
Abstract
Immune checkpoint blockade (ICB) necessitates a thorough understanding of intricate cellular interactions within the tumor microenvironment (TME). Mesenchymal stromal cells (MSCs) play a pivotal role in cancer generation, progression, and immunosuppressive tumor microenvironment. Within the TME, MSCs encompass both resident and circulating counterparts that dynamically communicate and actively participate in TME immunosurveillance and response to ICB. This review aims to reevaluate various facets of MSCs, including their potential self-transformation to function as cancer-initiating cells and contributions to the creation of a conducive environment for tumor proliferation and metastasis. Additionally, we explore the immune regulatory functions of tumor-associated MSCs (TA-MSCs) and MSC-derived extracellular vesicles (MSC-EVs) with analysis of potential connections between circulating and tissue-resident MSCs. A comprehensive understanding of the dynamics of MSC-immune cell communication and the heterogeneous cargo of tumor-educated versus naïve MSCs may unveil a new MSC-mediated immunosuppressive pathway that can be targeted to enhance cancer control by ICB.
Similar content being viewed by others
Background
Multiple intrinsic cascades of genomic and epigenetic events denoted as the hallmarks of cancer are believed to drive the malignant cell transformation and tumor progression [1, 2]. However, in addition to the well-defined cell intrinsic pre-cancer trends, increasing evidence indicates a potential systemic and/or tissue environmental factor leading to a global pre-cancerous status [3,4,5]. Such a pre-cancerous environmental status is triggered by imbalanced tissue homeostasis and architecture far before the malignant phenotype can be noticed [6]. Consistently, non-mutational epigenetic events and senescent cells are suggested to play an effective role in the tumor microenvironment (TME) [3], and cancer origination, aggressiveness, and metastasis are indicated to be the result of communications between pre-cancer cells or tumors and the extracellular environment [7]. This central dogma of cancer is evidenced by the well-defined pro-cancer chronic inflammation that generates pro-inflammatory cytokines and growth factors [7,8,9]. It is assumed that even at the very early tumor initiation phase, a variety of cell types from local and distant niches are recruited into the growing neoplasm to provide stromal support for the transformed cells [10]. Such a pre-cancerous stroma can further attract different regulatory molecules to mobilize an array of cell types to join in the "transforming niche," in which mesenchymal stromal cells (MSCs) may guide the process.
In adult humans, MSCs are generally identified by detecting several non-hematopoietic markers, e.g., CD29, CD44, CD73, CD90, CD105, and by the characteristics of differentiating towards diverse cell types, including adipocytes, osteoblasts, chondrocytes, and connective tissues [11]. In addition to the homing function of circulating MSCs, a vast number of tissue-resident MSCs are primarily located in perivascular sites and are capable of quickly responding to external stimuli to differentiate into pericytes to regulate vascular morphogenesis and biological functions [12, 13]. Due to their injury-homing properties, along with the relative enriched resources for isolation and in vitro expansion, priming MSCs or engineered MSC/MSC-EVs are extensively investigated for disease treatment [14], and regenerative and drug delivery therapies [15]. MSCs-regulated immune inhibition offers a desirable approach in adjuvant immunomodulation post-allogeneic transplantation [16, 17] and cancer control [18]. Companying with the wholesale clinical applications, MSCs from different tissue resources are cautioned by a potential risk of malignant initiation and cancer progression owning to their stem cell features and immunosuppressive potential. The tissue-resident MSCs include the TA-MSCs highlighted to be a critical element in TME [18, 19], and targeting TA-MSCs by different approaches including radiotherapy diminished tumor aggressiveness [20]. Given that myriad factors influence the phenotype and function of MSCs within the TME [21], it holds paramount significance to further elucidate the fundamental feature of MSCs beyond the communication and coordination with tumor and immune cells in the TME.
MSCs as the potential cancer-initiating cells
Animal MSCs self-transformation
Observations that lower organisms can regenerate multiple tissues and organs have led to the proposal of somatic stem cells (SCs) [22], which are generally believed to be a small fraction of somatic cells capable of self-renewing and are responsible for tissue regeneration and homeostasis [23, 24]. Experimental data suggest that SCs share some essential features with the clonogenic tumor cells or the tumor-initiating cells (TICs) illustrating the similarity between the two types of cells [25]. With this hypothesis, MSCs are assumed to be the original cellular resources holding the pre-cancerous trends when a certain level of cancer hallmark gene mutation is achieved through accumulated cell division times [26, 27]. Additionally, it has been identified that a single passage in culture extensively alters MSCs molecular signatures associated with cell cycling, differentiation, and immune response, highlighting the need to clarify the consequence of the transition [28]. However, unlike fully transformed cancer cells, MSCs demonstrate both pro- and anti-tumor functions [29], thus, the precise biological connections between MSCs and TICs remain to be further elucidated. Although vast evidence illustrates that mouse bone marrow-derived mesenchymal stromal cells (mBMSCs) promote tumor growth in cancer-carrying mice, the confidence level of true transformation of MSCs in vivo is still under debate, and DNA fingerprinting is suggested in such experimental settings [30,31,32,33]. Nonetheless, the following reports are cited supporting the concept that mMSCs can spontaneously undergo transformation in vitro and generate sarcoma in vivo [34, 35] (Fig. 1A). Using the tracing approach in aged mice, Houghton’s group demonstrates that MSCs were able to spontaneously transform with p53 mutation leading to fibrosarcoma generation. Of note, the transformed MSCs not only rooted the tumor mass but also generated the tumor-associated vasculature and stromal supports [36]. Similarly, CD44+/CD29+ mBMSCs transplanted into myocardial infarction and diabetic neuropathy mice resulted in the formation of malignant sarcoma with chromosomal aberrations including fusion, fragmentation, and ring formation [37]. mBMSCs can also acquire immortality with transformation behavior and fibrosarcoma formation via altered telomerase activity, c-myc expression [38], and Notch+/Hh−/Wnt− signaling pathway[39]. In addition, MSCs derived from cynomolgus monkeys were tumor-generative after long-term in vitro expansion [40]. Such pro-transformative tendencies in MSCs could be unique, as they are not observed in most other mouse stem cells, including hematopoietic and embryonic stem cells [41]. A recent report further demonstrated that with single-cell RNAseq and lineage tracing approaches mMSCs can undergo mesenchymal-epithelial transition (MET) and be incorporated into the re-epithelialized luminal surface of the repaired tissue [42]. However, it remains to be further characterized and verified how cancer cells could at least in part result from a potential intermediate cell type derived from mMSCs via MET.

MSCs serve as cancer-initiating cells via self-transformation and/or pro-cancerous niches. A In animal models, MSCs can spontaneously transform vitro and in vivo (sarcoma formation) by cytogenetic abnormalities, chromosomal aberrations, telomerase activity alterations, c-myc expression, p53 mutation, MET and Notch( +)/Hh(−)/Wnt(−) signaling pathway. In human, MSCs self-transformation depends on chromosome aneuploid, altered telomerase activity, LIN28B/LET-7, hTERT/H-ras, c-myc, p16, DNA hypomethylation, MET. B Different genotoxic and cellular stress conditions including UV, ionizing radiation, virus infection, and chemical carcinogens, lead to a pro-cancer niche with the recruitment of inflammatory cells, and the resident and/or circulating MSCs. Damages or loss of homeostasis of the local epithelial tissue enhance the homing of MSCs followed by attraction of an array of the inflammatory cells and their derived cytokines that triggers the oncogenic transformation of stem cell or progenitor cells initiating a malignant proliferative cancer niche in which the precise cellular resource of the pre-tumor cells remains unelucidated (cc, cancer cells)
Human MSCs self-transformation
Similarly, to animal models, the idea of human MSCs (hMSCs) being the original source of both epithelial and non-epithelial malignancy is still a topic of debate. Long-term in vitro expansion of hMSCs can induce senescence-associated phenotype with chromosomal alterations including aneuploidy and polyploidy but no self-transformation is detected [43, 44]. However, variable aneuploid clone proportions were identified in a large group of hMSCs suggesting a potential transform trend [45]. A pioneer work by Wang’s group provided the first in vivo evidence that BMSCs are the potential cancer originating cells in a mouse gastric tumor model [46]. Such evidence has been further strengthened by a series of studies supporting that the spontaneous transformation of hBMSCs can be enhanced by LIN28B expression leading to sarcoma formation in immunocompromised mice, suggesting a prognostic factor for clinic sarcoma patients [47]. The potential hMSCs tumorigenic potential was achieved as high as 45.8% by in vitro expansion [48] with a significantly proliferative capacity [48, 49]. In a set of 46 independent cultures of hMSCs, four batches of transformed MSCs were able to generate sarcoma-like tumors in immunodeficient mice [48]. The pathological features of the transformed cells include cells with spherical, cuboidal to spindly in shape, adherent, and exhibited contact-independent growth. Cytogenetic analysis showed chromosome aneuploidy and translocations with a higher level of telomerase activity compared with typical MSCs, which led to multiple solid tumors when transplanted into immune compromised host [30]. Furthermore, the transformation of hMSCs could be induced by the exogenous expression of hTERT, H-ras [50, 51]. In the course of transformation, senescence is avoided via upregulation of c-myc and repression of p16 which was accompanied with reduced mitochondrial metabolism and DNA damage repair capacity [52]. DNA hypomethylation was also indicated to occur late during stepwise MSCs transformation and was not indispensable during the process of transformation in vitro [53]. While MSC-originated tumorigenesis is mainly limited to sarcoma, it is possible that MET observed in cancer metastasis may be acquired during MSC self-transformation, leading to different epithelial source malignancy [46, 54,55,56]. Like the animal syngeneic MSC cancer model, it remains to be investigated whether human epithetical cancers could be rooted from hMSCs or hMSC-like intermediate forms of pre-cancer cell types via MET.
MSCs identified in the pro-cancerous niches
Cell transformation is tightly associated with chronic inflammation or induces an inflammatory response (tumor-elicited inflammation), both supporting that chronic inflammation is an extrinsic factor for malignant cell transformation in the pre-cancerous tissue niche [57]. Probably by a similar cluster of attractors, MSCs are shown to migrate to the tumor site resembling the migration toward injured tissue. MSCs gathered from the local tissue and/or the circulating MSCs are recruited and reside in the so-called inflammatory niche (Fig. 1B) generated from different injuries, including virus infection and mechanical stress such as cut and ionizing radiation, which is documented to be a pre-cancer environment. Inflammatory cytokines such as TNF-α and IL-1β confer MSCs the ability to release high levels of CCL2, CXCL8 and CCL5, which lead to exacerbated inflammatory and pro-cancerous profiles [58]. These results demonstrate the possibility that the circulating MSCs are actively involved in creating the pro-malignant inflammatory tissue environment motivating a favorable condition for cell transformation. It remains to be examined if an unknown transition type of MSCs could be the original TICsin the pre-cancerous niches. In addition, MSCs could regulate the ecological dynamics leading to the transition of the pre-cancerous niches to the fully transformed malignant tumor microenvironment.
MSCs contribute to pro-tumor ecological environment
The ecological dynamics of TME feature the evolution of tumor cells related to the host stromal environment [59,60,61,62]. Although it is unknown how MSCs contributed to the overall ecology of TME, increasing evidence indicates that MSCs promote tumor proliferation. Tumor growth is boosted by administration of MSCs into the systemic circulation of tumor-bearing animals [63]. Orthotopic gastric tumors can be enhanced if the host mice receive transplantation of syngeneic mBM-MSCs [64]. The tumor-boosting function is also observed by co-injection of tumor cells with MSCs isolated from human head and neck carcinoma [65], gastric cancer [66], and gliomas [67], which is potentially related to the immunosuppressive function of MSCs [68]. The MSC-attracting functions of TME are generated by the multiple tumor-secreted chemokines, cytokines, and growth factors (Fig. 2) for recruiting MSCs from bone marrow or adipose tissue towards tumor xenografts. VEGF [69], FGF2 [69], PGF [70], IL-6 [71], IL-8 [72], HGF [73], SDF-1 [74], IGF-1 [75], MCP-1 [76], uPA [77], PGE2 [78], TGF-β1 [79] among others have been identified as the tumor-MSC recruiting factors. Such MSC-attracting dynamics function to recruit MSCs and educate the MSCs in the TME. Liu et al. found that MSCs mediated C26 colon cancer growth with enhanced angiogenesis if MSCs were pre-stimulated with both IFN-γ and TNF-α rather than with either IFN-γ or TNF-α alone [80]. When arriving to TME, MSCs may acutely or chronically differentiate into TA-MSCs or cancer-associated fibroblasts (CAFs) by the tumor-guided education, further assisting the malignant progression [81]. Tumor-educated MSCs can further activate and release the chemoprotective and immunomodulatory factors including CXCL1, CXCL2, and IL-8, favoring tumor progression [82] in which MSCs generated CXCL2, VEGF, TGF-β, and IL-6 can further raise tumor aggressive phenotype by boosting tumor angiogenesis [83]. MSC-mediated tumor ecologic dynamics are also related to MSC-related TME immunosuppression [84]. MSCs co-injected with inflammatory breast cancer cells can stimulate the secretion of IL-6 from macrophages which is required for the colonization of the inflammatory breast cancer [85]. Such pro-tumor ecology is further illustrated by the communication between tumor cells and stromal cells with a feed-forward loop generating a metabolic synergy for tumor energy consumption demands [86]. The tumor-educated MSCs are actively involved in the metabolic reprogramming in the TME in which the mitochondria of MSCs play a central role in driving the MSC-boosted tumor progression via an energy-transferring mechanism [87], thereby increasing cell proliferation and invasion of breast cancer and glioblastoma cells [88, 89]. Suppression of MSCs migration capacity inhibits MSC-enhanced tumor aggressive phenotype [90] in which miR-126 is shown to inhibit SDF-1α expression and diminish MSC recruitment into TME [91]. Further elucidation of the mechanistic insights and key elements required for MSC recruitment is mandatory for the invention of therapeutic approaches to reverse the proliferative tumor ecology.

MSCs recruitment by tumor-secreted elements accelerate tumor proliferation. Tumor-secreted bioactive elements including chemokines (CXCL12, MCP-1), cytokines (IL-6, IL-8), growth factors (VEGF, FGF2, HGF, IGF-1, TGF-β1, PGF), and other factors (uPA, PGE2) favor the recruitment of MSCs into TME. In TME, MSCs can be further educated into TA-MSC and/or CAF, both promoting tumor cell proliferation via mitochondrial transfer and secreting factors including CXCL-1, CXCL-2, VEGF, TGF-β, IL-8, and IL-6. Simultaneously, immune cell response activated by inflammatory factors in TME is inhibited by recruited MSCs by cell–cell contact, soluble factors, miRNA, and EVs approaches, leading to the establishment of the immunosuppressive TME. accelerates
MSCs enhance tumor cell metastatic capacity and enrich metastatic tissue niche
MSCs enhance tumor metastatic capacity
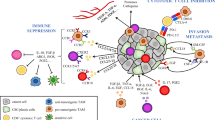
The tumor cells capable of colonization into the metastatic site can be boosted by MSC-derived cytokines (Fig. 3A). TA-MSCs and CAFs accelerate the progression of tumor cells towards a more aggressive phenotype, including invasive and pro-metastatic states [92, 93]. Myeloma cell-educated MSCs demonstrate altered differentiation and transcriptomics which fit into an efficient niche to support the survival and proliferation of the myeloma cells [94, 95]. Tumor-educated MSCs can promote epithelial-mesenchymal transition (EMT) for tumor metastasis [96, 97]. Breast cancer MDA-MB-231 cells pre-treated with mouse and human MSCs significantly increased lung metastasis [98]. CXCL16 is indicated to play a critical role in MSC-boosted tumor metastasis by facilitating MSC recruitment and conversion to CAFs that secrete CXCL12 to regulate EMT in tumor cells [99]. MSCs-derived CCL5 can raise tumor cell invasion and metastasis [100], while MSCs-released TGF-β leads to the force-dependent directional migration of invasive breast cancer cells [101]. Breast cancer metastasis is enhanced by HIF-dependent CXCL10 upregulation in MSCs [102], or by DDR2 expression in MSCs [103]. Human colorectal cancer-derived MSCs enhanced the growth and metastasis of colorectal cancer cells in vitro and in vivo via the IL-6/JAK2/STAT3 signaling pathway [104]. In addition to EMT, growth-accelerating genes including integrin α5 [105] and ionotropic purinergic signaling pathway [106] are enhanced in MSCs-mediated tumor cell metastatic potential.

MSCs boost tumor metastasis in both seed-to-soil and soil-to-seed cascades. A TA-MSCs and CAFs enhance the “seed” tumor cells by releasing cytokines including CCL12, CCL5, TGF-β, CXCL10, DDR2, IL-6, integrin α5, and MSCs-derived macrovesicles, conferring tumor cells with metastatic potential mainly by EMT, which boosts local recurrence and distant metastasis. B Meanwhile, MSCs in TME provide the adaptive “soil” to fit into the metastatic niche for the homing tumor cells. Firstly, paracrine factors and EVs secreted by primary tumor recruit MSCs to the pre-metastatic niches (PMN) where a mature niche is developed for the anchoring of homing tumor cells which is followed by MSCs-secreted factors facilitating the cancer cell colonization at PMN
MSCs enrich metastatic niche
The pre-metastatic niche (PMN) forms a permissive environment facilitating the metastatic cell implantation and providing a context for the selection of cells capable of surviving and thriving in the new tissue environment [107]. PMN is established by modulation of the local ecological conditions including cell repopulation and nutrient adjustment and by preconditioning BMSCs that migrate and prepare the parenchyma for cancer cell colonization [108]. The MSCs in the PMN provide the tissue-specific pro-transformation status of BRCA1/2-mutant mediated breast and ovary cancers [60]. Mesenchymal gene expression has been observed in tumor bone metastases, indicating that mesenchymal signals from the primary tumor stroma may promote distant metastasis [109]. In this regard, the MSCs already educated by the primary tumor cells may travel in the system and function to sustain and promote the PMN [82, 110]. Paracrine factors and EVs secreted by the primary tumor are involved in recruiting the BMSCs to the second site to develop a mature niche for tumor cell metastasis [111]. In prostate cancer bone marrow metastasis, the PMN is assumed to be established via exosome pyruvate kinase M2 to promote metastasis [112]. Exosomes secreted by prostate cancer cells enhance the activity of matrix metalloproteinase in the PMN leading to extracellular matrix remodeling required for the recruitment of bone marrow cells to the PMN [113]. BMSCs play an essential role in bone homeostasis; failures in their functionality can cause osteolysis [114] which favors PMN [115] to home circulating tumor cells [116]. A CXCL12-enriched bone marrow PMN is identified to enhance the clonal seeding of triple-negative breast cancer metastasis [117]. BMSCs-secreted IL-6, IL-8, LIF, GM-CSF, ICAM-1 and MMP-3 are involved in bone remodeling which further supports the metastatic cell colonization [111]. Together, these findings indicate that MSCs and circulating tumor cells and their released immunosuppressive cytokines, coordinatively create PMN for tumor metastasis (Fig. 3B).
MSCs contribute to immunosuppressive TME
MSCs target immune cells
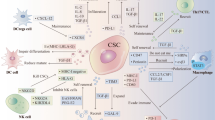
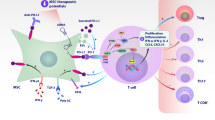
In addition to the well-defined immunosuppressive cells including Tregs, M2 macrophages, and MDSCs [118], the immunosuppressive tumor microenvironment is significantly influenced by TA-MSCs or CAFs that release molecules to inhibit immune surveillance or induce the EMT of tumor cells, resulting in tumor migration and invasion [99]. MSC-derived cytokines and growth factors promote an immunosuppressive environment that leads to inhibition of the adaptive immune system [63]. This immunosuppressive function of MSCs can be further enhanced in the TME through re-education, which could sustain a high level of various inflammatory factors and further recruit circulating MSCs and immunosuppressive cells. About 1–5% of MSCs were found in endometrial cancer and the population of MSCs in tumor tissues was correlated with the progressive status and expression level of the programmed death ligands PD-L1 and PD-L2 [119]. The MSCs residing in the TME can stimulate tumor growth by promoting immunosuppression [120], which is supported by many studies indicating that all immune cells, including T cells, B cells, macrophages, NK cells, and DC, can be targeted by MSCs leading to the immunosuppressive tumor status (Fig. 4, Table 1). In the TME of breast and prostate cancers, MSCs can defend tumor cells by upregulating Treg cells, promoting M2 polarization, and downregulating NK cells and cytotoxic T lymphocyte (CTL) [121, 122]. The immunosuppressive effect of MSCs is predominantly elicited by IFN-γ and the concomitant presence of any of TNFα, IL-1α, or IL-1β, which induce a high expression of inducible nitric oxide synthase by MSCs, inhibiting T cell response [123]. Furthermore, TA-MSCs can inhibit DCs' ability to promote the expansion of naïve CD4+ and CD8+ T cells, the secretion of IFN-γ, and the cytotoxic functions of T cells on tumor cells through an IL-10-STAT3 dependent pathway [124]. Immunosuppressive TA-MSCs are indicated to promote M2 polarization limiting the phagocytotic attack on tumor cells and favoring tumor progression [82]. TA-MSCs are also involved in educating macrophages by manipulating metabolic programs in differentially polarized macrophages [125]. Moreover, transferring MSCs-derived mitochondria to T cells caused Treg generation restricting the inflammatory response [126]. These results demonstrate that different immune functions are activated in the TME by targeting varied immune cells or subtypes of immune cell populations. Intriguingly, low-dose radiation-treated MSCs have been shown to reduce immune suppression, favoring the anti-tumor action of the immune system in mouse glioblastoma [127]. Together, TME-recruited MSCs generally create a pro-tumor immunosuppressive environment, and targeting TA-MSCs is an attractive approach to raising tumor response to ICB. In addition, TA-MSCs are specifically expanded by metastatic tumor cells and are more powerful than MSCs not educated by cancer cells in promoting tumor progression and dissemination accompanied by immune suppression [85, 128]. Further elucidation of the secretomics from circulating MSCs versus resident MSCs of normal and tumor tissues may provide more insights into MSCs-regulated tumor immunosuppression.

MSCs regulating TME immune cells. A Schematic is generated with the data of MSCs-mediated bioactive signals that are demonstrated to regulate specific clusters of immune cells and the consequences. B Experimental and clinical information were collected regarding the therapeutic approaches targeting the immune cells in TME of the cancer located in the indicated organs
Immunosuppressive MSC-EVs
MSC-EVs are nano-sized double-membraned vesicles acting as paracrine effectors of MSCs. The cargo of MSC-EVs contains a diverse range of bioactive molecules, such as proteins, miRNAs, and lipids with great potential in immune modulation, including targeting innate and adaptive immune cells like macrophages, granulocytes, mast cells, NK cells, DCs and lymphocytes [154, 155]. MSC-EVs are actively involved in cell–cell communication within the TME. Exosomes (EXOs) being the major type of EVs, play a critical role in regulating tumor proliferation, aggressive behavior of metastatic tumors, and chemoresistance and thus the potential therapeutic targets [156,157,158,159,160]. Increasing secretomics analysis has revealed a wide scale of immune regulating molecules in the cargo of MSC-EVs, including non-coding RNAs, miRNAs, long ncRNAs, transcription factors, and nucleic acids. EXOs secreted by tumor-educated MSCs can enhance breast cancer progression by inducing MDSC differentiation into immunosuppressive M2 macrophages. Inquiringly, MSC-EXOs but not EXOs from tumor cells contain TGF-β, C1q, and semaphorins, increasing the myeloid tolerogenic activity with PD-L1 overexpression in immature myelomonocytic precursors and committed CD206+ macrophages [143]. This result indicates that MSC-EXOs can promote MDSCs differentiation into protumor M2 macrophages leading to tumor immune evasion. Another study has revealed that MSC-EVs are capable of transporting miR-222 targeting ATF3, leading to AKT1 transcriptional suppression, and consequently enhancing malignant aggressiveness and immune escape [130]. Elevated delivery of miR-21-5p by MSC-EVs following hypoxia pre-challenge fosters lung cancer development through apoptosis reduction and facilitation of macrophage M2 polarization [145]. Hyaluronic acid (HA) secreted from TA-MSC-EVs is associated with GBM aggressiveness [159, 160]. Bioengineered MSC-EXOs are currently in pre-clinical and clinical testing stages [161, 162]. The development of more effective MSC-EV targets relies on further characteristics of the in vivo MSCs and the high heterogeneity including the great scale of various contents carried by the MSC-EVs from tumor-educated and non-educated MSCs. These studies will provide critical information to invent new therapeutic targets for enhancing cancer control by ICB.
Fusion of MSCs with other cells
It remains unclear why MSCs are fusing with other somatic cells under physiological and pathological conditions, although such fusion is an infrequent event. In vitro settings, fusion between MSCs and human breast epithelial MCF10A cells can be boosted by TNF-α mediated apoptotic response [163], whereas, EMT and malignant transformation are initiated from the fusion of MSCs with gastric epithelial cells [164]. The fusion of hMSCs delivered to the damaged murine heart is detected in the target organ and surrounding organ systems. The migration of hMSCs fusion products to distal organs is primarily located close to the vasculature, indicating that cell fusion requiring cell mobility is linked with the blood vessel velocity [165]. Experimental evidence also shows that the occurrence of cell fusion mainly depends on the density of the cells, the cell ratio of the parental populations, the components of the medium, and culture conditions [166]. In contrast, MSC-tumor cell fusion has been extensively studied. Spontaneous hybrid cells are identified in hMSCs co-cultured with an array of cancer cells. The hybrids seem to acquire a mixed property of functions inherited from both parental cell types (MSCs and cancer cells), including the expression of specific markers of the two cells, increased proliferation, migration capacity, and stemness [167]. The engulfment of MSCs by MDA-MB-231 breast cancer cells can enhance breast cancer cell metastatic potential, resulting in hybrid cells with mesenchymal-like, invasion, and stem cell traits [168]. In vivo tests further demonstrate that such MSC-cancer cell hybrids have elevated tumorigenicity and metastatic potential with enhanced tumor heterogeneity in breast cancer [168,169,170,171]. Interestingly, microarray-based mRNA profiling of the hybrids defined a cluster of genes for EMT and metastasis-associated S100A4 and ZEB1, with decreased expression of CK-18 [168, 171]. The MSC-engulfed breast cancer MDA-MB-231 cells demonstrated enhanced EMT and invasive potential [168]. Although the precise mechanisms underlying MSC-tumor cell fusion remain to be elucidated, hypoxic condition-induced cell apoptosis could be a prerequisite for the fusion [172], thus the phenomenon of MSC-tumor cell fusion could indicate a form of adaptive prosurvival function of tumor cells. The physiological and pathological fusion of MSC-normal and MSC-tumor cells require further investigation.
Targeting MSCs in cancer radiotherapy
A dual function of normal tissue radioprotection and inhibiting tumor cells by MSCs is suggested. MSCs are shown to repair radiation-induced normal tissue injuries [173] whereas targeting TA-MSCs is an attractive approach for the regulation of immune status in TME [120]. The idea of MSCs-mediated radiation damage repair is encouraged by the finding that radiation can activate MSCs metabolism, thus enhancing MSCs functional activity [174]. Preclinical studies have indeed demonstrated the potential of MSCs being recruited to the radiation-induced lesion sites, repairing tissue damage and supporting the regeneration of functional tissues [175], especially in the restoration of the radiation-injured intestine [176], lung [177], and skin [178] as well as to mitigate premature ovarian failure [179]. Following the aforementioned tumor-promoting function of TA-MSCs, it is thus to be carefully balanced on an MSCs-targeted approach in cancer radiotherapy. It is already demonstrated that in TME, irradiation-induced cytokines secreted by 4T1 cells, including TGF-β1, VEGF, and PDGF-BB, can facilitate MSCs chemotaxis towards the tumor site [180]. Furthermore, CCL2 can act as a factor in the IR-induced tropism of MSCs to provide pro-tumor gliomas TME [181]. Conversely, irradiated MSC-EXOs enhanced tumor radiation response, improving the control of melanoma cell growth and metastasis [182]. Similarly, MSCs could potentially promote the effect of radiotherapy in colorectal cancer by secreting TNF-α and IFN-γ [183]. Based on these reports, currently, the exact activities of the tissue-resident MSCs in TME under therapeutic irradiation, especially the TA-MSCs in recurrent and metastatic lesions after radiotherapy, need to be further elucidated. The heterodetic MSCs sub-populations, dynamic communication, as well as potential MSC-tumor or other stromal cell fusion in the irradiated TME, are currently unknown. It is thus expected that a specific subtype of TA-MSCs or evolution of TA-MSC could drive tumor aggressive growth or metastasis.
Conclusions and perspectives
This review describes the widescale functions of MSCs from different resources including their potential self-transformation and acquiring tumor-promoting functions, serving as TICs. A fundamental question that remains unanswered is whether the original malignant epithelial cell(s) arise through MET of MSCs derived from tissue residence and/or circulation. In an age of multi/single genome omics and spatial transcriptomics, reevaluating the fundamental traits and tracking of cancer-initiating MSCs could represent a significant breakthrough in our comprehension of cancer cell origin and potential prevention [184, 185]. Increasing evidence has revealed an important role in tumor immune regulation especially the tumor-infiltrated immune cells targeted by both circulating and tissue-resident MSCs. In this regard, further characterization of the heterogeneity of MSCs in TME may help to clarify the clusters responsible for carcinogenesis and progression [186]. These insights will also be informative for the application of MSCs and MSC-EVs in the fields of regenerative medicine using MSCs from different tissue resources, although current clinical evidence doesn't strongly suggest MSC-originated malignancy. Lastly, to enhance the effectiveness of cancer immunotherapy, it is imperative to gain a comprehensive understanding of the dynamic interactions between MSCs and various immune cells within the TME.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ATF3:
-
Activating transcription factor 3
- BMSCs:
-
Bone marrow-derived mesenchymal stromal cells
- CAFs:
-
Cancer-associated fibroblasts
- CCL:
-
Chemokine (C–C motif) ligand
- CXCL:
-
Chemokine (C-X-C motif) ligand
- CTL:
-
Cytotoxic T lymphocyte
- CK-18:
-
Keratin 18
- DCs:
-
Dendritic cells
- DDR2:
-
Discoidin domain receptor 2
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-mesenchymal transition
- EVs:
-
Extracellular vesicles
- EXOs:
-
Exosomes
- ELMO1:
-
Engulfment and cell motility protein 1
- FGF2:
-
Fibroblast growth factor 2
- FTEM:
-
Far tumor microenvironment
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HGF:
-
Hepatocyte-growth factor
- hMSCs:
-
Human mesenchymal stromal cells
- HIF:
-
Hypoxia-inducible factor
- IGF-1:
-
Insulin-like growth factor-1
- ICAM-1:
-
Intercellular adhesion molecule 1
- IL:
-
Interleukin
- IFN-γ:
-
Interferon-γ
- ITME:
-
Irradiated tumor microenvironment
- LIF:
-
Leukemia inhibitory factor
- NKs:
-
Natural killer cells
- MSCs:
-
Mesenchymal stromal cells, also termed as mesenchymal stem cells, mesenchymal stem-like cells
- MCP-1:
-
Monocyte chemotactic protein-1
- MDSCs:
-
Myeloid-derived suppressor cells
- MET:
-
Mesenchymal-epithelial transition
- mMSCs:
-
Mouse mesenchymal stromal cells
- NO:
-
Nitric oxide
- MSR1:
-
Macrophage scavenger receptor
- PDGF-BB:
-
Platelet-derived growth factor BB
- PGF:
-
Placental growth factor
- PGE2:
-
Prostaglandin E2
- PCa:
-
Prostate cancer
- PMN:
-
Pre-metastatic niche
- PD-L1:
-
Programmed death-ligand 1
- SCs:
-
Stem cells
- SDF-1:
-
Stromal-cell derived factor
- S100A4:
-
S100 calcium binding protein A4
- SIRPB1:
-
Signal regulatory protein beta 1
- TME:
-
Tumor microenvironment
- TA-MSCs:
-
Tumor-associated mesenchymal stromal cells
- TGF-β:
-
Transforming growth factor-β
- TICs:
-
Tumor-initiating cells, also termed cancer stem cells
- Tregs:
-
Regulatory T cells
- TNF-α:
-
Tumor necrosis factor α
- uPA:
-
Urokinase plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
- ZEB1:
-
Zinc finger E-box binding homeobox 1
References
Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T. The cancer cell map initiative: defining the hallmark networks of cancer. Mol Cell. 2015;58:690–8.
Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8.
Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31–46.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. 2017;8:248.
Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–9.
Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–31.
Sfanos KS, Yegnasubramanian S, Nelson WG, De Marzo AM. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol. 2018;15:11–24.
Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019;15:139–54.
Lee HY, Hong IS. Double-edged sword of mesenchymal stem cells: cancer-promoting versus therapeutic potential. Cancer Sci. 2017;108:1939–46.
Klimczak A, Kozlowska U. Mesenchymal stromal cells and tissue-specific progenitor cells: their role in tissue homeostasis. Stem Cells Int. 2016;2016:4285215.
Xu J, Gong T, Heng BC, Zhang CF. A systematic review: differentiation of stem cells into functional pericytes. FASEB J. 2017;31:1775–86.
O’Farrell FM, Attwell D. A role for pericytes in coronary no-reflow. Nat Rev Cardiol. 2014;11:427–32.
Zhou T, Yuan Z, Weng J, Pei D, Du X, He C, et al. Challenges and advances in clinical applications of mesenchymal stromal cells. J Hematol Oncol. 2021;14:24.
Caplan H, Olson SD, Kumar A, George M, Prabhakara KS, Wenzel P, et al. Mesenchymal stromal cell therapeutic delivery: translational challenges to clinical application. Front Immunol. 2019;10:1645.
Zhao K, Lin R, Fan Z, Chen X, Wang Y, Huang F, et al. Mesenchymal stromal cells plus basiliximab, calcineurin inhibitor as treatment of steroid-resistant acute graft-versus-host disease: a multicenter, randomized, phase 3, open-label trial. J Hematol Oncol. 2022;15:22.
Xia X, Chen W, Ma T, Xu G, Liu H, Liang C, et al. Mesenchymal stem cells administered after liver transplantation prevent acute graft-versus-host disease in rats. Liver Transpl. 2012;18:696–706.
Li W, Yang J, Zheng P, Li H, Zhao S. The origins and generation of cancer-associated mesenchymal stromal cells: an innovative therapeutic target for solid tumors. Front Oncol. 2021;11: 723707.
Frisbie L, Buckanovich RJ, Coffman L. Carcinoma-associated mesenchymal stem/stromal cells: architects of the pro-tumorigenic tumor microenvironment. Stem Cells. 2022;40:705–15.
Hernanda PY, Pedroza-Gonzalez A, van der Laan LJ, Broker ME, Hoogduijn MJ, Ijzermans JN, et al. Tumor promotion through the mesenchymal stem cell compartment in human hepatocellular carcinoma. Carcinogenesis. 2013;34:2330–40.
Harrell CR, Volarevic A, Djonov VG, Jovicic N, Volarevic V. Mesenchymal stem cell: a friend or foe in anti-tumor immunity. Int J Mol Sci. 2021;22:12429.
Poss KD. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nat Rev Genet. 2010;11:710–22.
McCulloch EA, Till JE. Perspectives on the properties of stem cells. Nat Med. 2005;11:1026–8.
Lloyd-Lewis B, Harris OB, Watson CJ, Davis FM. Mammary stem cells: premise, properties, and perspectives. Trends Cell Biol. 2017;27:556–67.
Donnenberg VS, Zimmerlin L, Rubin JP, Donnenberg AD. Regenerative therapy after cancer: what are the risks? Tissue Eng Part B Rev. 2010;16:567–75.
Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81.
Dong L, Lyu X, Faleti OD, He ML. The special stemness functions of Tbx3 in stem cells and cancer development. Semin Cancer Biol. 2019;57:105–10.
Hughes AM, Kuek V, Oommen J, Kotecha RS, Cheung LC. Murine bone-derived mesenchymal stem cells undergo molecular changes after a single passage in culture. Sci Rep. 2024;14:12396.
Norozi F, Ahmadzadeh A, Shahrabi S, Vosoughi T, Saki N. Mesenchymal stem cells as a double-edged sword in suppression or progression of solid tumor cells. Tumour Biol. 2016;37:11679–89.
Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J, et al. Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy. 2005;7:509–19.
Torsvik A, Rosland GV, Bjerkvig R, et al. Comment to: “Spontaneous transformation of adult mesenchymal stem cells from cynomolgus macaques in vitro” by Z. Ren et al. Exp. Cell Res. 317 (2011) 2950–2957: spontaneous transformation of mesenchymal stem cells in culture: facts or fiction? Exp Cell Res. 2012;2012(318):441–3.
de la Fuente R, Bernad A, Garcia-Castro J, Martin MC, Cigudosa JC. Retraction: spontaneous human adult stem cell transformation. Cancer Res. 2010;70:6682.
Rubio D, Garcia-Castro J, Martin MC, de la Fuente R, Cigudosa JC, Lloyd AC, et al. Spontaneous human adult stem cell transformation. Cancer Res. 2005;65:3035–9.
Tolar J, Nauta AJ, Osborn MJ, Panoskaltsis Mortari A, McElmurry RT, Bell S, et al. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells (Dayton, Ohio). 2007;25:371–9.
Popov BV, Petrov NS, Mikhailov VM, Tomilin AN, Alekseenko LL, Grinchuk TM, et al. Spontaneous transformation and immortalization of mesenchymal stem cells in vitro. Tsitologiia. 2009;51:91–102.
Li H, Fan X, Kovi RC, Jo Y, Moquin B, Konz R, et al. Spontaneous expression of embryonic factors and p53 point mutations in aged mesenchymal stem cells: a model of age-related tumorigenesis in mice. Cancer Res. 2007;67:10889–98.
Jeong JO, Han JW, Kim JM, Cho HJ, Park C, Lee N, et al. Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ Res. 2011;108:1340–7.
Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V, et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells. 2006;24:1095–103.
Xu S, De Becker A, De Raeve H, Van Camp B, Vanderkerken K, Van Riet I. In vitro expanded bone marrow-derived murine (C57Bl/KaLwRij) mesenchymal stem cells can acquire CD34 expression and induce sarcoma formation in vivo. Biochem Biophys Res Commun. 2012;424:391–7.
Ren Z, Wang J, Zhu W, Guan Y, Zou C, Chen Z, et al. Spontaneous transformation of adult mesenchymal stem cells from cynomolgus macaques in vitro. Exp Cell Res. 2011;317:2950–7.
Josse C, Schoemans R, Niessen NA, Delgaudine M, Hellin AC, Herens C, et al. Systematic chromosomal aberrations found in murine bone marrow-derived mesenchymal stem cells. Stem Cells Dev. 2010;19:1167–73.
Kirkwood PM, Gibson DA, Shaw I, Dobie R, Kelepouri O, Henderson NC, et al. Single-cell RNA sequencing and lineage tracing confirm mesenchyme to epithelial transformation (MET) contributes to repair of the endometrium at menstruation. Elife. 2022;11: e77663.
Wang Y, Zhang Z, Chi Y, Zhang Q, Xu F, Yang Z, et al. Long-term cultured mesenchymal stem cells frequently develop genomic mutations but do not undergo malignant transformation. Cell Death Dis. 2013;4: e950.
Tarte K, Gaillard J, Lataillade JJ, Fouillard L, Becker M, Mossafa H, et al. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood. 2010;115:1549–53.
Kim SY, Im K, Park SN, Kwon J, Kim JA, Choi Q, et al. Asymmetric aneuploidy in mesenchymal stromal cells detected by in situ karyotyping and fluorescence in situ hybridization: suggestions for reference values for stem cells. Stem Cells Dev. 2015;24:77–92.
Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71.
Vishnubalaji R, Elango R, Al-Toub M, Manikandan M, Al-Rikabi A, Harkness L, et al. Neoplastic transformation of human mesenchymal stromal cells mediated via LIN28B. Sci Rep. 2019;9:8101.
Pan Q, Fouraschen SM, de Ruiter PE, Dinjens WN, Kwekkeboom J, Tilanus HW, et al. Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Exp Biol Med (Maywood). 2014;239:105–15.
Rosland GV, Svendsen A, Torsvik A, Sobala E, McCormack E, Immervoll H, et al. Long-term cultures of bone marrow-derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res. 2009;69:5331–9.
Shima Y, Okamoto T, Aoyama T, Yasura K, Ishibe T, Nishijo K, et al. In vitro transformation of mesenchymal stem cells by oncogenic H-rasVal12. Biochem Biophys Res Commun. 2007;353:60–6.
Li N, Yang R, Zhang W, Dorfman H, Rao P, Gorlick R. Genetically transforming human mesenchymal stem cells to sarcomas: changes in cellular phenotype and multilineage differentiation potential. Cancer. 2009;115:4795–806.
Rubio D, Garcia S, Paz MF, De la Cueva T, Lopez-Fernandez LA, Lloyd AC, et al. Molecular characterization of spontaneous mesenchymal stem cell transformation. PLoS ONE. 2008;3: e1398.
Wild L, Funes JM, Boshoff C, Flanagan JM. In vitro transformation of mesenchymal stem cells induces gradual genomic hypomethylation. Carcinogenesis. 2010;31:1854–62.
Metwally AM, Li H, Houghton J. Alterations of epigenetic regulators and P53 mutations in murine mesenchymal stem cell cultures: a possible mechanism of spontaneous transformation. Cancer Biomark. 2021;32:327–37.
Balcik-Ercin P, Cayrefourcq L, Soundararajan R, Mani SA, Alix-Panabieres C. Epithelial-to-mesenchymal plasticity in circulating tumor cell lines sequentially derived from a patient with colorectal cancer. Cancers (Basel). 2021;13:5408.
Piperigkou Z, Koutsandreas A, Franchi M, Zolota V, Kletsas D, Passi A, et al. ESR2 drives mesenchymal-to-epithelial transition in triple-negative breast cancer and tumorigenesis in vivo. Front Oncol. 2022;12: 917633.
Denk D, Greten FR. Inflammation: the incubator of the tumor microenvironment. Trends Cancer. 2022;8:901–14.
Katanov C, Lerrer S, Liubomirski Y, Leider-Trejo L, Meshel T, Bar J, et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: prominent roles for TNF-α and the NF-κB pathway. Stem Cell Res Ther. 2015;6:87.
Eliason J, Rao A. Investigating ecological interactions in the tumor microenvironment using joint species distribution models for point patterns. bioRxiv. 2023;11: 567108.
Gatenby RA. Population ecology issues in tumor growth. Cancer Res. 1991;51:2542–7.
Kenny PA, Nelson CM, Bissell MJ. The ecology of tumors: by perturbing the microenvironment, wounds and infection may be key to tumor development. Scientist. 2006;20:30.
Revsine M, Wang L, Forgues M, Behrens S, Craig AJ, Liu M, et al. Lineage and ecology define liver tumor evolution in response to treatment. Cell Rep Med. 2024;5: 101394.
Cuiffo BG, Karnoub AE. Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh Migr. 2012;6:220–30.
Kasashima H, Yashiro M, Nakamae H, Masuda G, Kinoshita H, Morisaki T, et al. Bone marrow-derived stromal cells are associated with gastric cancer progression. Br J Cancer. 2015;113:443–52.
Kansy BA, Dißmann PA, Hemeda H, Bruderek K, Westerkamp AM, Jagalski V, et al. The bidirectional tumor–mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res Therapy. 2014;5:95.
Kim EK, Kim HJ, Yang YI, Kim JT, Choi MY, Choi CS, et al. Endogenous gastric-resident mesenchymal stem cells contribute to formation of cancer stroma and progression of gastric cancer. Korean J Pathol. 2013;47:507–18.
Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells. 2015;33:2400–15.
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–44.
Ritter E, Perry A, Yu J, Wang T, Tang L, Bieberich E. Breast cancer cell-derived fibroblast growth factor 2 and vascular endothelial growth factor are chemoattractants for bone marrow stromal stem cells. Ann Surg. 2008;247:310–4.
Chaturvedi P, Gilkes DM, Wong CC, Kshitiz, Luo W, Zhang H, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123:189–205.
Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. 2010;316:3417–24.
Bi LK, Zhou N, Liu C, Lu FD, Lin TX, Xuan XJ, et al. Kidney cancer cells secrete IL-8 to activate Akt and promote migration of mesenchymal stem cells. Urol Oncol. 2014;32:607–12.
Vogel S, Peters C, Etminan N, Börger V, Schimanski A, Sabel MC, et al. Migration of mesenchymal stem cells towards glioblastoma cells depends on hepatocyte-growth factor and is enhanced by aminolaevulinic acid-mediated photodynamic treatment. Biochem Biophys Res Commun. 2013;431:428–32.
Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells (Dayton, Ohio). 2009;27:857–65.
Li Y, Yu X, Lin S, Li X, Zhang S, Song Y-H. Insulin-like growth factor 1 enhances the migratory capacity of mesenchymal stem cells. Biochem Biophys Res Commun. 2007;356:780–4.
Dwyer RM, Potter-Beirne SM, Harrington KA, Lowery AJ, Hennessy E, Murphy JM, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13:5020–7.
Pulukuri SM, Gorantla B, Dasari VR, Gondi CS, Rao JS. Epigenetic upregulation of urokinase plasminogen activator promotes the tropism of mesenchymal stem cells for tumor cells. Mol Cancer Res MCR. 2010;8:1074–83.
Lu X, Han J, Xu X, Xu J, Liu L, Huang Y, et al. PGE2 promotes the migration of mesenchymal stem cells through the activation of FAK and ERK1/2 pathway. Stem Cells Int. 2017;2017:8178643.
Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells (Dayton, Ohio). 2016;34:2536–47.
Liu Y, Han ZP, Zhang SS, Jing YY, Bu XX, Wang CY, et al. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem. 2011;286:25007–15.
Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–5.
Le Naour A, Prat M, Thibault B, Mevel R, Lemaitre L, Leray H, et al. Tumor cells educate mesenchymal stromal cells to release chemoprotective and immunomodulatory factors. J Mol Cell Biol. 2020;12:202–15.
Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Therapy. 2013;4:70.
Papait A, Stefani FR, Cargnoni A, Magatti M, Parolini O, Silini AR. The multifaceted roles of MSCs in the tumor microenvironment: interactions with immune cells and exploitation for therapy. Front Cell Dev Biol. 2020;8:447.
Wolfe AR, Trenton NJ, Debeb BG, Larson R, Ruffell B, Chu K, et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget. 2016;7:82482–92.
Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. 2014;41:195–216.
Li C, Cheung MKH, Han S, Zhang Z, Chen L, Chen J, et al. Mesenchymal stem cells and their mitochondrial transfer: a double-edged sword. Biosci Rep. 2019;39:BSR20182417.
Caicedo A, Fritz V, Brondello J-M, Ayala M, Dennemont I, Abdellaoui N, et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci Rep. 2015;5:9073.
Nzigou Mombo B, Gerbal-Chaloin S, Bokus A, Daujat-Chavanieu M, Jorgensen C, Hugnot JP, et al. MitoCeption: transferring isolated human MSC mitochondria to glioblastoma stem cells. J Vis Exp. 2017:55245.
Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–8.
Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol. 2013;15:284–94.
Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16:31.
Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–9.
Fairfield H, Costa S, Falank C, Farrell M, Murphy CS, D’Amico A, et al. Multiple myeloma cells alter adipogenesis, increase senescence-related and inflammatory gene transcript expression, and alter metabolism in preadipocytes. Front Oncol. 2020;10: 584683.
Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–88.
Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res. 2006;12:5606–7.
Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D, et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015;75:924–9.
Albarenque SM, Zwacka RM, Mohr A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011;7:163–71.
Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63.
McAndrews KM, McGrail DJ, Ravikumar N, Dawson MR. Mesenchymal stem cells induce directional migration of invasive breast cancer cells through TGF-beta. Sci Rep. 2015;5:16941.
Chaturvedi P, Gilkes DM, Wong CC, Luo W, Zhang H, Wei H, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Investig. 2013;123:189–205.
Gonzalez ME, Martin EE, Anwar T, Arellano-Garcia C, Medhora N, Lama A, et al. Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Rep. 2017;18:1215–28.
Zhang X, Hu F, Li G, Li G, Yang X, Liu L, et al. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018;9:25.
Chen J, Ji T, Wu D, Jiang S, Zhao J, Lin H, et al. Human mesenchymal stem cells promote tumor growth via MAPK pathway and metastasis by epithelial mesenchymal transition and integrin α5 in hepatocellular carcinoma. Cell Death Dis. 2019;10:425.
Maffey A, Storini C, Diceglie C, Martelli C, Sironi L, Calzarossa C, et al. Mesenchymal stem cells from tumor microenvironment favour breast cancer stem cell proliferation, cancerogenic and metastatic potential, via ionotropic purinergic signalling. Sci Rep. 2017;7:13162.
Valenzuela Alvarez M, Gutierrez LM, Correa A, Lazarowski A, Bolontrade MF. Metastatic niches and the modulatory contribution of mesenchymal stem cells and its exosomes. Int J Mol Sci. 2019;20:1946.
Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–23.
Liu Q, Zhang H, Jiang X, Qian C, Liu Z, Luo D. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol Cancer. 2017;16:176.
Mei S, Alchahin AM, Tsea I, Kfoury Y, Hirz T, Jeffries NE, et al. Single-cell analysis of immune and stroma cell remodeling in clear cell renal cell carcinoma primary tumors and bone metastatic lesions. Genome Med. 2024;16:1.
Sanmartin MC, Borzone FR, Giorello MB, Pacienza N, Yannarelli G, Chasseing NA. Bone marrow/bone pre-metastatic niche for breast cancer cells colonization: the role of mesenchymal stromal cells. Crit Rev Oncol Hematol. 2021;164: 103416.
Dai J, Escara-Wilke J, Keller JM, Jung Y, Taichman RS, Pienta KJ, et al. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J Exp Med. 2019;216:2883–99.
Deep G, Jain A, Kumar A, Agarwal C, Kim S, Leevy WM, et al. Exosomes secreted by prostate cancer cells under hypoxia promote matrix metalloproteinases activity at pre-metastatic niches. Mol Carcinog. 2020;59:323–32.
Esposito M, Kang Y. Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther. 2014;141:222–33.
Borzone FR, Giorello MB, Martinez LM, Sanmartin MC, Feldman L, Dimase F, et al. Senescent mesenchymal stem/stromal cells in pre-metastatic bone marrow of untreated advanced breast cancer patients. Oncol Res. 2023;31:361–74.
Feldman LJ Sr, Fernández Vallone VB, Choi H, Labovsky V, Martinez LM, Bordenave RH, et al. Bone marrow mesenchymal stem cells: pre-metastatic niche for breast cancer. Blood. 2013;122:4859.
Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–73.
Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15:61.
Wang KH, Chang YH, Harnod T, Ding DC. Endometrial cancer-infiltrating mesenchymal stem cells exhibit immunosuppressive effects. Cell Transplant. 2022;31:9636897221104452.
Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol. 2018;9:262.
Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-beta. J Immunol (Baltimore, Md: 1950). 2010;184:5885–94.
Krueger TE, Thorek DLJ, Meeker AK, Isaacs JT, Brennen WN. Tumor-infiltrating mesenchymal stem cells: drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate. 2019;79:320–30.
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50.
Ghosh T, Barik S, Bhuniya A, Dhar J, Dasgupta S, Ghosh S, et al. Tumor-associated mesenchymal stem cells inhibit naive T cell expansion by blocking cysteine export from dendritic cells. Int J Cancer. 2016;139:2068–81.
Vasandan AB, Jahnavi S, Shashank C, Prasad P, Kumar A, Prasanna SJ. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci Rep. 2016;6:38308.
Court AC, Le-Gatt A, Luz-Crawford P, Parra E, Aliaga-Tobar V, Batiz LF, et al. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep. 2020;21: e48052.
Stefani FR, Eberstål S, Vergani S, Kristiansen TA, Bengzon J. Low-dose irradiated mesenchymal stromal cells break tumor defensive properties in vivo. Int J Cancer. 2018;143:2200–12.
Lindoso RS, Collino F, Vieyra A. Extracellular vesicles as regulators of tumor fate: crosstalk among cancer stem cells, tumor cells and mesenchymal stem cells. Stem Cell Investig. 2017;4:75.
Liotta F, Querci V, Mannelli G, Santarlasci V, Maggi L, Capone M, et al. Mesenchymal stem cells are enriched in head neck squamous cell carcinoma, correlates with tumour size and inhibit T-cell proliferation. Br J Cancer. 2015;112:745–54.
Li S, Yan G, Yue M, Wang L. Extracellular vesicles-derived microRNA-222 promotes immune escape via interacting with ATF3 to regulate AKT1 transcription in colorectal cancer. BMC Cancer. 2021;21:349.
Egan H, Treacy O, Lynch K, Leonard NA, O’Malley G, Reidy E, et al. Targeting stromal cell sialylation reverses T cell-mediated immunosuppression in the tumor microenvironment. Cell Rep. 2023;42: 112475.
Wang XS, Yu XJ, Wei K, Wang SX, Liu QK, Wang YG, et al. Mesenchymal stem cells shuttling miR-503 via extracellular vesicles enhance glioma immune escape. Oncoimmunology. 2022;11:1965317.
Ling W, Zhang J, Yuan Z, Ren G, Zhang L, Chen X, et al. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Can Res. 2014;74:1576–87.
Montesinos JJ, Mora-García MdL, Mayani H, Flores-Figueroa E, García-Rocha R, Fajardo-Orduña GR, et al. In vitro evidence of the presence of mesenchymal stromal cells in cervical cancer and their role in protecting cancer cells from cytotoxic T cell activity. Stem Cells Dev. 2013;22:2508–19.
de Lourdes M-G, Garcia-Rocha R, Morales-Ramirez O, Montesinos JJ, Weiss-Steider B, Hernandez-Montes J, et al. Mesenchymal stromal cells derived from cervical cancer produce high amounts of adenosine to suppress cytotoxic T lymphocyte functions. J Transl Med. 2016;14:302.
Marin-Aquino LA, Mora-Garcia ML, Moreno-Lafont MC, Garcia-Rocha R, Montesinos-Montesinos JJ, Lopez-Santiago R, et al. Adenosine increases PD-L1 expression in mesenchymal stromal cells derived from cervical cancer through its interaction with A(2A)R/A(2B)R and the production of TGF-beta1. Cell Biochem Funct. 2024;42: e4010.
Corradi G, Bassani B, Simonetti G, Sangaletti S, Vadakekolathu J, Fontana MC, et al. Release of IFNγ by acute myeloid leukemia cells remodels bone marrow immune microenvironment by inducing regulatory T cells. Clin Cancer Res. 2022;28:3141–55.
Fakhimi M, Talei AR, Ghaderi A, Habibagahi M, Razmkhah M. Helios, CD73 and CD39 induction in regulatory T cells exposed to adipose derived mesenchymal stem cells. Cell J. 2020;22:236–44.
Wang M, Chen B, Sun X-X, Zhao X-D, Zhao Y-Y, Sun L, et al. Gastric cancer tissue-derived mesenchymal stem cells impact peripheral blood mononuclear cells via disruption of Treg/Th17 balance to promote gastric cancer progression. Exp Cell Res. 2017;361:19–29.
Patel SA, Dave MA, Bliss SA, Giec-Ujda AB, Bryan M, Pliner LF, et al. Treg/Th17 polarization by distinct subsets of breast cancer cells is dictated by the interaction with mesenchymal stem cells. J Cancer Stem Cell Res. 2014;2014: e1003.
Galland S, Vuille J, Martin P, Letovanec I, Caignard A, Fregni G, et al. Tumor-derived mesenchymal stem cells use distinct mechanisms to block the activity of natural killer cell subsets. Cell Rep. 2017;20:2891–905.
Di Matteo S, Avanzini MA, Pelizzo G, Calcaterra V, Croce S, Spaggiari GM, et al. Neuroblastoma tumor-associated mesenchymal stromal cells regulate the cytolytic functions of NK cells. Cancers (Basel). 2022;15:19.
Biswas S, Mandal G, Roy Chowdhury S, Purohit S, Payne KK, Anadon C, et al. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J Immunol. 2019;203:3447–60.
Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT, et al. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia. 2016;18:142–51.
Ren W, Hou J, Yang C, Wang H, Wu S, Wu Y, et al. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J Exp Clin Cancer Res. 2019;38:62.
Cortes-Morales VA, Chavez-Sanchez L, Rocha-Zavaleta L, Espindola-Garibay S, Monroy-Garcia A, Castro-Manrreza ME, et al. Mesenchymal stem/stromal cells derived from cervical cancer promote M2 macrophage polarization. Cells. 2023;12:1047.
Louault K, Porras T, Lee M-H, Muthugounder S, Kennedy RJ, Blavier L, et al. Fibroblasts and macrophages cooperate to create a pro-tumorigenic and immune resistant environment via activation of TGF-β/IL-6 pathway in neuroblastoma. Oncoimmunology. 2022;11:2146860.
Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci USA. 2014;111:E2120–9.
Guilloton F, Caron G, Ménard C, Pangault C, Amé-Thomas P, Dulong J, et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood. 2012;119:2556–67.
Cascio S, Chandler C, Zhang L, Sinno S, Gao B, Onkar S, et al. Cancer-associated MSC drive tumor immune exclusion and resistance to immunotherapy, which can be overcome by Hedgehog inhibition. Sci Adv. 2021;7:eabi5790.
Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. 2012;11:812–24.
Giallongo C, Romano A, Parrinello NL, La Cava P, Brundo MV, Bramanti V, et al. Mesenchymal stem cells (MSC) regulate activation of granulocyte-like myeloid derived suppressor cells (G-MDSC) in chronic myeloid leukemia patients. PLoS ONE. 2016;11: e0158392.
Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget. 2016;7:85764–75.
Bazzoni R, Takam Kamga P, Tanasi I, Krampera M. Extracellular vesicle-dependent communication between mesenchymal stromal cells and immune effector cells. Front Cell Dev Biol. 2020;8: 596079.
Wong C, Stoilova I, Gazeau F, Herbeuval JP, Fourniols T. Mesenchymal stromal cell derived extracellular vesicles as a therapeutic tool: immune regulation, MSC priming, and applications to SLE. Front Immunol. 2024;15:1355845.
D’Agnano I, Berardi AC. Extracellular vesicles, a possible theranostic platform strategy for hepatocellular carcinoma—an overview. Cancers (Basel). 2020;12:261.
Golinelli G, Mastrolia I, Aramini B, Masciale V, Pinelli M, Pacchioni L, et al. Arming mesenchymal stromal/stem cells against cancer: has the time come? Front Pharmacol. 2020;11: 529921.
Lyu T, Wang Y, Li D, Yang H, Qin B, Zhang W, et al. Exosomes from BM-MSCs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance via upregulation of S100A4. Exp Hematol Oncol. 2021;10:24.
Lim EJ, Suh Y, Yoo KC, Lee JH, Kim IG, Kim MJ, et al. Tumor-associated mesenchymal stem-like cells provide extracellular signaling cue for invasiveness of glioblastoma cells. Oncotarget. 2017;8:1438–48.
Kudo-Saito C. Cancer-associated mesenchymal stem cells aggravate tumor progression. Front Cell Dev Biol. 2015;3:23.
Weng Z, Zhang B, Wu C, Yu F, Han B, Li B, et al. Therapeutic roles of mesenchymal stem cell-derived extracellular vesicles in cancer. J Hematol Oncol. 2021;14:136.
Liu X, Wei Q, Lu L, Cui S, Ma K, Zhang W, et al. Immunomodulatory potential of mesenchymal stem cell-derived extracellular vesicles: targeting immune cells. Front Immunol. 2023;14:1094685.
Melzer C, von der Ohe J, Hass R. In vitro fusion of normal and neoplastic breast epithelial cells with human mesenchymal stroma/stem cells partially involves tumor necrosis factor receptor signaling. Stem Cells (Dayton, Ohio). 2018;36:977–89.
He X, Li B, Shao Y, Zhao N, Hsu Y, Zhang Z, et al. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer. 2015;15:24.
Freeman BT, Kouris NA, Ogle BM. Tracking fusion of human mesenchymal stem cells after transplantation to the heart. Stem Cells Transl Med. 2015;4:685–94.
Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell Commun Signal. 2016;14:20.
Xue J, Zhu Y, Sun Z, Ji R, Zhang X, Xu W, et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer. 2015;15:793.
Chen YC, Gonzalez ME, Burman B, Zhao X, Anwar T, Tran M, et al. Mesenchymal stem/stromal cell engulfment reveals metastatic advantage in breast cancer. Cell Rep. 2019;27(3916–26): e5.
Mandel K, Yang Y, Schambach A, Glage S, Otte A, Hass R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013;22:3114–27.
Melzer C, von der Ohe J, Hass R. In vivo cell fusion between mesenchymal stroma/stem-like cells and breast cancer cells. Cancers (Basel). 2019;11:185.
Melzer C, von der Ohe J, Hass R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun Signal. 2018;16:2.
Noubissi FK, Harkness T, Alexander CM, Ogle BM. Apoptosis-induced cancer cell fusion: a mechanism of breast cancer metastasis. FASEB J. 2015;29:4036–45.
Nicolay NH, Lopez Perez R, Debus J, Huber PE. Mesenchymal stem cells—a new hope for radiotherapy-induced tissue damage? Cancer Lett. 2015;366:133–40.
Patten DA, Ouellet M, Allan DS, Germain M, Baird SD, Harper ME, et al. Mitochondrial adaptation in human mesenchymal stem cells following ionizing radiation. FASEB J. 2019;33:9263–78.
Rühle A, Xia O, Perez RL, Trinh T, Richter W, Sarnowska A, et al. The radiation resistance of human multipotent mesenchymal stromal cells is independent of their tissue of origin. Int J Radiat Oncol Biol Phys. 2018;100:1259–69.
Chang PY, Qu YQ, Wang J, Dong LH. The potential of mesenchymal stem cells in the management of radiation enteropathy. Cell Death Dis. 2015;6: e1840-e.
Xu T, Zhang Y, Chang P, Gong S, Shao L, Dong L. Mesenchymal stem cell-based therapy for radiation-induced lung injury. Stem Cell Res Therapy. 2018;9:18.
Fang Z, Chen P, Tang S, Chen A, Zhang C, Peng G, et al. Will mesenchymal stem cells be future directions for treating radiation-induced skin injury? Stem Cell Res Therapy. 2021;12:179.
El-Derany MO, Said RS, El-Demerdash E. Bone marrow-derived mesenchymal stem cells reverse radiotherapy-induced premature ovarian failure: emphasis on signal integration of TGF-beta, Wnt/beta-catenin and hippo pathways. Stem Cell Rev Rep. 2021;17:1429–45.
Klopp AH, Spaeth EL, Dembinski JL, Woodward WA, Munshi A, Meyn RE, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67:11687–95.
Thomas JG, Parker Kerrigan BC, Hossain A, Gumin J, Shinojima N, Nwajei F, et al. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J Neurosurg. 2018;128:287–95.
de Araujo FV, O’Valle F, Serrano-Saenz S, Anderson P, Andrés E, López-Peñalver J, et al. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol Cancer. 2018;17:122.
Feng H, Zhao J-K, Schiergens TS, Wang P-X, Ou B-C, Al-Sayegh R, et al. Bone marrow-derived mesenchymal stromal cells promote colorectal cancer cell death under low-dose irradiation. Br J Cancer. 2018;118:353–65.
Fu T, Dai LJ, Wu SY, Xiao Y, Ma D, Jiang YZ, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14:98.
Yu M, Sui K, Wang Z, Zhang X. MSCsDB: a database of single-cell transcriptomic profiles and in-depth comprehensive analyses of human mesenchymal stem cells. Exp Hematol Oncol. 2024;13:29.
Jia Q, Wang A, Yuan Y, Zhu B, Long H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol. 2022;11:24.
Funding
The study was supported in part by the Jiangsu Postdoctoral Research Funding Program 2021K206B (Y.Z.), the University of California Davis Cancer Center Cancer Immunology Pilot Grant Support (J.J.L.).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Wang, C. & Li, J.J. Revisiting the role of mesenchymal stromal cells in cancer initiation, metastasis and immunosuppression. Exp Hematol Oncol 13, 64 (2024). https://doi.org/10.1186/s40164-024-00532-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-024-00532-4