554.
Semenova EA, et al. Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep. 2016;16:631–43. https://doi.org/10.1016/j.celrep.2016.06.020.
Ferone G, et al. FGFR1 oncogenic activation reveals an alternative cell of origin of SCLC in Rb1/p53 mice. Cell Rep. 2020;30:3837–50. https://doi.org/10.1016/j.celrep.2020.02.052.
Chen J, et al. Lineage-restricted neoplasia driven by Myc defaults to small cell lung cancer when combined with loss of p53 and Rb in the airway epithelium. Oncogene. 2022;41:138–45. https://doi.org/10.1038/s41388-021-02070-3.
Chen HJ, et al. Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J Exp Med. 2019;216:674–87. https://doi.org/10.1084/jem.20181155.
Huang YH, et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018;32:915–28. https://doi.org/10.1101/gad.314815.118.
Wu Q, et al. YAP drives fate conversion and chemoresistance of small cell lung cancer. Sci Adv. 2021;7:1850. https://doi.org/10.1126/sciadv.abg1850.
Ireland AS, et al. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell. 2020;38:60–78. https://doi.org/10.1016/j.ccell.2020.05.001.
Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165-172. https://doi.org/10.1016/S1470-2045(14)71180-5.
Balla A, Khan F, Hampel KJ, Aisner DL, Sidiropoulos N. Small-cell transformation of ALK-rearranged non-small-cell adenocarcinoma of the lung. Cold Spring Harb Mol Case Stud. 2018. https://doi.org/10.1101/mcs.a002394.
Gazeu A, et al. Small-cell lung cancer transformation as a mechanism of resistance to pralsetinib in RET-rearranged lung adenocarcinoma: a case report. Clin Lung Cancer. 2023;24:72–5. https://doi.org/10.1016/j.cllc.2022.10.005.
Lin JJ, et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis Oncol. 2020;4:21. https://doi.org/10.1038/s41698-020-0127-9.
Offin M, et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J Thorac Oncol. 2019;14:1784–93. https://doi.org/10.1016/j.jtho.2019.06.002.
Lee JK, et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol. 2017;35:3065–74. https://doi.org/10.1200/JCO.2016.71.9096.
Shen Q, Qu J, Sheng L, Gao Q, Zhou J. Case report: transformation from non-small cell lung cancer to small cell lung cancer during anti-PD-1 therapy: a report of two cases. Front Oncol. 2021;11:619371. https://doi.org/10.3389/fonc.2021.619371.
Imakita T, Fujita K, Kanai O, Terashima T, Mio T. Small cell lung cancer transformation during immunotherapy with nivolumab: a case report. Respir Med Case Rep. 2017;21:52–5. https://doi.org/10.1016/j.rmcr.2017.03.019.
Sehgal K, et al. Small cell transformation of non-small cell lung cancer on immune checkpoint inhibitors: uncommon or under-recognized? J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2020-000697.
Zhang J, et al. Comprehensive genomic profiling of combined small cell lung cancer. Transl Lung Cancer Res. 2021;10:636–50. https://doi.org/10.2103/tlcr-20-1099.
Zhao X, et al. Combined small cell carcinoma of the lung: is it a single entity? J Thorac Oncol. 2018;13:237–45. https://doi.org/10.1016/j.jtho.2017.10.010.
McFadden DG, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156:1298–311. https://doi.org/10.1016/j.cell.2014.02.031.
Liu Q, et al. Proteogenomic characterization of small cell lung cancer identifies biological insights and subtype-specific therapeutic strategies. Cell. 2024;187:184–203. https://doi.org/10.1016/j.cell.2023.12.004.
Sivakumar S, et al. Integrative analysis of a large real-world cohort of small cell lung cancer identifies distinct genetic subtypes and insights into histologic transformation. Cancer Discov. 2023;13:1572–91. https://doi.org/10.1158/2159-8290.Cd-22-0620.
Febres-Aldana CA, et al. Rb tumor suppressor in small cell lung cancer: combined genomic and IHC analysis with a description of a distinct Rb-proficient subset. Clin Cancer Res. 2022;28:4702–13. https://doi.org/10.1158/1078-0432.CCR-22-1115.
Wildey G, et al. Retinoblastoma expression and targeting by CDK4/6 inhibitors in small cell lung cancer. Mol Cancer Ther. 2023;22:264–73. https://doi.org/10.1158/1535-7163.Mct-22-0365.
Fu C, et al. The transcription factor ZFHX3 is crucial for the angiogenic function of hypoxia-inducible factor 1alpha in liver cancer cells. J Biol Chem. 2020;295:7060–74. https://doi.org/10.1074/jbc.RA119.012131.
Sun X, et al. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005;37:407–12. https://doi.org/10.1038/ng1528.
Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. https://doi.org/10.1038/nature11125.
Roper N, et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat Commun. 2021;12:3880. https://doi.org/10.1038/s41467-021-24164-y.
Augert A, et al. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J Thorac Oncol. 2017;12:704–13. https://doi.org/10.1016/j.jtho.2016.12.011.
Lee JE, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. https://doi.org/10.7554/eLife.01503.
Jia D, et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 2018;8:1422–37. https://doi.org/10.1158/2159-8290.CD-18-0385.
Rudin CM, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer. 2019;19:289–97. https://doi.org/10.1038/s41568-019-0133-9.
Carney DN, AF G, Bepler G, Guccion JG, Marangos PJ, Moody TW, Zweig MH, Minna JD. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985;45:2913–23.
Zhang W, et al. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res. 2018;7:32–49. https://doi.org/10.2103/tlcr.2018.02.02.
Jiang L, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12:e1005895. https://doi.org/10.1371/journal.pgen.1005895.
Poirier JT, et al. Selective tropism of seneca valley virus for variant subtype small cell lung cancer. J Natl Cancer Inst. 2013;105:1059–65. https://doi.org/10.1093/jnci/djt130.
Borromeo MD, et al. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 2016;16:1259–72. https://doi.org/10.1016/j.celrep.2016.06.081.
McColl K, et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget. 2017;8:73745–56. https://doi.org/10.1863/oncotarget.20572.
Baine MK, et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J Thorac Oncol. 2020;15:1823–35. https://doi.org/10.1016/j.jtho.2020.09.009.
Gay CM, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell. 2021;39:346–60. https://doi.org/10.1016/j.ccell.2020.12.014.
Heeke S, et al. Tumor- and circulating-free DNA methylation identifies clinically relevant small cell lung cancer subtypes. Cancer Cell. 2024;42:225–37. https://doi.org/10.1016/j.ccell.2024.01.001.
Nabet BY, et al. Immune heterogeneity in small-cell lung cancer and vulnerability to immune checkpoint blockade. Cancer Cell. 2024;42:429–43. https://doi.org/10.1016/j.ccell.2024.01.010.
Cao L, et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell. 2021;184:5031–52. https://doi.org/10.1016/j.cell.2021.08.023.
Chen YJ, et al. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell. 2020;182:226–44. https://doi.org/10.1016/j.cell.2020.06.012.
Clark DJ, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell. 2019;179:964–83. https://doi.org/10.1016/j.cell.2019.10.007.
Poirier JT, et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34:5869–78. https://doi.org/10.1038/onc.2015.38.
Wooten DJ, et al. Systems-level network modeling of small cell lung cancer subtypes identifies master regulators and destabilizers. PLoS Comput Biol. 2019;15:e1007343. https://doi.org/10.1371/journal.pcbi.1007343.
Simpson KL, et al. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nat Cancer. 2020;1:437–51. https://doi.org/10.1038/s43018-020-0046-2.
Chen Y, et al. Integrative analysis of multi-omics data reveals the heterogeneity and signatures of immune therapy for small cell lung cancer. Clin Transl Med. 2021;11:e620. https://doi.org/10.1002/ctm2.620.
Zhang J, et al. Single-cell transcriptome identifies drug-resistance signature and immunosuppressive microenvironment in metastatic small cell lung cancer (Advanced Genetics 2/03). Adv Genet. 2022;3:2270021. https://doi.org/10.1002/ggn2.202270021.
Gong X, et al. Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019;9:248–63. https://doi.org/10.1158/2159-8290.CD-18-0469.
Lochmann TL, et al. Venetoclax is effective in small-cell lung cancers with high BCL-2 expression. Clin Cancer Res. 2018;24:360–9. https://doi.org/10.1158/1078-0432.CCR-17-1606.
Rudin CM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18:42–51. https://doi.org/10.1016/S1470-2045(16)30565-4.
Morgensztern D, et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin Cancer Res. 2019;25:6958–66. https://doi.org/10.1158/1078-0432.CCR-19-1133.
Augert A, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal. 2019. https://doi.org/10.1126/scisignal.aau2922.
Helfrich BA, et al. Barasertib (AZD1152), a small molecule aurora B inhibitor, inhibits the growth of SCLC cell lines in vitro and in vivo. Mol Cancer Ther. 2016;15:2314–22. https://doi.org/10.1158/1535-7163.MCT-16-0298.
Huang F, et al. Inosine monophosphate dehydrogenase dependence in a subset of small cell lung cancers. Cell Metab. 2018;28:369–82. https://doi.org/10.1016/j.cmet.2018.06.005.
Chalishazar MD, et al. MYC-driven small-cell lung cancer is metabolically distinct and vulnerable to arginine depletion. Clin Cancer Res. 2019;25:5107–21. https://doi.org/10.1158/1078-0432.CCR-18-4140.
Sen T, et al. CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res. 2017;77:3870–84. https://doi.org/10.1158/0008-5472.CAN-16-3409.
He T, et al. Targeting the mSWI/SNF complex in POU2F-POU2AF transcription factor-driven malignancies. Cancer Cell. 2024. https://doi.org/10.1016/j.ccell.2024.06.006.
Thomas A, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell. 2021;39:566–79. https://doi.org/10.1016/j.ccell.2021.02.014.
Byers LA, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. https://doi.org/10.1158/2159-8290.CD-12-0112.
Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34:360–94. https://doi.org/10.1101/gad.334516.119.
Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov. 2020;19:711–36. https://doi.org/10.1038/s41573-020-0076-6.
Sen T, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–61. https://doi.org/10.1158/2159-8290.CD-18-1020.
Jiao S, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20. https://doi.org/10.1158/1078-0432.CCR-16-3215.
Xiong J, Barayan R, Louie AV, Lok BH. Novel therapeutic combinations with PARP inhibitors for small cell lung cancer: a bench-to-bedside review. Semin Cancer Biol. 2022;86:521–42. https://doi.org/10.1016/j.semcancer.2022.07.008.
Park S, et al. Biomarker-driven phase 2 umbrella trial: clinical efficacy of olaparib monotherapy and combination with ceralasertib (AZD6738) in small cell lung cancer. Cancer. 2024;130:541–52. https://doi.org/10.1002/cncr.35059.
Woll P, et al. Olaparib as maintenance treatment in patients with chemosensitive small cell lung cancer (STOMP): a randomised, double-blind, placebo-controlled phase II trial. Lung Cancer. 2022;171:26–33. https://doi.org/10.1016/j.lungcan.2022.07.007.
Ai X, et al. Efficacy and safety of niraparib as maintenance treatment in patients with extensive-stage SCLC after first-line chemotherapy: a randomized, double-blind, phase 3 study. J Thorac Oncol. 2021;16:1403–14. https://doi.org/10.1016/j.jtho.2021.04.001.
de Bono J, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7:620–9. https://doi.org/10.1158/2159-8290.CD-16-1250.
Pietanza MC, et al. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol. 2018;36:2386–94. https://doi.org/10.1200/JCO.2018.77.7672.
Farago AF, et al. Combination olaparib and temozolomide in relapsed small-cell lung cancer. Cancer Discov. 2019;9:1372–87. https://doi.org/10.1158/2159-8290.CD-19-0582.
Krebs MG, et al. Olaparib and durvalumab in patients with relapsed small cell lung cancer (MEDIOLA): an open-label, multicenter, phase 1/2, basket study. Lung Cancer. 2023;180:107216. https://doi.org/10.1016/j.lungcan.2023.107216.
Thomas A, et al. Durvalumab in combination with olaparib in patients with relapsed SCLC: results from a phase II study. J Thorac Oncol. 2019;14:1447–57. https://doi.org/10.1016/j.jtho.2019.04.026.
Byers LA, et al. Veliparib in combination with carboplatin and etoposide in patients with treatment-naive extensive-stage small cell lung cancer: a phase 2 randomized study. Clin Cancer Res. 2021;27:3884–95. https://doi.org/10.1158/1078-0432.CCR-20-4259.
Owonikoko TK, et al. Randomized phase II trial of cisplatin and etoposide in combination with veliparib or placebo for extensive-stage small-cell lung cancer: ECOG-ACRIN 2511 study. J Clin Oncol. 2019;37:222–9. https://doi.org/10.1200/JCO.18.00264.
Paz-Ares L, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394:1929–39. https://doi.org/10.1016/S0140-6736(19)32222-6.
Lok BH, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res. 2017;23:523–35. https://doi.org/10.1158/1078-0432.CCR-16-1040.
Atrafi F, et al. A phase I dose-escalation study of veliparib combined with carboplatin and etoposide in patients with extensive-stage small cell lung cancer and other solid tumors. Clin Cancer Res. 2019;25:496–505. https://doi.org/10.1158/1078-0432.CCR-18-2014.
Willis SE, et al. Retrospective analysis of Schlafen11 (SLFN11) to predict the outcomes to therapies affecting the DNA damage response. Br J Cancer. 2021;125:1666–76. https://doi.org/10.1038/s41416-021-01560-1.
Allison Stewart C, et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget. 2017;8:28575–87. https://doi.org/10.1863/oncotarget.15338.
Owonikoko TK, et al. Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer. Cancer Med. 2014;3:1579–94. https://doi.org/10.1002/cam4.317.
Laird J, et al. Positron-emission tomographic imaging of a fluorine 18-radiolabeled poly(ADP-Ribose) polymerase 1 inhibitor monitors the therapeutic efficacy of talazoparib in SCLC patient-derived xenografts. J Thorac Oncol. 2019;14:1743–52. https://doi.org/10.1016/j.jtho.2019.05.032.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. https://doi.org/10.1016/j.molcel.2010.09.019.
Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–17. https://doi.org/10.1016/j.molcel.2017.05.015.
Saldivar JC, et al. An intrinsic S/G(2) checkpoint enforced by ATR. Science. 2018;361:806–10. https://doi.org/10.1126/science.aap9346.
Li X, et al. Immunogenicity of small-cell lung cancer associates with STING pathway activation and is enhanced by ATR and TOP1 inhibition. Cancer Med. 2023;12:4864–81. https://doi.org/10.1002/cam4.5109.
Thomas A, et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J Clin Oncol. 2018;36:1594–602. https://doi.org/10.1200/JCO.2017.76.6915.
Takahashi N, et al. Berzosertib plus topotecan vs topotecan alone in patients with relapsed small cell lung cancer: a randomized clinical trial. JAMA Oncol. 2023;9:1669–77. https://doi.org/10.1001/jamaoncol.2023.4025.
Schultz CW, et al. ATR inhibition augments the efficacy of lurbinectedin in small-cell lung cancer. EMBO Mol Med. 2023;15:e17313. https://doi.org/10.1525/emmm.202217313.
Krushkal J, et al. Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin Epigenetics. 2020;12:93. https://doi.org/10.1186/s13148-020-00876-8.
Christensen CL, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–22. https://doi.org/10.1016/j.ccell.2014.10.019.
Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134:1013–23. https://doi.org/10.1002/ijc.28226.
Gadhikar MA, et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12:1860–73. https://doi.org/10.1158/1535-7163.MCT-13-0157.
Hsu WH, et al. Checkpoint kinase 1 inhibition enhances cisplatin cytotoxicity and overcomes cisplatin resistance in SCLC by promoting mitotic cell death. J Thorac Oncol. 2019;14:1032–45. https://doi.org/10.1016/j.jtho.2019.01.028.
Thompson R, Meuth M, Woll P, Zhu Y, Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int J Oncol. 2012;40:194–202. https://doi.org/10.3892/ijo.2011.1187.
Teicher BA, et al. Small cell lung carcinoma cell line screen of etoposide/carboplatin plus a third agent. Cancer Med. 2017;6:1952–64. https://doi.org/10.1002/cam4.1131.
Byers LA, et al. A phase II trial of prexasertib (LY2606368) in patients with extensive-stage small-cell lung cancer. Clin Lung Cancer. 2021;22:531–40. https://doi.org/10.1016/j.cllc.2021.04.005.
Hong D, et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J Clin Oncol. 2016;34:1764–71. https://doi.org/10.1200/JCO.2015.64.5788.
Jones R, et al. A phase I/II trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in patients with advanced cancer. Clin Cancer Res. 2023;29:331–40. https://doi.org/10.1158/1078-0432.CCR-22-2074.
Sen T, et al. Combination treatment of the oral CHK1 inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J Thorac Oncol. 2019;14:2152–63. https://doi.org/10.1016/j.jtho.2019.08.009.
Zhao X, et al. Acquired small cell lung cancer resistance to Chk1 inhibitors involves Wee1 up-regulation. Mol Oncol. 2021;15:1130–45. https://doi.org/10.1002/1878-0261.12882.
Dammert MA, et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat Commun. 2019;10:3485. https://doi.org/10.1038/s41467-019-11371-x.
di Luserna Ghelli Rora A, Cerchione C, Martinelli G, Simonetti G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol. 2020;13:126. https://doi.org/10.1186/s13045-020-00959-2.
da Costa A, Chowdhury D, Shapiro GI, D’Andrea AD, Konstantinopoulos PA. Targeting replication stress in cancer therapy. Nat Rev Drug Discov. 2023;22:38–58. https://doi.org/10.1038/s41573-022-00558-5.
Fu S, Wang Y, Keyomarsi K, Meric-Bernstam F, Meric-Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs. 2018;27:741–51. https://doi.org/10.1080/13543784.2018.1511700.
Lallo A, et al. The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res. 2018;24:5153–64. https://doi.org/10.1158/1078-0432.CCR-17-2805.
Palve V, et al. The non-canonical target PARP16 contributes to polypharmacology of the PARP inhibitor talazoparib and its synergy with WEE1 inhibitors. Cell Chem Biol. 2022;29:202–14. https://doi.org/10.1016/j.chembiol.2021.07.008.
Taniguchi H, et al. WEE1 inhibition enhances the antitumor immune response to PD-L1 blockade by the concomitant activation of STING and STAT1 pathways in SCLC. Cell Rep. 2022;39:110814. https://doi.org/10.1016/j.celrep.2022.110814.
Lu Y, et al. Knocking down the expression of Aurora-A gene inhibits cell proliferation and induces G2/M phase arrest in human small cell lung cancer cells. Oncol Rep. 2014;32:243–9. https://doi.org/10.3892/or.2014.3194.
Oser MG, et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov. 2019;9:230–47. https://doi.org/10.1158/2159-8290.CD-18-0389.
Chi YH, et al. Discovery and synthesis of a pyrimidine-based aurora kinase inhibitor to reduce levels of MYC oncoproteins. J Med Chem. 2021;64:7312–30. https://doi.org/10.1021/acs.jmedchem.0c01806.
Sos ML, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci USA. 2012;109:17034–9. https://doi.org/10.1073/pnas.1207310109.
Li Y, et al. Aurora A kinase inhibition induces accumulation of SCLC tumor cells in mitosis with restored interferon signaling to increase response to PD-L1. Cell Rep Med. 2023;4:101282. https://doi.org/10.1016/j.xcrm.2023.101282.
Melichar B, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16:395–405. https://doi.org/10.1016/S1470-2045(15)70051-3.
Lim KH, et al. Phase 1 study combining alisertib with nab-paclitaxel in patients with advanced solid malignancies. Eur J Cancer. 2021;154:102–10. https://doi.org/10.1016/j.ejca.2021.06.012.
Owonikoko TK, et al. Randomized phase II study of paclitaxel plus alisertib versus paclitaxel plus placebo as second-line therapy for SCLC: primary and correlative biomarker analyses. J Thorac Oncol. 2020;15:274–87. https://doi.org/10.1016/j.jtho.2019.10.013.
de Sousa MJ, et al. Cyclin-dependent kinases 4/6 inhibitors in neuroendocrine neoplasms: from bench to bedside. Curr Oncol Rep. 2022;24:715–22. https://doi.org/10.1007/s11912-022-01251-x.
Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci. 2002;59:126–42. https://doi.org/10.1007/s00018-002-8410-1.
O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–30. https://doi.org/10.1038/nrclinonc.2016.26.
Lv S, et al. CDK4/6 inhibitors in lung cancer: current practice and future directions. Eur Respir Rev. 2024. https://doi.org/10.1183/16000617.0145-2023.
He S, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aal3986.
Hart LL, et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: results from a randomized, double-blind, placebo-controlled phase II study. Adv Ther. 2021;38:350–65. https://doi.org/10.1007/s12325-020-01538-0.
Daniel D, et al. Trilaciclib prior to chemotherapy and atezolizumab in patients with newly diagnosed extensive-stage small cell lung cancer: a multicentre, randomised, double-blind, placebo-controlled Phase II trial. Int J Cancer. 2021;148:2557–70. https://doi.org/10.1002/ijc.33453.
Weiss JM, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30:1613–21. https://doi.org/10.1093/annonc/mdz278.
Dhillon S. Trilaciclib: first approval. Drugs. 2021;81:867–74. https://doi.org/10.1007/s40265-021-01508-y.
Lai AY, et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2020-000847.
Holliday R. The inheritance of epigenetic defects. Science. 1987;238:163–70. https://doi.org/10.1126/science.3310230.
Khan P, et al. Epigenetic landscape of small cell lung cancer: small image of a giant recalcitrant disease. Semin Cancer Biol. 2022;83:57–76. https://doi.org/10.1016/j.semcancer.2020.11.006.
Mehta A, Dobersch S, Romero-Olmedo AJ, Barreto G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015;34:229–41. https://doi.org/10.1007/s10555-015-9563-3.
Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. https://doi.org/10.1016/j.cell.2004.12.012.
Takagi S, et al. LSD1 inhibitor T-3775440 inhibits SCLC cell proliferation by disrupting LSD1 interactions with SNAG domain proteins INSM1 and GFI1B. Cancer Res. 2017;77:4652–62. https://doi.org/10.1158/0008-5472.CAN-16-3502.
Chen HY, et al. Regulation of neuroendocrine plasticity by the RNA-binding protein ZFP36L1. Nat Commun. 2022;13:4998. https://doi.org/10.1038/s41467-022-31998-7.
Mohammad HP, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69. https://doi.org/10.1016/j.ccell.2015.06.002.
Qin Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene. 2019;38:390–405. https://doi.org/10.1038/s41388-018-0451-5.
Han Y, et al. Targeting LSD1 suppresses stem cell-like properties and sensitizes head and neck squamous cell carcinoma to PD-1 blockade. Cell Death Dis. 2021;12:993. https://doi.org/10.1038/s41419-021-04297-0.
Nguyen EM, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility complex class I antigen presentation and overcomes programmed death-ligand 1 blockade resistance in SCLC. J Thorac Oncol. 2022;17:1014–31. https://doi.org/10.1016/j.jtho.2022.05.014.
Hiatt JB, et al. Inhibition of LSD1 with bomedemstat sensitizes small cell lung cancer to immune checkpoint blockade and T-cell killing. Clin Cancer Res. 2022;28:4551–64. https://doi.org/10.1158/1078-0432.CCR-22-1128.
Ramaiah MJ, Tangutur AD, Manyam RR. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021;277:119504. https://doi.org/10.1016/j.lfs.2021.119504.
Wu Z, et al. YAP silencing by RB1 mutation is essential for small-cell lung cancer metastasis. Nat Commun. 2023;14:5916. https://doi.org/10.1038/s41467-023-41585-z.
Lin CA, et al. EGFR-mutant SCLC exhibits heterogeneous phenotypes and resistance to common antineoplastic drugs. J Thorac Oncol. 2019;14:513–26. https://doi.org/10.1016/j.jtho.2018.11.021.
Pan CH, et al. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer. 2016;16:857. https://doi.org/10.1186/s12885-016-2888-7.
Solta A, et al. Entinostat enhances the efficacy of chemotherapy in small cell lung cancer through s-phase arrest and decreased base excision repair. Clin Cancer Res. 2023;29:4644–59. https://doi.org/10.1158/1078-0432.CCR-23-1795.
de Marinis F, et al. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer. J Thorac Oncol. 2013;8:1091–4. https://doi.org/10.1097/JTO.0b013e318293d88c.
Balasubramaniam S, et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anticancer Drugs. 2018;29:457–65. https://doi.org/10.1097/CAD.0000000000000596.
Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106:243–7. https://doi.org/10.1038/bjc.2011.551.
Bracken AP, et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–35. https://doi.org/10.1093/emboj/cdg542.
Hubaux R, et al. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol. 2013;8:1102–6. https://doi.org/10.1097/JTO.0b013e318298762f.
Gardner EE, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell. 2017;31:286–99. https://doi.org/10.1016/j.ccell.2017.01.006.
Niu Y, et al. Long non-coding RNA TUG1 is involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2. Mol Cancer. 2017;16:5. https://doi.org/10.1186/s12943-016-0575-6.
Zhai G, et al. hTERT promoter methylation promotes small cell lung cancer progression and radiotherapy resistance. J Radiat Res. 2020;61:674–83. https://doi.org/10.1093/jrr/rraa052.
Villanueva L, Alvarez-Errico D, Esteller M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020;41:676–91. https://doi.org/10.1016/j.it.2020.06.002.
Ciminera AK, Jandial R, Termini J. Metabolic advantages and vulnerabilities in brain metastases. Clin Exp Metastasis. 2017;34:401–10. https://doi.org/10.1007/s10585-017-9864-8.
Kwon SH, et al. The highest metabolic activity on FDG PET is associated with overall survival in limited-stage small-cell lung cancer. Medicine. 2016;95:e2772. https://doi.org/10.1097/MD.0000000000002772.
Zhu D, et al. Prognostic significance of metabolic parameters measured by (18)F-fluorodeoxyglucose positron emission tomography/computed tomography in patients with small cell lung cancer. Lung Cancer. 2011;73:332–7. https://doi.org/10.1016/j.lungcan.2011.01.007.
Cargill KR, et al. Targeting MYC-enhanced glycolysis for the treatment of small cell lung cancer. Cancer Metab. 2021;9:33. https://doi.org/10.1186/s40170-021-00270-9.
Kong F, et al. Metformin use improves the survival of diabetic combined small-cell lung cancer patients. Tumour Biol. 2015;36:8101–6. https://doi.org/10.1007/s13277-015-3549-1.
Kim Y, et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol Immunother. 2021;70:961–5. https://doi.org/10.1007/s00262-020-02703-8.
Chan DA, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3:94–70. https://doi.org/10.1126/scitranslmed.3002394.
Wu KH, et al. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J Food Drug Anal. 2018;26:221–31. https://doi.org/10.1016/j.jfda.2017.03.009.
Krencz I, Sztankovics D, Danko T, Sebestyen A, Khoor A. Progression and metastasis of small cell lung carcinoma: the role of the PI3K/Akt/mTOR pathway and metabolic alterations. Cancer Metastasis Rev. 2021;40:1141–57. https://doi.org/10.1007/s10555-021-10012-4.
Kelly MP, et al. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer. 2012;106:324–32. https://doi.org/10.1038/bjc.2011.524.
Hu Q, et al. ASS1-mediated reductive carboxylation of cytosolic glutamine confers ferroptosis resistance in cancer cells. Cancer Res. 2023;83:1646–65. https://doi.org/10.1158/0008-5472.CAN-22-1999.
Zhou B, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022;7:95. https://doi.org/10.1038/s41392-022-00934-y.
Zhang H, Yang Y, Li X, Yuan X, Chu Q. Targeting the Notch signaling pathway and the Notch ligand, DLL3, in small cell lung cancer. Biomed Pharmacother. 2023;159:114248. https://doi.org/10.1016/j.biopha.2023.114248.
Sabari JK, Lok BH, Laird JH, Poirier JT, Rudin CM. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol. 2017;14:549–61. https://doi.org/10.1038/nrclinonc.2017.71.
Johnson ML, et al. Rovalpituzumab tesirine as a maintenance therapy after first-line platinum-based chemotherapy in patients with extensive-stage-SCLC: results from the phase 3 MERU study. J Thorac Oncol. 2021;16:1570–81. https://doi.org/10.1016/j.jtho.2021.03.012.
Blackhall F, et al. Efficacy and safety of rovalpituzumab tesirine compared with topotecan as second-line therapy in DLL3-High SCLC: results from the phase 3 TAHOE study. J Thorac Oncol. 2021;16:1547–58. https://doi.org/10.1016/j.jtho.2021.02.009.
Goebeler ME, Bargou RC. T cell-engaging therapies—BiTEs and beyond. Nat Rev Clin Oncol. 2020;17:418–34. https://doi.org/10.1038/s41571-020-0347-5.
Paz-Ares L, et al. Tarlatamab, a first-in-class DLL3-targeted bispecific T-cell engager, in recurrent small-cell lung cancer: an open-label, phase I study. J Clin Oncol. 2023;41:2893–903. https://doi.org/10.1200/JCO.22.02823.
Ahn MJ, et al. Tarlatamab for patients with previously treated small-cell lung cancer. N Engl J Med. 2023;389:2063–75. https://doi.org/10.1056/NEJMoa2307980.
Wang Z, et al. Investigation of the efficacy and feasibility of combined therapy of PD-L1-enhanced exogenous peripatetic adoptive natural killer (NK) cells in combination with antiangiogenic targeted therapy in the treatment of extensive-stage small cell lung cancer. Thorac Cancer. 2023;14:2877–85. https://doi.org/10.1111/1759-7714.15040.
Yen WC, et al. Targeting notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. 2015;21:2084–95. https://doi.org/10.1158/1078-0432.CCR-14-2808.
Smith DC, et al. A phase 1 dose escalation and expansion study of tarextumab (OMP-59R5) in patients with solid tumors. Invest New Drugs. 2019;37:722–30. https://doi.org/10.1007/s10637-018-0714-6.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. https://doi.org/10.1038/nrm3722.
Messaritakis I, et al. Bcl-2 expression in circulating tumor cells (CTCs) of patients with small cell lung cancer (SCLC) receiving front-line treatment. Lung Cancer. 2018;124:270–8. https://doi.org/10.1016/j.lungcan.2018.08.021.
Ramkumar K, et al. Targeting BCL2 overcomes resistance and augments response to aurora kinase B inhibition by AZD2811 in small cell lung cancer. Clin Cancer Res. 2023;29:3237–49. https://doi.org/10.1158/1078-0432.CCR-23-0375.
Wang WZ, et al. Predicting ROR1/BCL2 combination targeted therapy of small cell carcinoma of the lung. Cell Death Dis. 2021;12:577. https://doi.org/10.1038/s41419-021-03855-w.
Rudin CM, et al. Phase I study of G3139, a bcl-2 antisense oligonucleotide, combined with carboplatin and etoposide in patients with small-cell lung cancer. J Clin Oncol. 2004;22:1110–7. https://doi.org/10.1200/JCO.2004.10.148.
Rudin CM, et al. A pilot trial of G3139, a bcl-2 antisense oligonucleotide, and paclitaxel in patients with chemorefractory small-cell lung cancer. Ann Oncol. 2002;13:539–45. https://doi.org/10.1093/annonc/mdf124.
Langer CJ, et al. Randomized phase II study of carboplatin and etoposide with or without obatoclax mesylate in extensive-stage small cell lung cancer. Lung Cancer. 2014;85:420–8. https://doi.org/10.1016/j.lungcan.2014.05.003.
Baggstrom MQ, et al. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol. 2011;6:1757–60. https://doi.org/10.1097/JTO.0b013e31822e2941.
Rudin CM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–9. https://doi.org/10.1158/1078-0432.CCR-11-3090.
Pillai RN, et al. Interferon alpha plus 13-cis-retinoic acid modulation of BCL-2 plus paclitaxel for recurrent small-cell lung cancer (SCLC): an eastern cooperative oncology group study (E6501). Cancer Chemother Pharmacol. 2014;74:177–83. https://doi.org/10.1007/s00280-014-2427-7.
Rudin CM, et al. Randomized phase II study of carboplatin and etoposide with or without the bcl-2 antisense oligonucleotide oblimersen for extensive-stage small-cell lung cancer: CALGB 30103. J Clin Oncol. 2008;26:870–6. https://doi.org/10.1200/JCO.2007.14.3461.
Reck M, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83. https://doi.org/10.1093/annonc/mds213.
Antonia SJ, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–95. https://doi.org/10.1016/s1470-2045(16)30098-5.
Chung HC, et al. Pembrolizumab after two or more lines of previous therapy in patients with recurrent or metastatic SCLC: results from the KEYNOTE-028 and KEYNOTE-158 studies. J Thorac Oncol. 2020;15:618–27. https://doi.org/10.1016/j.jtho.2019.12.109.
Ott PA, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the phase Ib KEYNOTE-028 study. J Clin Oncol. 2017;35:3823–9. https://doi.org/10.1200/JCO.2017.72.5069.
Spigel DR, et al. Second-line nivolumab in relapsed small-cell lung cancer: CheckMate 331(☆). Ann Oncol. 2021;32:631–41. https://doi.org/10.1016/j.annonc.2021.01.071.
Gadgeel SM, et al. Phase II study of maintenance pembrolizumab in patients with extensive-stage small cell lung cancer (SCLC). J Thorac Oncol. 2018;13:1393–9. https://doi.org/10.1016/j.jtho.2018.05.002.
Owonikoko TK, et al. Nivolumab and ipilimumab as maintenance therapy in extensive-disease small-cell lung cancer: CheckMate 451. J Clin Oncol. 2021;39:1349–59. https://doi.org/10.1200/JCO.20.02212.
Reck M, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol. 2016;34:3740–8. https://doi.org/10.1200/JCO.2016.67.6601.
Rudin CM, et al. Pembrolizumab or placebo plus etoposide and platinum as first-line therapy for extensive-stage small-cell lung cancer: randomized, double-blind, phase III KEYNOTE-604 study. J Clin Oncol. 2020;38:2369–79. https://doi.org/10.1200/JCO.20.00793.
Goldman JW, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2021;22:51–65. https://doi.org/10.1016/S1470-2045(20)30539-8.
Wang J, et al. Adebrelimab or placebo plus carboplatin and etoposide as first-line treatment for extensive-stage small-cell lung cancer (CAPSTONE-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022;23:739–47. https://doi.org/10.1016/S1470-2045(22)00224-8.
Cheng Y, et al. Effect of first-line serplulimab vs placebo added to chemotherapy on survival in patients with extensive-stage small cell lung cancer: the ASTRUM-005 randomized clinical trial. JAMA. 2022;328:1223–32. https://doi.org/10.1001/jama.2022.16464.
Cheng Y, et al. Tislelizumab plus platinum and etoposide versus placebo plus platinum and etoposide as first-line treatment for extensive-stage SCLC (RATIONALE-312): a multicenter, double-blind, placebo-controlled, randomized, phase 3 clinical trial. J Thor Oncol. 2024. https://doi.org/10.1016/j.jtho.2024.03.008.
Donlon NE, Power R, Hayes C, Reynolds JV, Lysaght J. Radiotherapy, immunotherapy, and the tumour microenvironment: turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021;502:84–96. https://doi.org/10.1016/j.canlet.2020.12.045.
Ganti AKP, et al. Small cell lung cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;19:1441–64. https://doi.org/10.6004/jnccn.2021.0058.
Fan Y, et al. Camrelizumab plus apatinib in extensive-stage SCLC (PASSION): a multicenter, two-stage, phase 2 trial. J Thorac Oncol. 2021;16:299–309. https://doi.org/10.1016/j.jtho.2020.10.002.
Cheng Y, et al. Benmelstobart, anlotinib and chemotherapy in extensive-stage small-cell lung cancer: a randomized phase 3 trial. Nat Med. 2024. https://doi.org/10.1038/s41591-024-03132-1.
Rudin CM, et al. SKYSCRAPER-02: tiragolumab in combination with atezolizumab plus chemotherapy in untreated extensive-stage small-cell lung cancer. J Clin Oncol. 2023. https://doi.org/10.1200/JCO.23.01363.
Park S, et al. Durvalumab with chemoradiotherapy for limited-stage small-cell lung cancer. Eur J Cancer. 2022;169:42–53. https://doi.org/10.1016/j.ejca.2022.03.034.
Welsh JW, et al. Phase 1/2 trial of pembrolizumab and concurrent chemoradiation therapy for limited-stage SCLC. J Thorac Oncol. 2020;15:1919–27. https://doi.org/10.1016/j.jtho.2020.08.022.
Peters S, et al. Consolidation nivolumab and ipilimumab versus observation in limited-disease small-cell lung cancer after chemo-radiotherapy—results from the randomised phase II ETOP/IFCT 4–12 STIMULI trial. Ann Oncol. 2022;33:67–79. https://doi.org/10.1016/j.annonc.2021.09.011.
Senan S, et al. Design and rationale for a phase III, randomized, placebo-controlled trial of durvalumab with or without tremelimumab after concurrent chemoradiotherapy for patients with limited-stage small-cell lung cancer: the ADRIATIC study. Clin Lung Cancer. 2020;21:e84–8. https://doi.org/10.1016/j.cllc.2019.12.006.
Imfinzi significantly improved overall survival and progression-free survival for patients with limited-stage small cell lung cancer in ADRIATIC Phase III trial, <https://www.astrazeneca.com/media-centre/press-releases/2024/imfinzi-improved-os-and-pfs-in-limited-stage-sclc.html> (5 April 2024).
Y C, et al. Adebrelimab with concurrent chemoradiation (cCRT) for limited-stage small cell lung cancer (LS-SCLC): safety run-in results of a phase III trial. Eur Lung Cancer Congress. 2024. https://doi.org/10.1016/esmoop/esmoop102577.
Lang C, et al. Clinical and prognostic implications of CD47 and PD-L1 expression in surgically resected small-cell lung cancer. ESMO Open. 2022;7:100631. https://doi.org/10.1016/j.esmoop.2022.100631.
Weiskopf K, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126:2610–20. https://doi.org/10.1172/JCI81603.
Barkal AA, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19:76–84. https://doi.org/10.1038/s41590-017-0004-z.
Theruvath J, et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat Med. 2022;28:333–44. https://doi.org/10.1038/s41591-021-01625-x.
Nishiga Y, et al. Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat Cancer. 2022;3:1351–66. https://doi.org/10.1038/s43018-022-00456-0.
Andrzejczak A, Karabon L. BTLA biology in cancer: from bench discoveries to clinical potentials. Biomark Res. 2024;12:8. https://doi.org/10.1186/s40364-024-00556-2.
Sordo-Bahamonde C, et al. Beyond the anti-PD-1/PD-L1 era: promising role of the BTLA/HVEM axis as a future target for cancer immunotherapy. Mol Cancer. 2023;22:142. https://doi.org/10.1186/s12943-023-01845-4.
Yu Y, et al. Tifcemalimab combined with toripalimab and chemotherapy as 1st line treatment for extensive-stage small cell lung cancer (ES-SCLC): a phase Ib/II, open-label study. ASCO Ann Meet. 2024. https://doi.org/10.1200/JCO.2024.42.16_suppl.8089.
Kontos F, et al. B7–H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res. 2021;27:1227–35. https://doi.org/10.1158/1078-0432.CCR-20-2584.
Fabrizio FP, Muscarella LA, Rossi A. B7–H3/CD276 and small-cell lung cancer: What’s new? Transl Oncol. 2024;39:101801. https://doi.org/10.1016/j.tranon.2023.101801.
Saidak Z, et al. A pan-cancer analysis of the human tumor coagulome and its link to the tumor immune microenvironment. Cancer Immunol Immunother. 2021;70:923–33. https://doi.org/10.1007/s00262-020-02739-w.
Qiu MJ, et al. The expression of three negative co-stimulatory B7 family molecules in small cell lung cancer and their effect on prognosis. Front Oncol. 2021;11:600238. https://doi.org/10.3389/fonc.2021.600238.
Demaria O, et al. Harnessing innate immunity in cancer therapy. Nature. 2019;574:45–56. https://doi.org/10.1038/s41586-019-1593-5.
Andre P, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731–43. https://doi.org/10.1016/j.cell.2018.10.014.
Chiappori AA, et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.p53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol Immunother. 2019;68:517–27. https://doi.org/10.1007/s00262-018-2287-9.
Huang J, et al. Chemotherapy in combination with cytokine-induced killer cell transfusion: an effective therapeutic option for patients with extensive stage small cell lung cancer. Int Immunopharmacol. 2017;46:170–7. https://doi.org/10.1016/j.intimp.2016.12.005.
Ma B, et al. Sintilimab maintenance therapy post first-line cytokine-induced killer cells plus chemotherapy for extensive-stage small cell lung cancer. Front Oncol. 2022;12:852885. https://doi.org/10.3389/fonc.2022.852885.
Dash AS, Patel MR. Viroimmunotherapy of thoracic cancers. Biomedicines. 2017. https://doi.org/10.3390/biomedicines5010002.
Schenk EL, et al. A randomized double-blind phase II study of the seneca valley virus (NTX-010) versus placebo for patients with extensive-stage SCLC (ES SCLC) who were stable or responding after at least four cycles of platinum-based chemotherapy: north central cancer treatment group (Alliance) N0923 study. J Thorac Oncol. 2020;15:110–9. https://doi.org/10.1016/j.jtho.2019.09.083.
Kellish P, et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J Clin Invest. 2019;129:2279–92. https://doi.org/10.1172/JCI121323.
George J, et al. Evolutionary trajectories of small cell lung cancer under therapy. Nature. 2024;627:880–9. https://doi.org/10.1038/s41586-024-07177-7.
Wagner AH, et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat Commun. 2018;9:3787. https://doi.org/10.1038/s41467-018-06162-9.
Park S, et al. DNA damage response and repair pathway alteration and its association with tumor mutation burden and platinum-based chemotherapy in SCLC. J Thorac Oncol. 2019;14:1640–50. https://doi.org/10.1016/j.jtho.2019.05.014.
Yokouchi H, et al. Detection of somatic TP53 mutation in surgically resected small-cell lung cancer by targeted exome sequencing: association with longer relapse-free survival. Heliyon. 2020;6:e04439. https://doi.org/10.1016/j.heliyon.2020.e04439.
Udagawa H, et al. Genetic profiling-based prognostic prediction of patients with advanced small-cell lung cancer in large scale analysis. Lung Cancer. 2018;126:182–8. https://doi.org/10.1016/j.lungcan.2018.11.014.
Umemura S, et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J Thorac Oncol. 2014;9:1324–31. https://doi.org/10.1097/jto.0000000000000250.
Zhou H, et al. Multi-region exome sequencing reveals the intratumoral heterogeneity of surgically resected small cell lung cancer. Nat Commun. 2021;12:5431. https://doi.org/10.1038/s41467-021-25787-x.
Acknowledgements
Not applicable.
Funding
This study was supported by the Innovation Program of Shanghai Municipal Education Commission (Grant No. 2023ZKZD33) and the foundation of Shanghai Pulmonary Hospital (Grant No. FKLY20004, FKCX2304).
Author information
Authors and Affiliations
Contributions
Jing Zhang: Conceptualization, Data curation, Writing—original draft; Xiaoping Zeng: Data curation, Writing—original draft; Qiji Guo: Data curation, Writing—original draft; Zhenxin Sheng: Data curation, Writing—original draft; Yan Chen:Data curation; Shiyue Wan: Supervision; Lele Zhang: Supervision; Peng Zhang: Conceptualization, Data curation, Supervision, Funding acquisition, Writing—review & editing.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, J., Zeng, X., Guo, Q. et al. Small cell lung cancer: emerging subtypes, signaling pathways, and therapeutic vulnerabilities. Exp Hematol Oncol 13, 78 (2024). https://doi.org/10.1186/s40164-024-00548-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-024-00548-w