
Abstract
Marvelous advancements have been made in cancer therapies to improve clinical outcomes over the years. However, therapeutic resistance has always been a major difficulty in cancer therapy, with extremely complicated mechanisms remain elusive. N6-methyladenosine (m6A) RNA modification, a hotspot in epigenetics, has gained growing attention as a potential determinant of therapeutic resistance. As the most prevalent RNA modification, m6A is involved in every links of RNA metabolism, including RNA splicing, nuclear export, translation and stability. Three kinds of regulators, “writer” (methyltransferase), “eraser” (demethylase) and “reader” (m6A binding proteins), together orchestrate the dynamic and reversible process of m6A modification. Herein, we primarily reviewed the regulatory mechanisms of m6A in therapeutic resistance, including chemotherapy, targeted therapy, radiotherapy and immunotherapy. Then we discussed the clinical potential of m6A modification to overcome resistance and optimize cancer therapy. Additionally, we proposed existing problems in current research and prospects for future research.
Similar content being viewed by others

Introduction
Among more than 100 types of RNA modifications discovered in mammalian messenger RNA (mRNA) and noncoding RNA (ncRNA), m6A, which refers to methylation at the N6 position of adenosine, is reported as the most abundant type [1, 2]. Advanced techniques like m6A-seq and miCLIP-seq uncovered that m6A methylation targeted at consensus sequences like DRACH (D = G, A, or U; R = G or A; H = A, C, or U), mainly enriched in coding sequence (CDS) and 3’UTR region of mRNA, as well as other kinds of RNAs [1, 3]. m6A methylation has been verified to play a crucial role in determining RNA fates, including splicing, nuclear export, translation, stabilization and degradation, further regulating gene expression and cellular phenotypes [4]. m6A mediated-RNA epigenetic modification orchestrates many physiological activities, such as DNA repair, meiosis, tissue remodeling, and circadian rhythm, etc. [5, 6], while dysregulated m6A methylation participated in numerous pathologic processes, especially tumorigenesis, progression, metastasis and therapeutic resistance [7, 8].
In a global context, cancer remains the top threat to public health and imposes a severe economic burden on society [9]. Owing to the significant heterogeneity, cancer cells frequently display therapeutic resistance, which is the main obstacle for cancer treatment [10]. The rationales of chemoresistance can be basically categorized as reduced cellular accumulation, increased detoxification, increased DNA repair, decreased apoptosis, and autophagy [11]. And various studies have explicitly indicated that m6A modification could regulate drug resistance in these ways [12,13,14]. Cancer cells gradually develop radiotherapy resistance via enhancing DNA damage repair ability, altering expression of oncogene/tumor suppressors and cancer metabolism of cancer cell [15]. Correspondingly, there are evidences showing the regulatory roles of m6A methylation in these procedures. Moreover, therapeutic resistance appears as a thorny problem in immunotherapy, undoubtedly impeding its further development. Inspiringly, m6A modification not only exhibits the possibility to overcome the immunotherapy resistance, but also potentiate novel immunotherapy.
Overall, illuminating the influence of m6A methylation in therapeutic resistance has profound significance for exploring potential strategies to overcome resistance and develop more efficient therapeutic targets, ultimately optimizing the cancer therapy [4, 16]. In this review, we systematically summarize the latest studies, providing mechanistic insights into the formation of resistance under the regulation of m6A methylation, aiming at facilitating the development of potential therapeutic strategies. At same time, we point out the deficiency of current study and put forward some feasible solutions and promising research directions.
m6A methylation and demethylation
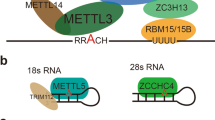
The m6A methylation is mainly catalyzed by methyltransferase complex (MTC), a heterodimer constituted by Methyltransferase-like 3 (METTL3) and METTL14, in which METTL3 exerts catalytic function and METTL14 provides structural support and recognizes target RNA [17]. Wilms tumor 1-associated protein (WTAP), vir like m6A methyltransferase associated (VIRMA, also known as KIAA1429), RNA-binding motif protein 15 (RBM15), and zinc finger CCCH domain-containing protein 13 (ZC3H13) function as cofactors, localizing MTC to target sites and initiating methylation [5]. The demethylation is executed by two demethylases, Obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) [18, 19]. FTO is the first m6A “eraser” to be discovered and mainly effects mRNA stability, translation and splicing [20, 21]. As the homologue of FTO, ALKBH5 can modulate RNA splicing, export and degradation [22]. Particularly, they have different substrate affinities. Which one of m6A and m6Am is the preferential target of FTO remains controversial, while ALKBH5 exclusively demethylates m6A [23].
“Reader” can specifically recognize and bind to m6A sites to control RNA fate, realizing epigenetic regulation in gene expression and biological phenotypes. YT521-B homology (YTH) proteins, including YTHDC1–2 and YTHDF1–3, are extensively involved in RNA metabolism. YTHDC1 has been verified to facilitate RNA splicing, nuclear export and decay [24,25,26], and YTHDC2 contributes to enhancing translation efficiency and mRNA decay [27, 28]. YTHDF1 can facilitate translation initiation by interacting with eukaryotic initiation factor 3 (eIF3), YTHDF3 not only exerts a synergistic effect with YTHDF1 in promoting translation, but also cooperates with YTHDF2 to induce mRNA degradation [29, 30]. The insulin-like growth factor 2 mRNA binding protein family, IGF2BP1-3 recognize m6A targets via K homology domains, promoting RNA stability and translation [31]. The heterogeneous nuclear ribonucleoproteins (HNRNPs) are confirmed to elicit alternative splicing and mediate co-translational regulation [32]. Moreover, fragile X mental retardation proteins (FMRPs) is indispensable for RNA export [33].
"Writers", "erasers" and “readers” work jointly to catalyze, remove and recognize m6A methylation, establishing a reversible and dynamic equilibrium. Except for mRNA, ncRNA including tRNA, rRNA, miRNA, lncRNA and circRNA, can all be m6A-modified, which further influences RNA splicing, translation, maturation, transport, localization, and RNA–protein interactions.
m6A and chemotherapy resistance
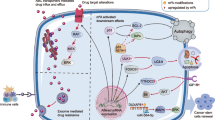
Chemotherapy is one of the most potent clinical strategies for cancer treatment, especially in patients with advanced malignancy who cannot undergo surgery [34]. Resistance to chemotherapeutic agents greatly limits the overall treatment efficiency and develops into an increasingly austere clinical tissue [11]. Remarkably, growing evidences emphasized m6A modification as a potential determinant of tumor response to chemotherapy (Table 1; Fig. 1).

The role and molecular mechanisms of m6A regulators in chemotherapy resistance
Platinum drugs
The first generation of platinum drugs, cisplatin was approved by FDA for testicular cancer in 1965, then the second-generation carboplatin and third-generation oxaliplatin were utilized worldwide for multiple types of tumors [79]. However, resistance to platinum drugs is commonly seen in a short time, and m6A has been verified to regulate resistance to cisplatin (DDP) and oxaliplatin (OXA).
In non-small cell lung cancer (NSCLC), there is no unanimous conclusion about whether METTL3 facilitates or suppresses resistance to cisplatin. It was revealed that METTL3 contributed to insensitivity to cisplatin by enhancing AKT serine/threonine kinase 1 (AKT1) protein expression, and upregulated METTL3 in chemo-resistant NSCLC samples was positively related with poor survival [35]. However, Ling et al. found that propofol enhanced cisplatin sensitivity dose-dependently via promoting METTL3-mediated m6A methylation. Propofol augments m6A enrichment on pri-miR-486-5p to promote its maturation and further inactivates the Ras-associated protein1 (RAP1)-NF-kappaB (NF-κB) axis, and knockdown of METTL3 reversed the promoting effect [36]. Besides, METTL3 was negatively regulated by miR-4443, which is enriched in exosomes of cisplatin-resistant NSCLC. The decreased METTL3 further upregulated ferroptosis suppressor protein 1 (FSP1), reducing the ferroptosis induced by DDP and thus conferring chemoresistance [37]. Ablation of YTHDF1 mediates cisplatin resistance in NSLCC via decreasing translation of Kelch-like ECH-associated protein 1 (KEAP1) in an oxidative stress state induced by DDP, which in turn activates the antioxidant ROS clearance system aldo–keto reductases 1C1 (AKR1C1), thus inhibiting hypoxia-induced apoptosis. Moreover, the lower YTHDf1 level correlates with a worse clinical outcome for NSCLC patients [38].
In gastric cancer (GC), the previous studies confirmed in accordance that METTL3 augmented the resistance to platinum drugs. METTL3 was reported to methylate Rho GTPase activating protein 5 (ARHGAP5) mRNA to facilitate its translation, conferring resistance to chemotherapeutic drugs, including cisplatin, doxorubicin hydrochloride, and 5-fluorouracil (5-Fu) [39]. Additionally, in CD133 + GC stem cells, METTL3 play a decisive role in oxaliplatin resistance. In mechanism, the upregulated METTTL3 could enhance the stability of PARP1 mRNA by recruiting YTHDF1 to the 3'-UTR, and PARP1 contributes to repairing the drug-induced DNA damage [12]. YTHDF2 participates in the destabilization of Cystathionine β-synthase (CBS) mRNA induced by CBS mRNA-destabilizing lncRNA (CBSLR), downregulated CBS reduces the methylation of ACSL4, protecting GC cells from ferroptosis in vivo and in vitro, thus leading to a poorer response to chemotherapy [40]. Moreover, musashi RNA binding protein 2 (MSI2), was supposed to promote DDP resistance, acting as a potential m6A reader. In mechanism, LNC942 facilitates the expression of MSI2 by inhibiting its ubiquitination. MSI2 stabilizes c-Myc mRNA in an m6A-dependent manner [41]. Moreover, LNC942 was previously reported to stabilize downstream targets by recruiting METTL14, indicating the participation of m6A [42].
In colorectal cancer (CRC), METTL3 was proposed to be significantly upregulated and sustain oxaliplatin resistance. Mechanism studies elucidated that METTL3 could increase CBX8 mRNA stability dependent on IGF2BP1, and then CBX8 promotes the transcription of leucine rich repeat containing G protein-coupled receptor 5 (LGR5) to maintain cancer stemness and chemoresistance [43]. METTL3 triggered m6A modification on Tumor necrosis factor receptor-associated factors (TRAF) mRNA and promoted its degradation, thus suppressing necroptosis and reducing OXA sensitivity. Meanwhile, M2 tumor-associated macrophages (TAMs) conduced to OXA resistance formation by enhancing m6a methylation [44]. In DDP-resistant CRC cell, overexpressed YTHDF1 promoted protein synthesis of glutaminase 1 (GLS1) via binding to the 3' UTR of GLS1 mRNA. Suppression of YTHDf1-mediated glutamine metabolism sensitizes CRC cell to cisplatin in vitro and in vivo [45]. Moreover, YTHDF3 facilitates translation of several resistance biomarker genes, such as ATPase copper transporting alpha (ATP7A), dual-specificity Y-phosphorylation-regulated kinase 1B (DYRK1B), via recognizing the 5' UTR of these m6A-marked mRNAs and recruiting eIF3A.
In ovarian cancer (OC), ALKBH5 was upregulated and exerted an important role in the chemoresistance both in vivo and in vitro. ALKBH5 was increased via forming a loop with the upstream transcription factor homeobox A10 (HOXA10), then they jointly activated the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway by m6A demethylation, thus promoting chemoresistance [47]. While Tripartite motif-containing protein (TRIM29) is supposed as an oncogene in OC related with the malignant characteristics, YTHDF1 binds to m6A sites on TRIM29 mRNA and facilitates its translation to enhance DDP resistance [48]. Additionally, comprehensive analysis of the transcriptome-wide m6A methylome of endometrioid OC showed that m6A enriched genes were significantly associated with resistance to platinum drug [80].
In seminoma, METTL3 was highly expressed in resistant seminoma cell and inhibition of METTL3 enhanced the response to cisplatin treatment. A recent study identified that METTL3-modified m6A methylation increased the expression of transcription factor-activating enhancer-binding protein 2C (TFAP2C), via IGF2BP1-mediated stabilization of its mRNA, further activated DNA repair genes WEE1 G2 checkpoint kinase (WEE1) and breast cancer type 1 (BRCA1) to promote resistance [49]. Additionally, overexpressed METTL3 confers resistance to DDP by enhancing autophagy. METTL3 could enhance the expression of ATG5, an autophagy elongation protein which increases autophagy to sustain cell viability and chemoresistance [13].
In oral squamous cell carcinoma (OSCC), the overexpression of ALKBH5 participates in the acquisition of resistance to DDP. In both resistant cell lines and clinical samples, highly expressed DDX3 directly elevates ALKBH5 level, compared with the sensitive counterparts [50]. Then ALKBH5 demethylated the transcripts of two transcription factors, forkhead box M1 (FOXM1) and Nanog homeobox (NANOG), to enhance their expression, both of which were previously reported as crucial for sustaining drug resistance [81, 82]. Also, it was verified that the upregulation of FTO was pivotal for arecoline‐induced OSCC stemness and chemoresistance to DDP [51].
In nasopharyngeal carcinoma (NPC), METTL3-mediated m6A modification was enriched in TRIM11 mRNA and stabilized it depending on IGF2BP2. TRIM11 facilitates the degradation of Daple by inducing ubiquitination, relieving the inhibition of Daple on Wnt/β-catenin signaling. Increased β-catenin directly bound to ABCC9 promoter to upregulate its expression, thus inducing chemoresistance via enhancing drug export [14]. Similarly, depletion of METTL3 in pancreas cancer results in higher sensitivity to several common anticancer drugs, such as gemcitabine, 5-FU and DDP in vitro, suggesting METTL3 as a potent target for improving therapeutic efficacy [83].
In bladder cancer, circ0008399 interacts with WTAP to facilitate the formation of MTC, stabilizing TNF alpha-induced protein 3 (TNFAIP3) mRNA by elevating its m6A modification level, which reduces the chemosensitivity to cisplatin [52]. Besides, in Nasal-type natural killer/T-cell lymphoma (NKTCL), WTAP was obviously upregulated and inactivate DDP by enhancing the mRNA stability of dual-specificity phosphatases 6 (DUSP6). Inhibition of WTAP decreased cell viability and reduced expression of drug resistance-associated protein MRP-1 and P-gp, indicating the therapeutic potential of WTAP in NKTCL [53]. VIRMA, another “writer”, was confirmed as an oncogene in Germ cell tumors (GCTs). VIRMA knockdown facilitated sensitivity to DDP, downregulated DNA damage repair players, XRCC4-like factor (XLF) and meiotic recombination 11 homolog 1 (MRE11) in a m6A-dependent way, thus enhancing DNA damage with increased expression of gamma histone variant H2AX (γH2AX) and growth arrest and DNA damage inducible alpha (GADD45A/B) [54].
Temozolomide
Temozolomide (TMZ), another alkylating agent apart from platinum drugs, is the main first-line chemotherapeutic drug in the standard regimen of advanced gliomas, combined with radiotherapy [84]. However, the overall clinical efficacy of this regimen in Glioblastoma (GBM) remains disappointing, on account of inherent or induced resistance to TMZ treatment [55].
It was demonstrated that METTL3 level was elevated in GBM and promoted resistance to TMZ, both in vitro and in vivo. In mechanism, METTL3-mediated m6A modification enhances the expression of O6-methylguanine–DNA methyltransferase (MGMT) and alkylpurine–DNA–N-glycosylase (APNG) to repair DNA damage [55]. Moreover, Li et al. underlined the interplay of m6A modification and histone modification in forming resistance. TMZ treatment upregulated METTL3 through increasing chromatin accessibility at the METTL3 locus via SRY-box transcription factor 4 (SOX4). And METTL3 depletion could reduce m6A level on the enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) mRNA, a histone modification-related gene, leading to mRNA degradation [85].
Intriguingly, the demethylase FTO was also reported to enhance the resistance to TMZ in GBM. FTO-mediated demethylation protected phosphoinositide dependent kinase-1 (PDK1) mRNA from degradation, while X-inactive specific transcript (JPX) significantly modulated the FTO/PDK1 interaction, leading to progression and chemoresistance of GBM [56]. Significantly, the FTO inhibitor R-2HG exhibits a synergistic effect with TMZ [86]. Besides, circ_0072083 could elevate ALKBH5 level by negatively regulating miR-1252-5p, and thus promote NANOG expression via reducing m6A modification-dependent degradation, while NANOG is an important biomarker for chemoresistance [57].
Gemcitabine
Gemcitabine (GEM), a pyrimidine analogue, used widely in several tumors. Especially, its application has revolutionized the treatment of pancreatic cancer (PC), improving the survival rate up to 20% [87]. The chemoresistance to GEM, emerging as an urgent matter, has been revealed to be regulated by m6A modification.
METTL3 was supposed to play a positive role in regulating sensitivity to GEM. m6A methylation sustain RNA stability of DBH-AS1 in PC cells, which sensitizes PC cell to GEM by sequestering miR-3163 and thus upregulating USP44. Particularly, downregulation of DBH-AS1 was observed in GEM-resistant PC tissues and cells, and closely related with low expression of METTL3 [58]. Opposite to METTL3, the METTL14 was upregulated in GEM-resistant PC cells, induced by transcriptional factor p65 and downstream enhanced the expression of cytidine deaminase (CDA) to inactivate GEM. Suppression of METTL14 obviously enhance the sensitivity of GEM both in vivo and in vitro, indicating METTL14 as a promising target for improving chemotherapeutic effects in PC [59].
Interestingly, demethylase ALKBH5 also tend to promote sensitivity to GEM, and decreased ALKBH5 correlates with poor prognosis in pancreatic ductal adenocarcinoma (PDAC). The mechanism studies showed that overexpression of ALKBH5 could promote the transcription Wnt inhibitory factor 1 (WIF-1) via demethylation, thus inhibited Wnt pathway to sensitize cancer cells to chemotherapy [60]. Moreover, the increased m6A level on ANRIL recruited serine/arginine-rich splicing factor 3 (SRSF3) to regulates its splicing, facilitating the expression of ANRIL-L isoforms, which forms a complex with EZH2 and Ring1B to enhance DNA homologous recombination repair (HR), thus promote drug resistance [61].
Impact on 5-FU resistance
5-FU is an analogue of uracil, incorporating into nucleic acids to interfere with nucleotide metabolism [88]. It has been widely involved in common chemotherapy regimens, including doublet PF (cisplatin and 5-FU), for many neoplasms treatment [89]. 5-FU is the first choice for CRC in the palliative and adjuvant settings, despite the unsatisfying response rate and frequent resistance [90].
Several studies have identified METTL3 as a driving force in the resistance to 5-FU. METTL3 was confirmed to inhibit the sensitivity through miR181b5p in cancer-associated fibroblasts (CAFs) secreted exosomes. The mechanistic study found that m6A modification enhanced the recognition of pri-miR181d by DiGeorge Syndrome Critical Region 8 (DGCR8) to promote the miR181b5p process. The increased miR181d5p interact with 3′-UTR of neurocalcin delta (NCALD) mRNA to suppress NCALD expression, inducing resistance to 5-FU in CRC cells [62]. It was revealed that METTL3 could enhance the stability of LBX2-AS1 dependent on IGF2BP1, upregulated LBX2-AS1 further increased AKT1 level by sponging miR-422a, correlated with poor response to 5-FU in CRC paitients [63]. Similarly, METTL3 promoted SEC62 expression via stabilizing its mRNA depending on IGF2BP1. SEC62 could protect β-catenin from degradation by directly binding to β-catenin and competitively inhibiting APC-β-catenin interaction, thus activate Wnt/β-catenin signaling to regulate stemness and chemoresistance to 5-Fu and oxaliplatin [64]. Moreover, methylation on p53 pre-mRNA catalyzed by METTL3 promotes a preferential splicing, inducing p53 R273H mutant protein, and giving rise to multidrug resistance in CRC cells, such as Oxa and 5-FU [65].
Nishizawa et al. proposed that inhibition of c-Myc-driven YTHDF1 transactivation suppresses CRC progression and triggers sensitization to anticancer drugs, such as OXA and 5-FU. And patients with high YTHDF1 levels were correlated with significantly poorer overall survival [91]. In accordance, another research showed that YTHDF1 was downregulated by miR-136-5p to suppress tumor progression and chemoresistance to 5-FU and OXA, while miR-136-5p was targeted by circPTK2 and declined in CRC cell lines and tissues [92].
Adriamycin/Doxorubicin
Doxorubicin (DOX) is one of anthracycline antibiotics, exerting anti-tumor effects by intercalating into DNA and disrupting DNA repair subsequently [93], or through oxidative stress [94]. DOX remains one of the main treatments for early and advanced breast cancer (BC), and many other malignancies. Except for lethal cardiotoxicity, resistance is the major obstacle in chemotherapeutic approaches.
Several researches have elucidated different pathways for METTL3 to provoke resistance to DOX in BC. Li et al. have observed highly expressed METTL3 in DOX-resistant breast cancer cells. They supposed that METTL3 promoted expression of metastasis associated lung adenocarcinoma transcript 1 (MALAT1, which recruited E2F transcription factor 1 (E2F1) to activate transcription of anterior gradient 2 (AGR2), thus desensitize BC cells to DOX [66]. METTL3 could enhance pri-miR-221-3p maturation, miR-221-3p further suppresses HIPK2 and upregulates the downstream Che-1, which leads to increased multidrug resistance protein (MDR1) and breast cancer resistance protein (BCRP), indicating enhanced chemoresistance [67]. METTL3-mediated m6A modifications facilitate the expression of estrogen receptor related receptor γ (ERRγ) by triggering the splicing of ERRγ pre-mRNA, and ERRγ interacts with p65 to promote ABCB1 transcription, encoding P-gp to decrease the sensitivity to Taxol (Tax) and doxorubicin (Dox). ERRγ/p65 complex also binds to CPT1B promoter to increase its expression, accelerating fatty acid β-oxidation (FAO) and thus promoting resistance [68]. Moreover, de novo synthesis of fatty acids was supposed to be associated with chemoresistance in BC. Inhibition of FTO significantly attenuates fatty acid synthesis, which obviously sensitize BC cells to chemotherapy [95].
Also, attention has been given to other m6A modifiers about their influences on resistance to DOX in BC. WTAP enhanced the stability of lncRNA DLGAP1 antisense RNA 1 (DLGAP1-AS1) and overexpressed DLGAP1-AS1 promoted chemoresistance in vitro and correlated with unfavorable prognosis of BC patients. Furthermore, DLGAP1-AS1 motivated WTAP, by sponging miR-299-3p to relieve its suppression on WTAP, forming a feedback loop [69]. YTHDF1 was supposed as a novel target to deal with chemoresistance in BC and a putative tumor promotor [96]. The silico analysis showed that YTHDF1 was overexpressed in BC tissues, which facilitated DNA damage repair and resistance to Adriamycin, Cisplatin, and Olaparib. To achieve this, YTHDF1 enhances E2F8 mRNA stability dependent on METTL14, as E2F8 was identified to promote cell proliferation in BC by effecting G1/S phase transition [70]. Wang et al. reported that FTO and STAT3 were highly expressed in DOX-resistant BC cells and STAT3 directly binds to the promotor region of FTO to regulate its transcription. FTO knockdown could increase the chemosensitivity to doxorubicin and reverse the opposite effects of STAT3 overexpression [71].
Aside from breast cancer, m6A methylation was discovered to modulate DOX resistance in other cancers. Yang et al. figured out that upregulated IGF2BP3 in CRC could promote ABCB1 expression by increasing mRNA stability, thus induced resistance to DOX in vitro and in vivo [72]. In osteosarcoma (OS), ablation of METTL3 and METTL14 could upregulate the TRIM7 expression via inhibiting YTHDF2-medaited degradation. TRIM7, as a ubiquitin ligase, inhibits BRMS1 through ubiquitination, resulting in increased resistance to ADR and MTX in vitro and in vivo [73]. In DOX-resistant CML cells, LINC00470 promoted the degradation of phosphatase and tensin homolog (PTEN) mRNA via METTL3-modified methylation, which acts as a tumor suppressor by inhibiting Notch and PI3K‐AKT‐Mtor signaling. Knockdown of METTL3 rescued PTEN expression, thus inhibiting the leukemia cells viability and restoring the chemosensitivity [74].
Other chemotherapeutic drugs
Wang et al. have demonstrated an unexplored role of adipocyte-derived exosomes to protect multiple myeloma (MM) cells against apoptosis induced by chemotherapy drugs such as bortezomib, melphalan, and carfilzomib. Herein, METTL7A promotes the enrichment of lncRNAs into exosomes to exert oncogenic roles, and the process is enhanced by MM cells through EZH2-mediated METTL7A methylation. This finding indicates a potential strategy to improve drug resistance by blocking this vicious exosome-mediated cycle [75]. In acute myeloid leukemia (AML), METTL3 was upregulated in resistant AML cells compared to the counterparts. METTL3 could promote chemoresistance to cytarabine (Ara-C) by facilitating expression of MYC [76]. However, bone marrow mesenchymal stem cells (MSCs) of AML have decreased global m6A levels and expressions of METTL3, and overexpressed METTL3 render MSCs sensitive to penicillin and streptomycin. Downregulated METTL3 increases AKT level and enhances adipogenesis, thereby contributing to chemoresistance [77]. In T-cell acute lymphoblastic leukemia (T-ALL), ALKBH5 could enhance the expression of ubiquitin-specific protease 1 (USP1) by stabilizing its transcripts. USP1 targeted at Aurora B and deubiquitinated it, facilitating resistance to dexamethasone in vitro and in vivo [78]. Using integrated bioinformatics analysis, the highly expressed RAB39B in DLBCL was revealed to be associated with 14 m6A regulators and resistance to several chemotherapy drugs including dexamethasone, doxorubicin, etoposide, vincristine, and cytarabine, as well as the poor survival in DLBCL patients [97]. A novel YTHDF3-based model was constructed to effectively predict the sensitivity of several therapeutic agents for breast cancer, including doxorubicin, paclitaxel, methotrexate, and vinorelbine [98]. Through silico analysis, researchers have supposed HNRNPC as a predictor of paclitaxel resistance in ovarian cancer [99]. Zhang et al. built an effective classifier to predict chemotherapy benefit of SCLC based on 7 m6A regulator-based. And in vitro experiments demonstrated that ZCCHC4, G3BP1, and RBMX may be potential therapeutic targets for overcoming chemoresistance in SCLCs [100].
m6A and targeted therapy resistance
Targeted therapy is a kind of groundbreaking therapeutics by interfering with specific molecules to impede caner growth, progression and metastasis. A range of targeted medicines have been approved by FDA, displaying favorable clinical benefits in the treatment of various cancer types including liver, colorectal, lung, breast and ovarian cancers. Nevertheless, resistance to targeted therapy is frequently acquired, principally resulted from on-target mutations, bypass alterations, and cellular plasticity [101]. Meanwhile, m6A methylation has been confirmed to universally influence targeted molecules or pathways, regulating therapeutic resistance (Table 2, Fig. 2).

The role and molecular mechanisms of m6A regulators in Targeted therapy resistance
Sorafenib
Sorafenib is a multiple-target tyrosine kinase inhibitor (TKI), which can inhibit Ras/Raf/MEK/ERK signaling pathways to restrain cancer cells proliferation, and target at receptors like vascular endothelial growth factor receptor (VEGFR) 2, platelet-derived growth factor receptor (PDGFR-β) to inhibit angiogenesis [122]. The application of sorafenib has significantly improved the outcomes of advanced HCC patients [123].
For both METTL3 and METTL14, there were contradictory findings about their regulatory roles in resistance to sorafenib. For the promotive role, the highly expressed METTL3 significantly enhanced the stability of lncRNA DUXAP8, which competitively binds to miR-584-5p via ceRNA mechanism, thus promotes MAPK1 expression and activates MAPK/ERK pathway, contributing to resistance acquisition in HCC [102]. METTL3 could upregulate NIFK-AS1, and NIFK-AS1 suppressed the expression of drug transporters Organic anion-transporting polypeptide 1B (OATP1B1) and OATP1B3 to inhibit sorafenib uptake, thus desensitize HCC cells. HCC patients with low level of NIFK-AS1 exhibited benefits to treatment [103]. In addition, m6A-circRNA interaction has been found in sustaining the resistance. METTL3/14 promoted circRNA SORE expression by increasing its stability, which acts as a sponge to sequester miR-103a-2-5p and miR-660-3p to activate the Wnt/beta-catenin pathway, contributing to resistance [104]. On the contrary, Lin et al. proposed that knockdown METLL3 can promote the resistance through FOXO3-mediated autophagy. METTL3 depletion attenuates the m6A level at 3′UTR of FOXO3 mRNA and depresses the YTHDF1-mediated stabilization, then downregulated FOXO3 induced the transcription of a series of autophagy-related genes to enhance autophagy in HCC cell lines [105]. Moreover, mechanism study showed that METTL14 could enhance HNF3γ mRNA stability via IGF2BPs, and thus induced transactivation of OATP1B1 and OATP1B3 expression, and reduced METTL14 in HCC cells contributes to developing resistance [106]. Except for the two principal writers, another methylase, RBM15B was also supposed to promote resistance to sorafenib in HCC. RBM15B interacts with the TRAM2 mRNA to enhance its stability, increased TRAM2 promotes the expression and activation of YAP and TAZ, activating the Hippo signaling pathway. Knockdown of RBM15B and TRAM2 significantly reversed the resistance, while overexpression exerted the opposite effects [107].
Gefitinib
Overexpression of EGFR is ubiquitous in solid tumor and related with tumorigenesis, progression and invasion. EGFR-TKI treatment have provided tremendous therapeutic benefits for NSCLC patients with activating mutations of EGFR [124]. For LC patients with the in-frame deletion in EGFR-exon 19 or single-point mutation of exon 21, the standard first-line treatment is first-generation (gefitinib, erlotinib), or second-generation (afatinib) TKIs [125].
In NSCLC, existing evidences elucidated that METTL3 played a positive role in acquiring the resistance to gefitinib. METTL3 induced the upregulation of SNHG17 via stabilizing its mRNA, and SNHG17 recruited EZH2 to the promoter region of large tumor suppressor kinase 2 (LATS2) to suppress it expression, which was a well-known tumor suppressor involved in the Hippo cascade, then epigenetically repressed LATS2 promote resistance to gefitinib in LUAD [108]. Also, METTL3 can contribute to the resistance via increasing autophagy. The overexpressed METTL3 upregulated genes of autophagy pathway such as ATG5 and ATG7, while β-elemene inhibited the expression of METTL3 with highly selectivity, thus reversed the resistance via disrupting autophagy [109]. Besides, Tang et al. reported increased expression of “writer” KIAA1429 in resistant NSCLC cells, accompanied with high m6A enrichment. KIAA149 promote the resistance in vitro, via targeting the 3'-UTR of HOXA1 mRNA to enhance its stability in an m6A-depnendent way [110].
FTO has also been found to be upregulated in NSCLC and conducive to resistance to gefitinib. The gefitinib-resistant NSCLC patients exhibit increased FTO expression and decreased m6A level in the serum exosomes, and deletion of FTO significantly enhances the sensitivity. FTO facilitates ABCC10 expression via inhibiting the YTHDF2-mediated mRNA decay, promoting resistance in vitro and in vivo [111]. Significantly, the FTO inhibitor meclofenamic acid (MA) was identified to overcome drug resistance, via restraining FTO-mediated demethylation of m6A-modified MYC, and further downregulating the membrane drug efflux proteins, BCRP and MRP-7 [112].
Other targeted therapies
Erlotinib is the first-generation EGFR inhibitor with similar molecular structures and pharmacologic mechanisms to gefitinib. In LUAD, YTHDF2-mediated inhibition of lncRNA TUSC7 provokes the resistance to erlotinib, as TUSC7 could sponge miR-146a and inhibit Notch signaling to decrease the cancer progression and improve sensitivity to erlotinib [113]. Osimertinib was the first 3rd-generation EGFR-TKIs to be approved for metastatic EGFR-mutant NSCLC. In osimertinib-resistant LUAD cells, metformin could upregulate METTL3 by promoting the bindings of DNA methyltransferase-3a/b (DNMT3a/b) to METTL3 promoter. Then METTL3-mediated m6A significantly facilitated pri-Let-7b maturation via NKAP and HNRNPA2B1, thus inactivating Notch signaling and re-captivating osimertinib treatment [114].
Cetuximab (CTX), a monoclonal antibody targeting EGFR, approved by FDA for treating metastatic CRC in 2004 [126]. The m6A reader HNRNPA2B1 was proposed to sustain resistance to CTX. Mechanistically, HNRNPA2B1 interacts with MIR100HG and recognizes the m6A sites of TCF7L2 mRNA to improve its stability, while TCF7L2 acts as an essential transcriptional coactivator of the Wnt/beta-catenin signaling [115]. Intriguingly, METTL3 was found to facilitate extracellular vesicle (EV)-mediated spread of the resistance by modulating miRNA metabolism. Knockdown of METTL3 decreased the cellular and extracellular levels of m6A-modified miRNAs, such as miR-100 and miR-125b, which were previously proved to induce resistance to CTX, and thus the EVs showed less capability of inducing the transfer of the resistance [116].
Apatinib, a selective TKI targeting at VEGFR-2 to anti-angiogenesis, applied in GC, HCC, and NSCLC and so on [127]. In HCC, suppression of METTL3 induced by S-adenosyl homocysteine or siRNA could activate p53, further sensitizing HCC cells to apatinib through apoptosis [117]. Sunitinib is a multi-target, anti-angiogenic TKI, which greatly improves the overall survival of metastatic RCC patients. The resistance to sunitinib in RCC was supposed to be linked with high expression level of TRAF1, as METTL14-mediated m6A modification enhances TRAF1 mRNA stability in a IGF2BP2-dependent manner [118].
PLX4032, a selective and potent inhibitor of BRAF V600E mutation, inhibits tumor growth in melanoma. Bhattarai et al. indicated that METTL3 could induce resistance to PLX4032 in melanoma via EGFR-mediated rebound activation. METTL3-mediated m6A modifications promoted the translation efficiency of EGFR mRNA, reactivating the RAF/MEK/ERK pathway to trigger resistance [119]. Crizotinib is a kinase inhibitor targeting c-MET/ALK/ROS1 used as the first-line therapy for NSCLC with ALK mutations. In ALK mutation-free and c-MET highly-expressed NSCLC cell lines, chidamide could enhance the sensitivity to crizotinib through decreasing expression of METTL3 and WTAP. Reduced m6A level on c-MET mRNA downregulated its expression, subsequently restored the response to crizotinib. And the c-MET ligand hepatocyte growth factor (HGF) could synergize with chidamide [120]. Poly(ADP-ribose) polymerase inhibitors (PARPi) such as Olaparib have demonstrated substantial therapeutic benefits for treating EOC patients with BRCA1/2 mutation [128]. In BRCA-mutated EOC cells, downregulated FTO and ALKBH5 elevated m6A level of FZD10 transcripts and stabilized it via IGF2BP2, further stimulated Wnt/β-catenin pathway, causing resistance to Olaparib and Rucaparib [121].
m6A and radiotherapy resistance
Radiotherapy remains a mainstay of cancer treatment regimens, which kills cancer cells through damaging the DNA. Developing reliable predictive biomarkers for radio-resistance is significant for personalized radiation therapy [129]. Notably, METTL3 was previously verified to influence ultraviolet-induced DNA damage responses [130]. Depletion of METTL3 in pancreatic cancer cells lead to higher sensitivity to a series of anticancer therapy including irradiation (IR) [83]. In GBM, overexpressed METTL3 binds to SOX2 transcripts and sustains their stability, contributing to increased DNA repair, thus promoting the resistance of glioma stem-like cells to γ-irradiation [131]. Knockdown of METTL3 influenced the localization of DNA polymerase kappa to DNA damage sites in U2OS and HeLa cells, displaying insufficient repair and higher sensitivity to ultraviolet [130].
In cervical squamous cell carcinoma (CSCC), upregulated FTO could promote β-catenin expression via demethylating its transcripts and further positively regulate the excision repair cross-complementation group 1 (ERCC1), contributing to the resistance to cisplatin and irradiation [132]. Researchers speculated that highly expressed ALKBH5 in GBM stem cells promoted radiotherapy resistance via modulating homologous recombination (HR). Deficiency of ALKBH5 induced inhibition of checkpoint kinases 1 (CHK1) and recombinase Rad51, both of which are key players in DNA damage repair [133]. Furthermore, the researchers previously revealed that FOXM1 interacted with MELK to block radiotherapy response [134], and in the present study, IR-induced upregulation of FOXM1 level was attenuated by ALKBH5 inhibition, implicating other possible pathway of ALKBH5-mediated radio-resistance in GBM [133]. Taken together, these findings demonstrated a fundamental need for further exploring the regulatory roles in resistance to radiotherapy (Table 3, Fig. 3).

The role and molecular mechanisms of m6A regulators in radiotherapy and immunotherapy resistance
m6A and immunotherapy resistance
Immunotherapy is one of the most promising therapeutic methods, which aims at combating tumors by stimulating and enhancing the host immune system, including immune checkpoint blockade (ICB), cell therapy, cancer vaccine and so on [141]. However, some patients exhibit unfavorable responses to immunotherapy, which restricts the further applications and development. Mounting evidences suggested that m6A modification could modulate anti-tumor immunity and tumor microenvironment (TME), influencing the therapeutic response to immunotherapy (Table 3; Fig. 3).
Immune checkpoint blockade
Among diverse immunotherapy strategies, ICB therapy is an unprecedented anti-tumor therapy by blocking inherent immune inhibiting factors, such as programmed cell death 1 (PD-1) or their ligands PD-L1, and cytotoxic T-lymphocyte antigen 4 (CTLA-4) [142]. Advances have been made in the clinical implements, such as PD-1 inhibitors pembrolizumab and nivolumab which are approved by FDA for advanced melanoma and NSCLC [143]. Nevertheless, for low‐mutation‐burden cancer patients, the failure of ICB or relapse are still common [144, 145]. Increasing researches revealed that m6A modification markedly affected the therapeutic resistance against ICB.
In mismatch‐repair‐proficient or microsatellite instability‐low (pMMR‐MSI‐L) tumors with low mutation burden, which constitutes about 85% of CRC, depletion of METTL3/14 could enhance ICB responses through increasing infiltration of cytotoxic CD8 + T cells and promoting secretion of IFN-γ, CXCL9, and CXCL10 in TME in vivo [135]. Mechanistically, METTL3/14 loss promoted IFN-γ-STAT1-IRF1 signaling through stabilizing the STAT1 and IRF1 mRNA via YTHDF2 [135]. Paradoxically, METTL14 was proposed to increase sensitivity to anti-PD1 immunotherapy in cholangiocarcinoma (CCA). METTL14 could enrich m6A in the 3'UTR of the siah E3 ubiquitin protein ligase 2 (SIAH2) mRNA and lead to YTHDF1-mediated decay. Therefore, deficiency of SIAH2 elevated PD-L1 expression level by reducing its K63-linked ubiquitination, and further sensitized tumor to ICB [136].
The previous study has confirmed that therapeutic efficacy of PD-L1 checkpoint blockade was enhanced in YTHDF1(−/−) mice. Depletion of YTHDF1 suppresses the translation of lysosomal cathepsins in DCs, downregulated cathepsins attenuates antigen degradation and thus enhances cross-presentation to CD8 + T cells, further increased IFN-γ expression in CD8 + T cells upregulates PD-L1 expression in tumor cells [137]. Accordingly, patients with low YTHDF1 level show more CD8 + T cell infiltration, implicating the potential of YTHDF1 inhibition in overcoming low response to ICB [137].
FTO was supposed to negatively regulate anti-PD-1 immunotherapy response in melanoma. Knockdown of FTO elevated the m6A level in transcripts of several critical melanoma-promoting genes, including PD-1, C-X-C motif chemokine receptor 4 (CXCR4), and SOX10, then accelerated RNA decay mediated by YTHDF2, restoring IFN-γ response in vitro and sensitizing anti-PD-1 treatment in vivo [138]. Moreover, Tsuruta et al. found that FTO could upregulate PD-L1 expression, positively regulating anti-PD-1 blockade response in CRC. Contrary to previous recognition that PD-L1 was upregulated by IFN-γ signaling [146], they indicated that FTO regulates PD-L1 expression in an IFN-γ-independent manner in HCT-116 cells [139].
Immune evasion resulting from upregulated immune checkpoint genes was deemed as the origin of hypomethylating agents (HMAs)-induced drug resistance [147]. HMAs, such as Azacitidine (AZA) and Decitabine (DAC), are wildly used in AML or myelodysplastic syndrome (MDS) treatment [148, 149]. Su et al. found that DAC treatment also cause reduction of global m6A abundance in AML cells, possibly related with the overexpressed FTO. They observed that FTO inhibition induced by small-molecule compounds, downregulated the immune checkpoint gene leukocyte immunoglobulin-like receptor subfamily B4 (LILRB4), with a greater tendency than PD-L1/2, further sensitized leukemia cells to T cell cytotoxicity and overcame HMA-induced immune evasion [150]. Although the low expression of PD-L1/2 in AML impedes application of anti-PD-1 blockade, these findings underlying the significance of combining FTO inhibitors or anti-LILRB4 agents and HMAs for myeloid malignancies. Moreover, FTO was supposed to mediated immune evasion via rewiring tumor glycolytic metabolism to restrict effector T cells functions. FTO-dependant demethylation facilitates the expression of several basic leucine zipper (bZIP) family transcription factors such as JunB and C/EBPβ, and then upregulates glycolysis-associated genes, which further establishes a metabolic advantage over T cells. Herein, inhibition of FTO promotes T cell infiltration and synergizes with anti-PD-L1 blockade in melanoma [151].
Via analyzing clinical data, ALKBH5 was identified as a predictive biomarker of anti–PD-1 blockade ressistance in melanoma. In mechanism, ALKBH5 deletion reduces MCT4 expression and lactate content in tumor interstitial fluids, which ultimately suppressed the Treg and polymorphonuclear myeloid derived cell [140]. In addition, the silico analysis showed that the m6A regulator ELAVL1 played a crucial role in the therapeutic response of anti-PD-L1 therapy in GBM [152]. In addition, a great deal of bioinformatic researches have underscored the close correlation between m6A methylation and immunotherapy response in various cancer [153,154,155]. For instance, via thoroughly analyzing the epigenetic modification patterns of 1518 GC patients, a subtype with high FTO level was identified to correlate with poor survival and immunotherapy resistance, which are potentially benefited from combination FTO inhibition and ICB [155].
Implications and future directions
With growing studies underlying the mechanisms of how m6A modification influences cancer therapeutic resistance, targeting at m6A to optimize cancer therapy becomes increasingly appealing. There are several promising research directions refueling the scientific interests.
Predictive biomarkers for therapy responses
More effective assessment approaches to predict potential therapeutic resistance and evaluate treatment effects have been demanded for a long time. As presented above, aberrant expressions of m6A regulators are ubiquitously involved in various resistance, providing theoretic fundament for predicting treatment responses. Amounting evidences identified that alteration of global m6A levels and regulators expression are closely related with therapy response, for example, upregulated METTL3 level was identified in TMZ resistant GBM tissue [64]. Besides next-generation sequencing (NSG) technology, tumor-derived organoids and network-based machine learning are also conducive to predicting drug efficiency or tumor resistance [156]. Recent years have witnessed plentiful mechanistic studies and bioinformatics analysis identifying the prognostic potential of m6A regulators in various cancer. However, there is still a long way from clinic applications.
Targeting m6A regulators for overcoming resistance
Specific inhibitors of m6A regulators have demonstrated exciting anti-tumor effects, including inhibitors of METTL3, FTO, ALKBH5, IGF2BP1 [157], while the therapeutic effects of METTL3 activators remain unverified [158]. Especially, researches have underscored the potential of several specific inhibitors to overcome therapeutic resistance, supporting the combined applications with present therapeutics.
Rhein is the first natural FTO inhibitor which competitively binds to the catalytic domain of FTO and displays therapeutic efficacy in leukemia mice [159]. The NSAID, meclofenamic acid 2 (MA2) was found to inhibit FTO and thus suppress glioblastoma progression [160]. The small-molecule compounds CHTB and N-CDPCB were identified via crystal structure screening, providing new binding sites for targeting FTO [161, 162]. R-2-hydroxyglutarate (R-2HG) inhibits progression of AML in vitro an in vivo through inhibiting FTO, synergizing with a series of first-line chemotherapy agents such as all-trans retinoic acid (ATRA), Azacitidine, Decitabine, and Daunorubicin [86]. Also, R-HG displays growth-suppressive effects in GBM cell lines and cooperates with TMZ [86]. Subsequently, Huang et al. have reported that tricyclic benzoic acid FB23-2 could suppress the proliferation and enhance the differentiation/apoptosis of AML cells [163]. Su et al. identified that two small molecular inhibitors, CS1, and CS2, can sensitize leukemia cells to T cell cytotoxicity, with potential to overcome HMA-induced immune evasion [150]. For immunotherapy, another inhibitor Dac5 relived the constraints on T cells activation and functions imposed by FTO, which improves the ICB efficacy in melanoma [151]. Recent advances showed that the FB23 analog, FB23-13a exerted a stronger anti-proliferative effect on AML cells both in vitro and in vivo [164]. Recent researches made favorable progressions in more tumor types rather than AML and glioma. A small-molecule compound, 18097 significantly restrained growth and colonization of breast cancer cells [165]. Significantly, the oxetane class of FTO inhibitors, FTO-43 demonstrated anti-tumor potency comparable to 5-FU in GC, glioblastoma and AML models [166]. And Qin et al. proposed compound C6, a 1,2,3-triazole analogues, as an orally antitumor agent for esophageal cancer [167]. Additionally, the technologies also keep evolving. researchers have synthesized a hybrid FTO inhibitor by merging fragments from previously reported inhibitors with anti-leukemia activity [168]. FTO inhibitor-loaded GSH-bioimprinted nanocomposites (GNPIPP12MA) synergized GSH depletion to anti-leukemogenesis, which augmented the therapeutic efficacy of the PD-L1 blockade [169].
The initial small molecule METTL3 inhibitors were discovered via the high-throughput docking into the SAM binding site, which are indeed adenosine analogues [170]. However, due to the binding promiscuity and poor permeability, non-nucleoside inhibitors were developed. A small-molecule METTL3 inhibitor, UZH1a demonstrated potent growth inhibition of AML [171]. UZH2, the analogues of UZH1a, exhibits antitumor effects in AML and prostate cancer cell lines [172]. Remarkably, in vitro and in vivo experiments identified potent anti-leukemia efficacy of STM2457, without influencing normal hematopoiesis [173]. The specific ALKBH5 inhibitor ALK-04 was found to improve anti-PD-1 therapy efficiency in melanoma [140]. Selberg et al. discovered two compounds selectively inhibited ALKBH5, with anti-proliferative effects in specific leukemia cell lines [174]. Moreover, BTYNB was identified as a potent and selective IGF2BP1 inhibitor, exhibiting growth-suppressive effects in melanoma and OC cells [175].
Collectively, m6A regulator-targeted drugs have gained lots of attention as a promising therapeutic strategy. For the specific inhibitors verified to improve therapeutic effects, further pre-clinical experiments should be boosted. For those novel durgs, their potentials to synergize present therapeutics and overcome resistance should be explored. Also, more endeavors should be put into developing more potent and selective inhibitors or activators (Table 4; Fig. 4).

The applications of m6A regulator-targeted inhibitors in cancers
Potential of m6A regulation in immunotherapy
It has been widely recognized that m6A modifications can regulate the fate and biological behaviors of tumor cell. At the same time, m6A exhibits prominent influences on immune cells, providing opportunities for developing immunotherapy. Both METTL3 and YTHDF2 are positive regulators of NK cell functions, including maintaining the homeostasis, maturation, survival, anti-tumor and anti-viral activity. Researchers have successfully enhanced the proliferation and cytotoxicity of NK cells in vitro by modulating m6A regulators, providing support for adoptive NK cell-based immunotherapy, such as chimeric antigen receptor (CAR) NK cells and induced pluripotent stem cell (iPSC)-derived NK cells [176, 177]. For T cells, METTL3 exerts an essential role in regulating the homeostasis and differentiation [178], influencing the anti-tumor responses in a bidirectional complex manner. On the one hand, depletion of METTL3 facilitates anti-tumor immunity by restraining Treg cells, on the other hand, reduced METTL3 in CD4 + T cell impairs humoral immunity by suppressing the differentiation and functional maturation of T follicular helper cell [179, 180]. Additionally, tumor-intrinsic FTO could suppress the activation and effector states of CD8 + T cells [151]. For macrophages, METTL3 exerts positive control on its polarization and immune functions, ablating METTL3 can promote tumor growth and metastasis. Besides, METTL3 depletion in macrophages reduced the efficacy of PD-1 blockade therapy and levels of lipopolysaccharide (LPS)-induced TNF-α [181, 182]. Also, METTL14 was found to regulate the functions of immunomodulatory ligands in macrophages, and METTL14 deficiency in macrophage1919s inhibited the anti-tumor function of CD8 + T cells and promoted tumor growth [183]. Taken together, these findings highlight the significant roles of m6A regulators in immune cells.
Furthermore, m6A modifications have the promising potential to remodel the TME, influencing cancer progression and responsiveness to immunotherapy. METTL3 plays a dual role as either an oncogene or a tumor suppressor gene in different types of cancers, with upregulated or downregulated expression. To complicate matters further, the level of METTL3 was significantly related with infiltration of various immune cells, in positive or negative correlation. For example, downregulation of METTL3 in testicular germ cell tumors (TGCT) tissue is positively correlated with levels of tumor-infiltrating CD8 + T cells, CD4 + T cells, and NK cells [184], whereas the increased METTL3 level in CRC is negatively related with infiltration of CD8 + T cells and secretion of IFN-γ, CXCL9, and CXCL10 [135]. Such complicated situation also exists in several other m6A regulators, such as METTL14, ALKBH5, and YTHDFs [185]. Significantly, except for affecting infiltration of immune cells, m6A exerts influences on TME by regulating the expression of immune checkpoint genes. For example, depletion of FTO induces suppression of LILRB4 in AML [150], and ALKBH5 facilitates expression of PD-L1 in intrahepatic cholangiocarcinoma (ICC) [186].
Overall, the above studies emphasize the potential of targeting m6A modifications to enhance the efficacy of current immunotherapy, as well as to develop novel strategies. Developing specific inhibitors or activators is undoubtedly meaningful. As mentioned above, agents capable of improving anti-tumor immunity have already been reported [150, 151], and the combined application has been verified to sensitize tumor cells to ICB [138, 140]. Additionally, modulating m6A modifications appears as a promising direction in adoptive cell therapy, including CAR NK cells, iPSC-derived NK cells and CAR T cell therapy. Since nanoparticles (NPs) have emerged as promising carriers in cancer precision therapy [187], targeted delivery of NP-encapsulated m6A regulators or target genes into TAMs could be a research direction of great value.
Discussion
The regulatory roles of m6A modification in controlling cancer therapeutic response/resistance have gained increasing attention in recent years. Significantly, targeting m6A modification system is highly conducive to improve cancer therapy efficacy. Compared with existing review, our study is much more comprehensive and systematic, not only supplementing the latest progresses, but also compensating the shortcomings of previous studies. Resistance to chemotherapy, targeted therapy, radiotherapy and immunotherapy were respectively overviewed, with adequate attention paid on immunotherapy. Furthermore, the clinical implications of m6A were highly valued to prevent resistance and develop new therapeutic strategies.
However, with breakthroughs made in this field, there are still contradictions and uncertainties in relevant rationales: (i) in the same cancer, the expression of m6A writers and erasers are regulated in the same direction, leading to discrepancy in m6A concentration alteration. For instance, both METTL3 and FTO are upregulated in NSCLC tissue. Liu et al. have reported significantly overexpressed METTL3 [116], while another research showed increased FTO expression and decreased m6A level in the serum exosomes [118]; (ii) in the same cancer, m6A writers and erasers display similar effects on therapy resistance. Take the resistance to TMZ in GBM as example, it was demonstrated that METTL3 contributed to the resistance by enhancing DNA damage repair [63]. Intriguingly, FTO was also reported to enhance the resistance via protecting PDK1 mRNA from degradation [65]; (iii) for the same type of regulators, they have opposite regulatory roles for the same therapy. For instance, METTL3 and METTL14 were reported to regulate sorafenib resistance of HCC reversely. METTL3 exerts positive regulation by inhibiting OATP1B1 and OATP1B3 expression to decrease sorafenib uptake [103]. On the contrary, METTL14 induced transactivation of OATP1B1 and OATP1B3 to sensitize HCC cells [106]; (iv) for the same regulator, it has contrary influences on the same therapy. Represented by the most learned METTL3, it was revealed that METTL3 promoted resistance to cisplatin by stimulating AKT1 in NSCLC [35]. However, Ling et al. found that METTL3 participated in the re-sensitization to cisplatin mediated by propofol [36].
Thus, in order to clarify these uncertainties and deepen the understanding of therapeutic resistance, more researches of m6A regulation are required. There are some reasonable approaches, including: (1) developing a precise and efficient editing tool to fine-tuning m6A modification of specific targets, a gene, a group of cells and even a specific m6A stie; (2) exploring the target selectivity of m6A regulators in different cellular context, different cancer cells from different tissue origins; (3) developing highly selectively m6A-targeted agents and exploring the feasibility of combined application; (4) mining the potential of m6A modifications in immunotherapy, developing targeted agents to activate endogenous anti-tumor immunity and remodel TME.
Conclusion
In virtue of the ubiquitous regulation of m6A on the RNA fates, m6A modification plays s significant role in regulating cancer therapeutic resistance, which lays the foundation for circumventing resistance and optimizing cancer treatment. Although numerous advancements have been achieved in exploring the underlying mechanisms and clinic implications in this field, our knowledge is in infancy. New approaches and techniques, more large-scale and deeper researches are required, and more attention should be given to the potential of emerging immunotherapy.
Availability of data and materials
Not Applicable.
Abbreviations
- 5-Fu:
-
5-Fluorouracil
- ALKBH3/5:
-
AlkB homolog 3/5 RNA demethylase
- AKR1C1:
-
Aldo–keto reductases 1C1
- AML:
-
Acute myeloid leukemia
- APNG:
-
Alkylpurine–DNA–N-glycosylase
- AKT1:
-
AKT serine/threonine kinase 1
- ATP7A:
-
ATPase copper transporting alpha
- ATRA:
-
All-trans retinoic acid
- BC:
-
Breast cancer
- BRCA1:
-
Breast cancer type 1
- BCRP:
-
Breast cancer resistance protein
- CAFs:
-
Cancer-associated fibroblasts
- CAR:
-
Chimeric antigen receptor
- CBSLR:
-
CBS mRNA-destabilizing lncRNA
- CCA:
-
Cholangiocarcinoma
- CDA:
-
Cytidine deaminase
- CDS:
-
Coding sequence
- CHK1:
-
Checkpoint kinases 1
- CRC:
-
Colorectal cancer
- CSCC:
-
Cervical squamous cell carcinoma
- CTLA-4:
-
Cytotoxic t-lymphocyte antigen 4
- CTX:
-
Cetuximab
- CXCR4:
-
C-X-C motif chemokine receptor 4
- DDP:
-
Cisplatin
- DUSP6:
-
Dual-specificity phosphatases 6
- DGCR8:
-
DiGeorge Syndrome Critical Region 8
- DOX:
-
Doxorubicin
- DNMT3a/b:
-
DNA methyltransferase-3a/b
- DYRK1B:
-
Dual-specificity Y-phosphorylation-regulated kinase 1B
- E2F1:
-
E2F transcription factor 1
- EGFR:
-
Epidermal growth factor receptor
- eIF:
-
Eukaryotic initiation factor
- ERCC1:
-
Excision repair cross-complementation group 1
- ERRγ:
-
Estrogen receptor related receptor γ
- FAO:
-
Fatty acid β-oxidation
- FMRPs:
-
Fragile X mental retardation proteins
- FOXM1:
-
Forkhead box M1
- FSP1:
-
Ferroptosis suppressor protein 1
- FTO:
-
Fat mass and obesity-associated protein
- GC:
-
Gastric cancer
- GADD45A/B:
-
DNA damage inducible alpha/beta
- GBM:
-
Glioblastoma
- GLS1:
-
Glutaminase 1
- HGF:
-
Hepatocyte growth factor
- HCC:
-
Hepatocellular carcinoma
- HuR:
-
Human antigen R
- IGF2BP:
-
IGF2 mRNA binding protein
- JAK2:
-
Janus kinase 2
- LATS2:
-
Large tumor suppressor kinase 2
- LC:
-
Liver cancer
- LILRB4:
-
Leukocyte immunoglobulin-like receptor subfamily B4
- LGR5:
-
Leucine rich repeat containing G protein-coupled receptor 5
- m6A:
-
N6-methyladenosine
- MALAT1:
-
Metastasis associated lung adenocarcinoma transcript 1
- MDR1:
-
Multidrug resistance protein
- METTL3/14/16:
-
Methyltransferase-like 3/14/16
- MGMT:
-
O6-methylguanine–DNA methyltransferase
- MRE11:
-
Meiotic recombination 11 homolog 1
- MSCs:
-
Mesenchymal stem cells
- MTC:
-
Methyltransferase complex
- NANOG:
-
Nanog homeobox
- NF-κB:
-
NF-kappaB
- NSCLC:
-
Non-small cell lung cancer
- NPC:
-
Nasopharyngeal carcinoma
- OATP1B:
-
Organic anion-transporting polypeptide 1B
- OC:
-
Ovarian cancer
- OSCC:
-
Oral squamous cell carcinoma
- PARPi:
-
Poly(ADP-ribose) polymerase inhibitors
- OXA:
-
Oxaliplatin
- PC:
-
Pancreatic cancer
- PDK1:
-
Phosphoinositide dependent kinase-1
- PD-1:
-
Programmed cell death 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGFR-β:
-
Platelet-derived growth factor receptor
- PTEN:
-
Phosphatase and tensin homolog
- NF-κB:
-
RBM15/15B: RNA-binding motif protein 15/15B
- R-2HG:
-
R-2-hydroxyglutarate
- RAP1:
-
Ras-associated protein1
- SIAH2:
-
Siah E3 ubiquitin protein ligase 2
- SOX4:
-
SRY-box transcription factor 4
- SRSF3:
-
Serine/arginine-rich splicing factor 3
- STAT3:
-
Signal transducer and activator of transcription 3
- Tax:
-
Taxol
- TKI:
-
Tyrosine kinase inhibitor
- TMZ:
-
Temozolomide
- TRAF:
-
Tumor necrosis factor receptor-associated factors
- TRIM29:
-
Tripartite motif-containing protein
- TFAP2C:
-
Transcription factor-activating enhancer-binding protein 2C
- VEGFR:
-
Vascular endothelial growth factor receptor
- VIRMA/KIAA1429:
-
Virlike m6A methyltransferase-associated
- WIF-1:
-
Wnt inhibitory factor 1
- WTAP:
-
WT1-associated protein
- XLF:
-
XRCC4-like factor
- YTHD:
-
YTH domain family of protein
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- γH2AX:
-
Gamma histone variant H2AX
References
Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42.
Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Huang H, Weng H, Chen J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–88.
Jiang X, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
He PC, He C. m(6) A RNA methylation: from mechanisms to therapeutic potential. EMBO J. 2021;40: e105977.
Luo J, Liu H, Luan S, He C, Li Z. aberrant regulation of mRNA m(6)A modification in cancer development. Int J Mol Sci. 2018;19:2515.
Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81–94.
Zhou J, et al. The drug-resistance mechanisms of five platinum-based antitumor agents. Front Pharmacol. 2020;11:343.
Li H, et al. METTL3 promotes oxaliplatin resistance of gastric cancer CD133+ stem cells by promoting PARP1 mRNA stability. Cell Mol Life Sci. 2022;79:135.
Chen H, et al. The m6A methyltransferase METTL3 regulates autophagy and sensitivity to cisplatin by targeting ATG5 in seminoma. Transl Androl Urol. 2021;10:1711–22.
Zhang R, et al. TRIM11 facilitates chemoresistance in nasopharyngeal carcinoma by activating the beta-catenin/ABCC9 axis via p62-selective autophagic degradation of Daple. Oncogenesis. 2020;9:45.
Shriwas O, Mohapatra P, Mohanty S, Dash R. The impact of m6A RNA modification in therapy resistance of cancer: implication in chemotherapy, radiotherapy, and immunotherapy. Front Oncol. 2020;10: 612337.
Xu Z, et al. N6-methyladenosine RNA modification in cancer therapeutic resistance: current status and perspectives. Biochem Pharmacol. 2020;182: 114258.
Liu J, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Bartosovic M, et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res. 2017;45:11356–70.
Zuidhof HR, Calkhoven CF. Oncogenic and tumor-suppressive functions of the RNA demethylase FTO. Cancer Res. 2022;82:2201–12.
Tang C, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115:E325–33.
Wei J, et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71:973-985 e975.
Xiao W, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Roundtree IA, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017. https://doi.org/10.7554/eLife.31311.
Shima H, et al. S-Adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–63.
Wojtas MN, et al. Regulation of m(6)A transcripts by the 3’→5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68:374-387.e312.
Mao Y, et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10:5332.
Orouji E, Peitsch WK, Orouji A, Houben R, Utikal J. Oncogenic role of an epigenetic reader of m(6)A RNA modification: YTHDF1 in Merkel cell carcinoma. Cancers (Basel). 2020;12:202.
Shi H, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Huang H, et al. Publisher correction: recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2020;22:1288.
Zhou KI, et al. Regulation of co-transcriptional Pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76:70-81.e79.
Edens BM, et al. FMRP modulates neural differentiation through m(6)A-dependent mRNA nuclear export. Cell Rep. 2019;28:845-854.e845.
Carceles-Cordon M, et al. Cellular rewiring in lethal prostate cancer: the architect of drug resistance. Nat Rev Urol. 2020;17:292–307.
Shi L, et al. Methyltransferase-like 3 upregulation is involved in the chemoresistance of non-small cell lung cancer. Ann Transl Med. 2022;10:139.
Ling Q, Wu S, Liao X, Liu C, Chen Y. Anesthetic propofol enhances cisplatin-sensitivity of non-small cell lung cancer cells through N6-methyladenosine-dependently regulating the miR-486-5p/RAP1-NF-kappaB axis. BMC Cancer. 2022;22:765.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276: 119399.
Shi Y, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10:4892.
Zhu L, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019;10:383.
Yang H, et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J Adv Res. 2022;37:91–106.
Zhu Y, et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin Transl Med. 2022;12: e703.
Sun T, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39:5358–72.
Zhang Y, et al. m(6)A modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18:185.
Lan H, et al. Tumor-associated macrophages promote oxaliplatin resistance via METTL3-mediated m(6)A of TRAF5 and necroptosis in colorectal cancer. Mol Pharm. 2021;18:1026–37.
Chen P, et al. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol Ther Oncolytics. 2021;20:228–39.
Zhao Y, et al. YTHDF3 facilitates eIF2AK2 and eIF3A recruitment on mRNAs to regulate translational processes in oxaliplatin-resistant colorectal cancer. ACS Chem Biol. 2022;17:1778–88.
Nie S, et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J Exp Clin Cancer Res. 2021;40:284.
Hao L, et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. 2021;1868: 118878.
Wei J, et al. METTL3 potentiates resistance to cisplatin through m(6) A modification of TFAP2C in seminoma. J Cell Mol Med. 2020;24:11366–80.
Shriwas O, et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m(6)A-demethylation of FOXM1 and NANOG. Apoptosis. 2020;25:233–46.
Li X, et al. Fat mass and obesity-associated protein regulates tumorigenesis of arecoline-promoted human oral carcinoma. Cancer Med. 2021;10:6402–15.
Wei W, et al. Circ0008399 interaction with WTAP promotes assembly and activity of the m(6)A methyltransferase complex and promotes cisplatin resistance in bladder cancer. Cancer Res. 2021;81:6142–56.
Ma H, et al. m6A methyltransferase Wilms’ tumor 1-associated protein facilitates cell proliferation and cisplatin resistance in NK/T cell lymphoma by regulating dual-specificity phosphatases 6 expression via m6A RNA methylation. IUBMB Life. 2021;73:108–17.
Miranda-Goncalves V, et al. The component of the m(6)A writer complex VIRMA is implicated in aggressive tumor phenotype, DNA damage response and cisplatin resistance in germ cell tumors. J Exp Clin Cancer Res. 2021;40:268.
Shi J, et al. METTL3 promotes the resistance of glioma to temozolomide via increasing MGMT and ANPG in a m(6)A dependent manner. Front Oncol. 2021;11: 702983.
Li XD, et al. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021;112:4543–52.
Ding C, et al. Warburg effect-promoted exosomal circ_0072083 releasing up-regulates NANGO expression through multiple pathways and enhances temozolomide resistance in glioma. J Exp Clin Cancer Res. 2021;40:164.
Ye X, et al. Increased m(6)A modification of lncRNA DBH-AS1 suppresses pancreatic cancer growth and gemcitabine resistance via the miR-3163/USP44 axis. Ann Transl Med. 2022;10:304.
Zhang C, et al. m(6)A methyltransferase METTL14-mediated upregulation of cytidine deaminase promoting gemcitabine resistance in pancreatic cancer. Front Oncol. 2021;11: 696371.
Tang B, et al. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol Cancer. 2020;19:3.
Wang ZW, et al. SRSF3-mediated regulation of N6-methyladenosine modification-related lncRNA ANRIL splicing promotes resistance of pancreatic cancer to gemcitabine. Cell Rep. 2022;39: 110813.
Pan S, et al. N6methyladenosine upregulates miR181d5p in exosomes derived from cancerassociated fibroblasts to inhibit 5FU sensitivity by targeting NCALD in colorectal cancer. Int J Oncol. 2022. https://doi.org/10.3892/ijo.2022.5304.
Ma YN, et al. LncRNA LBX2-AS1 promotes colorectal cancer progression and 5-fluorouracil resistance. Cancer Cell Int. 2021;21:501.
Liu X, et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/beta-catenin pathway. J Exp Clin Cancer Res. 2021;40:132.
Uddin MB, et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134–45.
Li S, Jiang F, Chen F, Deng Y, Pan X. Effect of m6A methyltransferase METTL3 -mediated MALAT1/E2F1/AGR2 axis on adriamycin resistance in breast cancer. J Biochem Mol Toxicol. 2022;36: e22922.
Pan X, Hong X, Li S, Meng P, Xiao F. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp Mol Med. 2021;53:91–102.
Chen Z, et al. N6-methyladenosine-induced ERRγ triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics. 2020;10:3382–96.
Huang T, et al. N(6)-methyladenosine (m(6)A)-mediated lncRNA DLGAP1-AS1enhances breast canceradriamycin resistance through miR-299-3p/WTAP feedback loop. Bioengineered. 2021;12:10935–44.
Ye L, et al. Upregulation of E2F8 promotes cell proliferation and tumorigenicity in breast cancer by modulating G1/S phase transition. Oncotarget. 2016;7:23757–71.
Wang Y, et al. Fat mass and obesity-associated protein (FTO) mediates signal transducer and activator of transcription 3 (STAT3)-drived resistance of breast cancer to doxorubicin. Bioengineered. 2021;12:1874–89.
Yang Z, et al. Binding of RNA m6A by IGF2BP3 triggers chemoresistance of HCT8 cells via upregulation of ABCB1. Am J Cancer Res. 2021;11:1428–45.
Zhou C, et al. N6-Methyladenosine modification of the TRIM7 positively regulates tumorigenesis and chemoresistance in osteosarcoma through ubiquitination of BRMS1. EBioMedicine. 2020;59: 102955.
Lai X, et al. Dysregulation of LINC00470 and METTL3 promotes chemoresistance and suppresses autophagy of chronic myelocytic leukaemia cells. J Cell Mol Med. 2021;25:4248–59.
Wang Z, et al. Induction of m(6)A methylation in adipocyte exosomal LncRNAs mediates myeloma drug resistance. J Exp Clin Cancer Res. 2022;41:4.
Wang A, et al. Tumor-suppressive MEG3 induces microRNA-493-5p expression to reduce arabinocytosine chemoresistance of acute myeloid leukemia cells by downregulating the METTL3/MYC axis. J Transl Med. 2022;20:288.
Pan ZP, et al. METTL3 mediates bone marrow mesenchymal stem cell adipogenesis to promote chemoresistance in acute myeloid leukaemia. FEBS Open Bio. 2021;11:1659–72.
Gong H, Liu L, Cui L, Ma H, Shen L. ALKBH5-mediated m6A-demethylation of USP1 regulated T-cell acute lymphoblastic leukemia cell glucocorticoid resistance by Aurora B. Mol Carcinog. 2021;60:644–57.
Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–27.
Yang L, et al. Comprehensive analysis of the transcriptome-wide m6A methylome in endometrioid ovarian cancer. Front Oncol. 2022;12: 844613.
Yang N, et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene. 2017;36:3428–40.
Zbinden M, et al. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. Embo J. 2010;29:2659–74.
Taketo K, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621–9.
Tong S, et al. Comprehensive pharmacogenomics characterization of temozolomide response in gliomas. Eur J Pharmacol. 2021;912: 174580.
Li F, et al. Interplay of m(6) A and histone modifications contributes to temozolomide resistance in glioblastoma. Clin Transl Med. 2021;11: e553.
Su R, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90-105 e123.
Heinemann V. Gemcitabine: progress in the treatment of pancreatic cancer. Oncology. 2001;60:8–18.
Noordhuis P, et al. 5-Fluorouracil incorporation into RNA and DNA in relation to thymidylate synthase inhibition of human colorectal cancers. Ann Oncol. 2004;15:1025–32.
Budach V. TPF sequential therapy: when and for whom? Oncologist. 2010;15(Suppl 3):13–8.
Vodenkova S, et al. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206: 107447.
Nishizawa Y, et al. Oncogene c-Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2018;9:7476–86.
Jiang Z, et al. Circular RNA protein tyrosine kinase 2 (circPTK2) promotes colorectal cancer proliferation, migration, invasion and chemoresistance. Bioengineered. 2022;13:810–23.
Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–8.
Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol. 2006;41:389–405.
Wang X, et al. Fatty acid receptor GPR120 promotes breast cancer chemoresistance by upregulating ABC transporters expression and fatty acid synthesis. EBioMedicine. 2019;40:251–62.
Sun Y, et al. YTHDF1 promotes breast cancer cell growth, DNA damage repair and chemoresistance. Cell Death Dis. 2022;13:230.
Xu C, Liang T, Liu J, Fu Y. RAB39B as a chemosensitivity-related biomarker for diffuse large B-cell lymphoma. Front Pharmacol. 2022;13: 931501.
Liu J, et al. A novel YTHDF3-based model to predict prognosis and therapeutic response in breast cancer. Front Mol Biosci. 2022;9: 874532.
Wang Q, Zhang Q, Li Q, Zhang J, Zhang J. Clinicopathological and immunological characterization of RNA m(6) A methylation regulators in ovarian cancer. Mol Genet Genomic Med. 2021;9: e1547.
Zhang Z, et al. m(6)A regulators as predictive biomarkers for chemotherapy benefit and potential therapeutic targets for overcoming chemotherapy resistance in small-cell lung cancer. J Hematol Oncol. 2021;14:190.
Aldea M, et al. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021;11:874–99.
Liu Z, et al. m6A modification-mediated DUXAP8 regulation of malignant phenotype and chemotherapy resistance of hepatocellular carcinoma through miR-584-5p/MAPK1/ERK pathway axis. Front Cell Dev Biol. 2021;9: 783385.
Chen YT, Xiang D, Zhao XY, Chu XY. Upregulation of lncRNA NIFK-AS1 in hepatocellular carcinoma by m(6)A methylation promotes disease progression and sorafenib resistance. Hum Cell. 2021;34:1800–11.
Xu J, et al. N(6)-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating beta-catenin signaling. Mol Cancer. 2020;19:163.
Lin Z, et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. Embo j. 2020;39: e103181.
Zhou T, et al. m6A RNA methylation-mediated HNF3gamma reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Signal Transduct Target Ther. 2020;5:296.
Tan C, et al. YY1-targeted RBM15B promotes hepatocellular carcinoma cell proliferation and Sorafenib resistance by promoting TRAM2 expression in an m6A-dependent manner. Front Oncol. 2022;12: 873020.
Zhang H, et al. m6A methyltransferase METTL3-induced lncRNA SNHG17 promotes lung adenocarcinoma gefitinib resistance by epigenetically repressing LATS2 expression. Cell Death Dis. 2022;13:657.
Liu S, et al. The mechanism of m(6)A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by beta-elemene. Cell Death Dis. 2020;11:969.
Tang J, Han T, Tong W, Zhao J, Wang W. N(6)-methyladenosine (m(6)A) methyltransferase KIAA1429 accelerates the gefitinib resistance of non-small-cell lung cancer. Cell Death Discov. 2021;7:108.
Xiao P, et al. Exosomal delivery of FTO confers gefitinib resistance to recipient cells through ABCC10 regulation in an m6A-dependent manner. Mol Cancer Res. 2021;19:726–38.
Chen H, Jia B, Zhang Q, Zhang Y. Meclofenamic acid restores gefinitib sensitivity by downregulating breast cancer resistance protein and multidrug resistance protein 7 via FTO/m6A-demethylation/c-Myc in non-small cell lung cancer. Front Oncol. 2022;12: 870636.
Li K, et al. M6A associated TSUC7 inhibition contributed to Erlotinib resistance in lung adenocarcinoma through a notch signaling activation dependent way. J Exp Clin Cancer Res. 2021;40:325.
Li K, et al. Stimulation of Let-7 Maturation by metformin improved the response to tyrosine kinase inhibitor therapy in an m6a dependent manner. Front Oncol. 2021;11: 731561.
Liu H, et al. Interaction of lncRNA MIR100HG with hnRNPA2B1 facilitates m(6)A-dependent stabilization of TCF7L2 mRNA and colorectal cancer progression. Mol Cancer. 2022;21:74.
Abner JJ, et al. Depletion of METTL3 alters cellular and extracellular levels of miRNAs containing m(6)A consensus sequences. Heliyon. 2021;7: e08519.
Ke W, Zhang L, Zhao X, Lu Z. p53 m(6)A modulation sensitizes hepatocellular carcinoma to apatinib through apoptosis. Apoptosis. 2022;27:426–40.
Chen Y, et al. N(6)-methyladenosine-modified TRAF1 promotes sunitinib resistance by regulating apoptosis and angiogenesis in a METTL14-dependent manner in renal cell carcinoma. Mol Cancer. 2022;21:111.
Bhattarai PY, Kim G, Poudel M, Lim SC, Choi HS. METTL3 induces PLX4032 resistance in melanoma by promoting m(6)A-dependent EGFR translation. Cancer Lett. 2021;522:44–56.
Ding N, You A, Tian W, Gu L, Deng D. Chidamide increases the sensitivity of non-small cell lung cancer to Crizotinib by decreasing c-MET mRNA methylation. Int J Biol Sci. 2020;16:2595–611.
Fukumoto T, et al. N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79:2812–20.
Wilhelm SM, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109.
Palmer DH. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:2498 (author reply 2498-2499).
Sim EH, Yang IA, Wood-Baker R, Bowman RV, Fong KM. Gefitinib for advanced non-small cell lung cancer. Cochrane Database Syst Rev. 2018;1:CD006847.
Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17:38.
Goldberg RM. Cetuximab. Nat Rev Drug Discov. 2005;4(5):S10-11.
Scott LJ. Apatinib: a review in advanced gastric cancer and other advanced cancers. Drugs. 2018;78:747–58.
Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: a trailblazing and transformative journey. Clin Cancer Res. 2018;24:4062–5.
Domina EA, Philchenkov A, Dubrovska A. Individual response to ionizing radiation and personalized radiotherapy. Crit Rev Oncog. 2018;23:69–92.
Xiang Y, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–6.
Visvanathan A, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Zhou S, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Kowalski-Chauvel A, et al. The m6A RNA demethylase ALKBH5 promotes radioresistance and invasion capability of glioma stem cells. Cancers (Basel). 2020;13:40.
Gouaze-Andersson V, et al. FGFR1/FOXM1 pathway: a key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget. 2018;9:31637–49.
Wang L, et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39: e104514.
Zheng H, et al. Decreased expression of programmed death ligand-L1 by seven in absentia homolog 2 in cholangiocarcinoma enhances T-cell-mediated antitumor activity. Front Immunol. 2022;13: 845193.
Han D, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4.
Yang S, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Tsuruta N, et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;530:235–9.
Li N, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159–70.
Zhang Z, et al. The importance of N6-methyladenosine modification in tumor immunity and immunotherapy. Exp Hematol Oncol. 2022;11:30.
Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864–72.
Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560–75.
Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Mimura K, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53.
Orskov AD, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. 2015;6:9612–26.
Dombret H, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291–9.
Yun S, et al. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenetics. 2016;8:68.
Su R, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79-96 e11.
Liu Y, et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021;33:1221-1233 e1211.
Pan Y, Xiao K, Li Y, Li Y, Liu Q. RNA N6-methyladenosine regulator-mediated methylation modifications pattern and immune infiltration features in glioblastoma. Front Oncol. 2021;11: 632934.
Zhu J, et al. Integrative analysis of m6A RNA methylation regulators and the tumor immune microenvironment in non-small-cell lung cancer. Dis Markers. 2022;2022:2989200.
Zhang H, et al. N6-methyladenosine-related lncRNAs as potential biomarkers for predicting prognoses and immune responses in patients with cervical cancer. BMC Genom Data. 2022;23:8.
Yuan C, et al. Crosstalk of histone and RNA modifications identified a stromal-activated subtype with poor survival and resistance to immunotherapy in gastric cancer. Front Pharmacol. 2022;13: 868830.
Kong J, et al. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat Commun. 2020;11:5485.
Lan Q, et al. The Emerging roles of RNA m(6)A methylation and demethylation as critical regulators of tumorigenesis, drug sensitivity, and resistance. Cancer Res. 2021;81:3431–40.
Selberg S, et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. 2019;26:3762-3771.e3765.
Chen B, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
Huang Y, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
He W, et al. Identification of a novel small-molecule binding site of the fat mass and obesity associated protein (FTO). J Med Chem. 2015;58:7341–8.
Qiao Y, et al. A novel inhibitor of the obesity-related protein FTO. Biochemistry. 2016;55:1516–22.
Huang Y, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677–91.
Liu Z, et al. Structure-activity relationships and antileukemia effects of the tricyclic benzoic acid FTO inhibitors. J Med Chem. 2022;65:10638–54.
Xie G, et al. A novel inhibitor of N (6)-methyladenosine demethylase FTO induces mRNA methylation and shows anti-cancer activities. Acta Pharm Sin B. 2022;12:853–66.
Huff S, et al. Rational design and optimization of m(6)A-RNA demethylase FTO inhibitors as anticancer agents. J Med Chem. 2022;65:10920–37.
Qin B, et al. Discovery of novel mRNA demethylase FTO inhibitors against esophageal cancer. J Enzyme Inhib Med Chem. 2022;37:1995–2003.
Prakash M, et al. Identification of potent and selective inhibitors of fat mass obesity-associated protein using a fragment-merging approach. J Med Chem. 2021;64:15810–24.
Cao K, et al. Glutathione-bioimprinted nanoparticles targeting of N6-methyladenosine FTO demethylase as a strategy against leukemic stem cells. Small. 2022;18: e2106558.
Bedi RK, et al. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020;15:744–8.
Moroz-Omori EV, et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem. 2021;16:3035–43.
Dolbois A, et al. 1,4,9-Triazaspiro[5.5]undecan-2-one derivatives as potent and selective METTL3 inhibitors. J Med Chem. 2021;64:12738–60.
Li J, Gregory RI. Mining for METTL3 inhibitors to suppress cancer. Nat Struct Mol Biol. 2021;28:460–2.
Selberg S, Seli N, Kankuri E, Karelson M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega. 2021;6:13310–20.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A Novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–27.
Ma S, et al. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J Exp Med. 2021. https://doi.org/10.1084/jem.20210279.
Song H, et al. METTL3-mediated m(6)A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat Commun. 2021;12:5522.
Li HB, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–42.
Tong J, et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28:253–6.
Yao Y, et al. METTL3-dependent m(6)A modification programs T follicular helper cell differentiation. Nat Commun. 2021;12:1333.
Yin H, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394.
Tong J, et al. Pooled CRISPR screening identifies m(6)A as a positive regulator of macrophage activation. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abd4742.
Dong L, et al. The loss of RNA N(6)-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8(+) T cell dysfunction and tumor growth. Cancer Cell. 2021;39:945-957 e910.
Luo Y, Sun Y, Li L, Mao Y. METTL3 may regulate testicular germ cell tumors through EMT and immune pathways. Cell Transplant. 2020;29:963689720946653.
Li X, Ma S, Deng Y, Yi P, Yu J. Targeting the RNA m(6)A modification for cancer immunotherapy. Mol Cancer. 2022;21:76.
Qiu X, et al. M(6)A demethylase ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res. 2021;81:4778–93.
Wang Y, Sun S, Zhang Z, Shi D. Nanomaterials for cancer precision medicine. Adv Mater. 2018;30: e1705660.
Acknowledgements
Not applicable.
Funding
This study was supported by the Scientific Research Project of Anhui Provincial Education Department (2022AH020079), the Youth Innovation Project of University of Science and Technology of China (WK9110000203), the Natural Science Foundation of Anhui Province (2008085MH241).
Author information
Authors and Affiliations
Contributions
W-WL and Z-YZ collected the related literature. W-WL wrote the manuscript. HW and FW participated in the design of the review and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, WW., Zhang, ZY., Wang, F. et al. Emerging roles of m6A RNA modification in cancer therapeutic resistance. Exp Hematol Oncol 12, 21 (2023). https://doi.org/10.1186/s40164-023-00386-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00386-2