
Abstract
Background
Economically important milk production traits including milk volume, milk fat and protein yield vary considerably across dairy goats in New Zealand. A significant portion of the variation is attributable to genetic variation. Discovery of genetic markers linked to milk production traits can be utilised to drive selection of high-performance animals.
A previously reported genome wide association study across dairy goats in New Zealand identified a quantitative trait locus (QTL) located on chromosome 19. The most significantly associated single nucleotide polymorphism (SNP) marker for this locus is located at position 26,610,610 (SNP marker rs268292132). This locus is associated with multiple milk production traits including fat, protein and volume. The predicted effect of selection for the beneficial haplotype would result in an average production increase of 2.2 kg fat, 1.9 kg protein and 73.6 kg milk yield.
An outstanding question was whether selection for the beneficial allele would co-select for any negative pleiotropic effects. An adverse relationship between milk production and udder health traits has been reported at this locus. Therefore, a genome wide association study was undertaken looking for loci associated with udder traits.
Results
The QTL and production associated marker rs268292132 was identified in this study to also be associated with several goat udder traits including udder depth (UD), fore udder attachment (FUA) and rear udder attachment (RUA). Our study replicates the negative relationship between production and udder traits with the high production allele at position 19:26,610,610 (SNP marker rs268292132) associated with an adverse change in UD, FUA and RUA.
Conclusions
Our study has confirmed the negative relationship between udder traits and production traits in the NZ goat population. We have found that the frequency of the high production allele is relatively high in the NZ goat population, indicating that its effect on udder conformation is not significantly detrimental on animal health. It will however be important to monitor udder conformation as the chromosome 19 locus is progressively implemented for marker assisted selection. It will also be of interest to determine if the gene underlying the production QTL has a direct effect on mammary gland morphology or whether the changes observed are a consequence of the increased milk volume.
Similar content being viewed by others


Background
Genetic gain focused on production for the dairy goat population in New Zealand is limited largely to within farm selection and some buck transfers between farms. This has resulted in considerable unrealised marker driven selection potential which led to the search for and discovery of quantitative trait loci (QTL) for milk production traits in a subset of the population. In particular, the most significant QTL identified was located on chromosome 19 associated with all three production traits including fat yield, protein yield and milk volume [1]. A QTL at the same location was also reported in the French [2, 3] and British dairy goat populations [4]. A pleiotropic effect for this QTL has been described on a number of udder conformation traits including udder attachment and udder depth [3, 4]. It appears that udders in animals with higher milk production determined by the chromosome 19 locus are characterised by greater udder depth with apparent weaker and narrower udder attachment. It is important to evaluate potential negative pleiotropic effects before large scale genetic selection. There are examples where the potential introduction of alleles may have or have resulted in the introduction of negative characteristics. For example, in cattle the non-synonymous A > C mutation in a splice enhancer region of the bovine Diacylglycerol O-Acyltransferase 1 (DGAT1) gene dramatically reduced milk fat content but caused scouring (non-bloody watery diarrhoea), impaired growth rate and flattened intestinal microvilli [5]. Furthermore, the non-synonymous C636R mutation in bovine Mannose Receptor C Type 2 (MRC2) gene increased muscularity in meat breeds but caused Crooked Tail Syndrome [6, 7].
Goat udder traits and teat morphology play important roles in milk quality (somatic cell count), milking ability/ease of milking [8] and overall milk production [9]. Goat udder circumference [10, 11] and udder depth [12] have been positively correlated with milk volume. Goats with greater udder depth also had a higher somatic cell count, a negative indicator of milk quality, udder health and marker of mastitis [13]. Teat size and shape was likewise indicative of mammary gland health with shorter and narrower teats associated with a lower somatic cell count [13, 14]. These factors influence the animal’s productive lifespan or longevity [15]. Extensive selection pressure for milk production traits has been shown to exert a negative effect on udder and teat traits [3, 4, 16]. It is thereby important to consider these traits in animal selection. Udder health and conformation traits have not been assessed at scale in the New Zealand goat herd. This paper reports for the first time a genome wide investigation for goat udder traits of the New Zealand goat population using Illumina GoatSNP50 BeadChip (Illumina Inc., San Diego, CA, USA) and identifies caprine SNP marker rs268292132 as a viable selection tool for future marker assisted breeding programmes.
Methods
Animals and phenotypic measurements
A total of 1058 animals from 2 farms (439 animals from farm A, 619 animals from farm B) with mixed age (mean age: 4.72 years, range: 2–9 years) and of primiparous or multiparous status (mean parity: 3.72 years, range: 1–8) were scored for udder traits in February and March 2020. The New Zealand goat herd is a mixed breed population comprised of approximately 85% Saanen, 5% Toggenburg, 5% Nubian/Anglo-Nubian, 4.5% Alpine and 0.5% Sable [17]. The udder traits measured included udder depth (UD), fore udder attachment (FUA), rear udder attachment (RUA), udder furrow (UF), teat placement (TP) and teat angle (TA). Udder traits were scored on a point system from 1 to 9 described in Table 1 with definition of traits as used in McLaren et al. [18]. Additionally, the following milk production traits, milk volume, fat yield and protein yield were measured for a subset of 336 animals out of the 1058-animal cohort (141 animals from farm A, 195 animals from farm B) as part of standard herd testing procedures using Fourier transform infrared spectroscopy. Animals were of mixed age (mean age: 6.49, range: 6–9) and primiparous or multiparous status (mean parity: 5.49, range: 5–8). The animals were from the cohort described in Scholtens et al [1]. Milk production traits were modelled by extrapolation to a 305-day lactation length period, restricted to animals with lactation length > 200 days.
Genotypes
Skin samples from the 1058 phenotyped animals were collected and genotyped with the Illumina GoatSNP50 BeadChip with 53,347 SNP markers (Illumina Inc., San Diego, CA, USA) [19]. Markers with ambiguous or missing genotypes in more than 5% of goats, or with minor allele frequency < 0.01 were omitted. After filtering, 51,477 markers were retained.
Genome-wide association analysis
Genome wide association mapping with an applied mixed linear model accounting for differences in population structure was performed using genome-wide complex trait analysis tool (GCTA) v1.93.2 [20, 21]. The following mixed linear model was used in our study:
y is the phenotype of interest, a is the mean term, b is the fixed (additive) effect of the SNP, x is the SNP genotype, g− is the effect of population structure estimated by calculating genetic relationship matrices (GRM) between all animals [21]. Mixed linear model association analysis, leave one chromosome out (MLMA-LOCO) approach was used where the chromosome on which the significant candidate SNP is located are excluded from the GRM calculations to avoid double fitting of candidate variants. e is the residual effect.
A SNP was considered significantly associated to a trait of interest at the whole genome level if their -log10 P-values exceeded -log10(5 × 10− 8). SNP marker trait association plots against the ARS1 goat reference [22] were constructed using the ggplot R package [23].
Calculation of allelic effect sizes for marker rs268292132
Allelic effect sizes for marker rs268292132 were calculated for production and udder traits in the 1058 animal population using a linear regression model adjusted for age and farm origin. A one-way analysis of variance (ANOVA) test between the means (α = 0.05) was performed to characterise allelic effect differences between rs268292132 genotype groups presented in Fig. 2. The Shapiro-Wilk normality assumption test was performed on all data presented. Multiple pairwise comparisons adjusted for false discovery rate were utilised to highlight statistically significant differences in the data presented.
Whole genome sequencing for variant identification
Whole genome sequencing was performed on 302 goats consisting of 48 animals in the cohort used for GWAS, 45 additional animals originating from farms A and B and 209 animals distributed across four other farms. The animals were selected to capture the genetic diversity of each farm. Skin samples were collected from these animals and sequenced on the Illumina HiSeq XTen platform (Illumina Inc., San Diego, CA, USA). For each animal, 700–1000 million paired end reads with read pair lengths of 150 nucleotides were generated. An average sequencing depth of 35 × was achieved. Read sequence quality was assessed using FastQC (FastQC v0.11.7). Reads were mapped to the ARS1 goat reference genome assembly using Burrows Wheeler aligner [24]. Duplicate read pairs were marked using picard MarkDuplicates (Picard v2.1.0., https://broadinstitute.github.io/picard/). Reads aligned around small insertions/deletions were realigned using the Genome Analysis Toolkit (GATK v 3.8, RealignerTargetCreator and IndelRealigner tools) [25, 26]. Probabilities for single nucleotide and indel variants were discovered individually in each alignment using GATK’s HaplotypeCaller tool [27] and genotyped jointly across the entire cohort with GATK’s GenotypeGVCFs tool. Pairwise linkage disequilibrium R2 values between variants were calculated using PLINK software [28]. The Ensembl Variant Effect Predictor (VEP v90.3) software (https://www.ensembl.org/vep) was used to annotate variants with their calculated variant consequences based on the ARS1 reference General Feature Format (GFF) file (http://ftp.ensembl.org/pub/release-100/gff3/capra_hircus/Capra_hircus.ARS1.100.gff3.gz).
Results
A chromosome 19 QTL associated with goat udder health traits in the NZ goat population
We report a single genome wide significant QTL for several udder health traits including udder depth, fore udder attachment and rear udder attachment (Table 2 & Fig. 1). The QTL extend from chromosome 19 positions 24 - 29 Mb. A QTL signal was also detected for teat angle and udder furrow at the same locus but did not reach genome wide significance. No QTL association was observed for the teat placement phenotype. Fitting the most significant QTL SNP (rs268292132) as a covariate in the association analysis resulted in the ablation of the entire QTL signal, indicative of a single haplotype responsible for the signal.

Manhattan plots of SNP marker association across chromosomes 1 to 29 plus X for Fore udder attachment (A), Rear udder attachment (B), Teat angle (C), Teat placement (D), Udder depth (E) and Udder furrow (F) for 1058 animals. Population structure was accounted for by estimating genetic relationship matrices (GRM) between all animals. Genome wide significance threshold is indicated by a dashed line at P-value of 5.0 × 10−8. SNP markers of genome wide significance are coloured in orange
An overlapping locus was previously identified to be associated with milk production traits in the French [3], British [4] and New Zealand goat populations [1]. The SNP most significantly associated with milk production in the New Zealand study (rs268292132) was also the most significantly associated SNP with the above udder traits in this study (Table 2 & Fig. 1).
Whole genome sequence analysis revealed two candidate non-synonymous mutations in the proteasome 20S subunit Beta 6 (PSMB6) and the sex hormone binding globulin (SHBG) genes
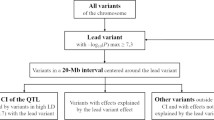
Whole genome sequence analysis of 302 animals which included 48 animals in the GWAS cohort highlighted 340/40,966 variants within the 24 - 29 Mb chromosome 19 QTL interval with high linkage disequilibrium, R2 > 0.8 with the most significant SNP marker, rs268292132. Of these, no high impact variants with severe consequence on protein sequence including transcript ablation, splice acceptor, splice donor, stop gain, frameshift, stop lost and transcript amplification variants were identified (Full breakdown of the 340 variants listed in Additional file 1 & Additional file 3). Two non-synonmous mutations and one inframe deletion was predicted to alter protein sequence. The in-frame deletion in candidate gene, SENP3 at position 27,036,323 (NC_030826.1:g. 27,036,324_27,036,326del), detected at an alternative allele frequency of 0.553 and linkage disequilibrium R2 value of 0.877 with the most significant SNP marker deletes a conserved glutamate (E89del). The variant has not been reported in other goat populations (Additional file 1 and Table 3). A non-synonymous variant in the candidate gene PSMB6 at position 26,370,181 (NC_030826.1:g. 26,370,181C > T, rs647861802), observed at an alternative allele frequency of 0.566 and linkage disequilibrium R2 value of 0.887 with the most significant SNP marker encodes a substitution of conserved valine 221 to isoleucine (ENSCHIP00000027128.1:p.V222I, GERP conservation score = 0.9). The variant has been reported in the NextGen collaborative research project at an allele frequency of 0.038 (Additional file 1 and Table 3). A G > T transversion at position 27,087,979 in the SHBG gene (NC_030826.1:g. 27,087,979G > T, rs672568929) exists at an alternative allele frequency of 0.555 and linkage disequilibrium R2 value of 0.87 with the most significant SNP marker in the genome-sequenced animals. This variant encodes a substitution of serine 267/277/281/166 (alternative transcripts) to isoleucine (ENSCHIP00000021646.1:p.S267I/ ENSCHIP00000021662.1:p.S277I/ENSCHIP00000021669.1:p.S281I/ENSCHIP00000021683.1:p.S166I/ENSCHIP00000021685.1:p.S166I). This variant has been reported in the NextGen collaborative research project at an allele frequency of 0.028 (Additional file 1 and Table 3).
An adverse relationship between milk production traits and udder conformation
Allelic effect sizes for marker rs268292132 adjusted for age and farm origin were calculated in our GWAS population. An average difference of 3.66 kg fat (P-value = 3.09 × 10− 4), 4.12 kg protein (P-value = 2.58 × 10− 8) and 159.52 L volume (P-value = 7.22 × 10− 10) was recorded when comparing opposing homozygotes (Fig. 2 & Table 4). The production gains were accompanied by a decrease of 1.14 score points in udder depth (P-value = 9.31 × 10− 18), 0.99 score points in fore udder attachment (P-value = 5.05 × 10− 7) and 1.19 score points in rear udder attachment (P-value = 1.08 × 10− 12) when selecting for the high production T allele (Fig. 2 & Table 4).

Allelic effect sizes for marker rs268292132 genotype groups. Production traits, Milk Fat (A), Milk Protein (B), Milk volume (C) and udder traits, Fore udder attachment (D), Rear Udder Attachment (E), Teat Angle (F), Teat Placement (G), Udder Depth (H), Udder Furrow (I) were plotted as least squared means with 95% confidence intervals. Asterisks indicate pairwise significant differences between genotype groups (P-value < 0.5, multiple pairwise comparisons adjusted for false discovery rate; α = 0.05)
Discussion
A genome wide association study with an applied mixed model to account for differences in population structure was performed in 1058 dairy goats in New Zealand. The study highlighted a QTL located on chromosome 19 (24–29 Mb) associated with udder depth, fore udder attachment and rear udder attachment. This QTL was also identified in a previous study of the dairy goat population in New Zealand to be associated with milk production traits; fat yield, protein yield and milk volume (as described in Scholtens et al. [1]). Our findings are in agreement with trait effects of an overlapping QTL previously reported in French dairy goats for milk fat content, milk protein content, udder depth and rear udder attachment [2, 3] and British dairy goats for milk volume, udder depth and udder attachment [4]. The pleiotropic effect of this locus on both production and udder phenotypes combined with a high density of genes within the region (22.5 genes/Mb) has made it difficult to nominate candidate genes for the observed traits. Martin and colleagues suggested several fatty acid and lipid metabolism pathway genes including phospholipase D2 (PLD2), gamma-glutamyltransferase 6 (GGT6) and the arachiodonate lipoxygenase (ALOX) family of genes [3]. Mucha and colleagues also noted a potential role for protein stability and lipid homeostasis gene, asialoglycoprotein receptor 2 (ASGR2) in udder formation [4]. Our analysis from 302 whole genome sequenced animals showed no protein-altering variants for PLD2, GGT6, ASGR1/ASGR2 and ALOX genes.
Whole genome sequence analysis revealed 340 variants within the 5 Mb QTL with linkage disequilibrium R2 > 0.8 with the most significant SNP marker rs268292132. After removing markers not predicted to change protein sequence, two non-synonmous variants and one inframe deletion variant were of interest. We propose two candidate non-synonymous variants including a valine to isoleucine variant (V222I) in the proteasome 20S subunit 6 (PSM6B) gene, and a serine to isoleucine variant (S267I/S277I/S281I/S166I) in the sex hormone binding globulin (SHBG) gene. SIFT scores were predicted at 0.3 and 0.01 respectively suggesting a likely deleterious effect for the SHBG mutation. Genetic variation in human PSM6B was associated with LDL cholesterol and total cholesterol levels in a meta-analysis of 617,303 individuals of multi-ethnic backgrounds with 32 million genotyped and imputed variants [29]. PSMB6 encodes a core component of the 20S proteasome complex involved in intracellular protein degradation. However, its role in lipid metabolism is unclear. In the same GWAS study, genetic variants in SHBG were associated with triglyceride levels [29]. SHBG regulates the plasma metabolic clearance rate of steroid hormones such as oestrogen [30,31,32], although like PSMB6 a strong functional link with lipid metabolism is not yet determined. Additionally, a GAG glutamate inframe deletion (E89del) was detected in the candidate gene Sentrin-specific protease 3 (SENP3). Limited knowledge exists in the literature on the mechanistic role of SENP3 in milk production or morphological udder development. Given its widespread roles in protein stability, chromatin organisation, transcription, DNA repair, autophagy, protein localisation and homeostasis, a number of potential mechanisms maybe of interest [33]. Attributing causality to either of these mutations will require further genotyping of these variants within a study population that contain sufficient meioses to separate out causative haplotypes and appropriate functionality testing. Despite directing the majority of our focus to variants predicted to impact protein sequence as a direct link to functionality, we acknowledge that non-coding and regulatory variants are also of interest. Indeed our whole genome analysis have revealed two splice region variants, one 5′ UTR variant and five 3′ UTR variants within the QTL that exists in high linkage disequilibrium with the most significant QTL SNP marker. Future studies involving these variants in eQTL analyses or chromatin accessibility analyses such as ChIP-seq or ATAC-seq or experimental CRISPR-cas9 variant models could provide additional insight into biological causality. Moreover, future investigation and characterisation of large copy number variants in our dataset could additionally reveal candidate variants not detectable by SNP chip methods.
Selection on the chromosome 19 QTL for milk production traits will hold economic value in dairy industries. Markers capturing the QTL effect may be utilised in marker-assisted breeding programmes to drive selection of the next generation of high-performance animals. Employing selection with the most significant marker, rs268292132 (chr19:26,610,610) was predicted to increase fat yield, protein yield and milk volume in our study population. However, a potential adverse relationship between production traits and udder conformation has been indicated for this locus [3, 4]. Our study replicated the observations of a shift in the negative direction for udder depth, fore udder attachment and rear udder attachment scores. It is unclear whether the negative correlation between production and udder traits are a consequence of increased milk production and udder volume retention giving rise to a perceived increase in udder depth with weak and narrow udder attachment, or a true biological deterioration of udder morphology independent of the production increase. In the future, it will be interesting to determine whether the gene underlying the QTL has a direct effect on mammary gland morphology. Both alleles at the rs268292132 locus are common in the dairy goat population, with the high-milk production allele approaching fixation on some farms after long-term phenotypic selection for both milk production and conformation traits. These observations imply that the magnitude of the udder effects may be well-balanced with the increase in milk production. A comprehensive analysis of udder shape deterioration versus the production increase across the wider population will be required to optimise selection and breeding programmes for marker rs268292132 as these traits affect production [9], milk quality (somatic cell count) [13], milking ability [8] and total number of days in production [15].
The overarching goal of this research is development of a marker-assisted breeding programme that can be implemented to accelerate productivity gain. This study identified use of the rs268292132 SNP as a viable selection marker for marker assisted breeding programmes to increase production.
Conclusion
Genome wide association analyses across the dairy goat population in New Zealand have discovered a 24-29 Mb QTL on caprine chromosome 19 that has a pleiotropic effect on milk production and udder conformation traits. A negative correlation between production and udder conformation traits was observed in the New Zealand goat population which corroborates reports in French and British dairy goat herds. Close monitoring of udder health is recommended for future selection programmes involving this locus. Marker assisted breeding programmes with markers effectively identifying this QTL can be utilised to select the next generation of high-performance animals to accelerate productivity gain.
Availability of data and materials
Supporting data is available through contact with the authors.
Abbreviations
- QTL:
-
Quantitative trait loci
- eQTL:
-
Expression quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome wide association study
- UD:
-
Udder depth
- FUA:
-
Fore udder attachment
- RUA:
-
Rear udder attachment
- UF:
-
Udder furrow
- TP:
-
Teat placement
- TA:
-
Teat angle
- DGAT1 :
-
Diacylglycerol o-acyltransferase 1
- MRC2 :
-
Mannose receptor C type 2
- GCTA:
-
Genome-wide complex trait analysis tool
- GRM:
-
Genetic relationship matrices
- MLMA-LOCO:
-
Mixed linear model association analysis, leave one chromosome out
- ANOVA:
-
Analysis of variance
- PSMB6 :
-
Proteasome 20S subunit beta 6
- SHBG :
-
Sex hormone binding globulin
- PLD2 :
-
Phospholipase D2
- GGT6 :
-
Gamma-glutamyltransferase 6
- ALOX :
-
Arachiodonate lipoxygenase
- ASGR2 :
-
Asialoglycoprotein receptor 2
- SENP3 :
-
Sentrin-specific protease 3
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- CRISPR-Cas9:
-
Clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9
- ATAC-seq:
-
Assay for transposase-accessible chromatin using sequencing.
References
Scholtens M, Jiang A, Smith A, Littlejohn M, Lehnert K, Snell R, et al. Genome-wide association studies of lactation yields of milk, fat, protein and somatic cell score in New Zealand dairy goats. J Anim Sci Biotechnol. 2020;11:55. https://doi.org/10.1186/s40104-020-00453-2.
Martin P, Palhiere I, Maroteau C, Bardou P, Canale-Tabet K, Sarry J, et al. A genome scan for milk production traits in dairy goats reveals two new mutations in DGAT1 reducing milk fat content. Sci Rep. 2017;7(1):1872. https://doi.org/10.1038/s41598-017-02052-0.
Martin P, Palhiere I, Maroteau C, Clement V, David I, Klopp GT, et al. Genome-wide association mapping for type and mammary health traits in french dairy goats identifies a pleiotropic region on chromosome 19 in the Saanen breed. J Dairy Sci. 2018;101(6):5214–26. https://doi.org/10.3168/jds.2017-13625.
Mucha S, Mrode R, Coffey M, Kizilaslan M, Desire S, Conington J. Genome-wide association study of conformation and milk yield in mixed-breed dairy goats. J Dairy Sci. 2018;101(3):2213–25. https://doi.org/10.3168/jds.2017-12919.
Lehnert K, Ward H, Berry SD, Ankersmit-Udy A, Burrett A, Beattie EM, et al. Phenotypic population screen identifies a new mutation in bovine DGAT1 responsible for unsaturated milk fat. Sci Rep. 2015;5(1):8484. https://doi.org/10.1038/srep08484.
Fasquelle C, Sartelet A, Li W, Dive M, Tamma N, Michaux C, et al. Balancing selection of a frame-shift mutation in the MRC2 gene accounts for the outbreak of the crooked tail syndrome in belgian blue cattle. PLoS Genet. 2009;5(9):e1000666. https://doi.org/10.1371/journal.pgen.1000666.
Sartelet A, Klingbeil P, Franklin CK, Fasquelle C, Geron S, Isacke CM, et al. Allelic heterogeneity of crooked tail syndrome: result of balancing selection? Anim Genet. 2012;43(5):604–7. https://doi.org/10.1111/j.1365-2052.2011.02311.x.
Marnet PG, McKusick BC. Regulation of milk ejection and milkability in small ruminants. Livest Prod Sci. 2001;70(1):125–33. https://doi.org/10.1016/S0301-6226(01)00205-6.
Gall C. Relationship between body conformation and production in dairy goats1. J Dairy Sci. 1980;63(10):1768–81. https://doi.org/10.3168/jds.S0022-0302(80)83136-5.
Montaldo H, Martínez-Lozano FJ. Phenotypic relationships between udder and milking characteristics, milk production and California mastitis test in goats. Small Rumin Res. 1993;12(3):329–37. https://doi.org/10.1016/0921-4488(93)90068-S.
Keskin S, Kor A, Karaca S, Lu H. A study of relationships between milk yield and some udder traits by using of path analysis in akkeci goats. J Anim Vet Adv. 2005;4(5):547–50.
Capote J, Argüello A, Castro N, López JL, Caja G. Short communication: correlations between udder morphology, milk yield, and milking ability with different milking frequencies in dairy goats. J Dairy Sci. 2006;89(6):2076–9. https://doi.org/10.3168/jds.S0022-0302(06)72276-7.
Rupp R, Clément V, Piacere A, Robert-Granié C, Manfredi E. Genetic parameters for milk somatic cell score and relationship with production and udder type traits in dairy alpine and saanen primiparous goats. J Dairy Sci. 2011;94(7):3629–34. https://doi.org/10.3168/jds.2010-3694.
Schulz J, Fahr RD, Finn G, Naumann I. Physical examination and palpation findings of mammary glands and indications of udder health in goat milk. Tierarztl Prax Ausg G Grosstiere Nutztiere. 1999;27(2):92–8.
Castaneda-Bustos VJ, Montaldo HH, Valencia-Posadas M, Shepard L, Perez-Elizalde S, Hernandez-Mendo O, et al. Linear and nonlinear genetic relationships between type traits and productive life in US dairy goats. J Dairy Sci. 2017;100(2):1232–45. https://doi.org/10.3168/jds.2016-11313.
Manfredi E, Piacere A, Lahaye P, Ducrocq V. Genetic parameters of type appraisal in saanen and alpine goats. Livest Prod Sci. 2001;70(3):183–9. https://doi.org/10.1016/S0301-6226(01)00180-4.
Orr M. Farming dairy goats: Introduction. 2010 [Available from: https://www.lifestyleblock.co.nz/lifestyle-file/livestock-a-pets/goats/dairygoats/item/846-farming-dairy-goats-introduction].
McLaren A, Mucha S, Mrode R, Coffey M, Conington J. Genetic parameters of linear conformation type traits and their relationship with milk yield throughout lactation in mixed-breed dairy goats. J Dairy Sci. 2016;99(7):5516–25. https://doi.org/10.3168/jds.2015-10269.
Tosser-Klopp G, Bardou P, Bouchez O, Cabau C, Crooijmans R, Dong Y, et al. International goat genome C. design and characterization of a 52K SNP chip for goats. PloS one. 2014;9(1):e86227.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. https://doi.org/10.1016/j.ajhg.2010.11.011.
Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46(2):100–6. https://doi.org/10.1038/ng.2876.
Bickhart DM, Rosen BD, Koren S, Sayre BL, Hastie AR, Chan S, et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet. 2017;49(4):643–50. https://doi.org/10.1038/ng.3802.
Wickham H. Ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016. https://doi.org/10.1007/978-3-319-24277-4.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From fastq data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1110):11.0.1–0.33.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. https://doi.org/10.1038/ng.806.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the million veteran program. Nat Genet. 2018;50(11):1514–23.
Cousin P, Calemard-Michel L, Lejeune H, Raverot G, Yessaad N, Emptoz-Bonneton A, et al. Influence of SHBG gene pentanucleotide TAAAA repeat and D327N polymorphism on serum sex hormone-binding globulin concentration in hirsute women. J Clin Endocrinol Metab. 2004;89(2):917–24. https://doi.org/10.1210/jc.2002-021553.
Eriksson AL, Lorentzon M, Mellstrom D, Vandenput L, Swanson C, Andersson N, et al. SHBG gene promoter polymorphisms in men are associated with serum sex hormone-binding globulin, androgen and androgen metabolite levels, and hip bone mineral density. J Clin Endocrinol Metab. 2006;91(12):5029–37. https://doi.org/10.1210/jc.2006-0679.
Selva DM, Hogeveen KN, Innis SM, Hammond GL. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. J Clin Invest. 2007;117(12):3979–87. https://doi.org/10.1172/JCI32249.
Long X, Zhao B, Lu W, Chen X, Yang X, Huang J, et al. The critical roles of the sumo-specific protease SENP3 in human diseases and clinical implications. Front Physiol. 2020;11:558220. https://doi.org/10.3389/fphys.2020.558220.
Crooks GE, Hon G, Chandonia JM, Brenner SE. Weblogo: A sequence logo generator. Genome Res. 2004;14(6):1188–90. https://doi.org/10.1101/gr.849004.
Acknowledgements
The authors would like to gratefully acknowledge the support of farmers who provided access to animals and milking records, and the provision of computing resources by the New Zealand eScience Infrastructure.
Funding
This project was co-funded by the Dairy Goat Co-operative, Ministry of Business, Innovation & Employment (3709153), and the Ministry for Primary Industries Sustainable Food and Fibre Futures Fund (5000835).
Author information
Authors and Affiliations
Contributions
RGS, KL, S-AT and CGP considered the study and obtained funding. AA-U measured on farm phenotypes. AJ, KL, MDL and RGS performed phenotype and genotype analysis. AJ wrote the manuscript. AA-U, S-AT, MS, MDL, NL-V, CGP, RGS, and KL revised and approved of the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Animal genotyping were approved by The University of Auckland animal ethics committee (Auckland, New Zealand) ethics permit 001763. Collection of animal phenotypes and milk sampling were approved by AgResearch ethics committee (Hamilton, New Zealand) ethics permits 14828 and 14889.
Consent for publication
N/A
Competing interests
KL CGP and AA-U received compensation from the Dairy Goat Co-operative (DGC) and RGS obtained research funding from the DGC. S-AT is an employee of the DGC. AJ was supported by the DGC Russ Moroney scholarship.
Supplementary Information
Additional file 1 Supplementary 1
Full protein sequence alignment between species across several taxa indicating the degree of conservation around the SENP3 E89del (A), PSMB6 V222I (B) and SHBG S267I/S277I/S281I/S166I (C) variant sites (outlined in red). Multiple sequence alignment was performed with ClustalW. NCBI or UniProt accession numbers provided in brackets. Sequence logos were created using WebLogo web based application [34]
Additional file 2 Supplementary 2
Summary of the 340 whole genome sequence variants identified from 302 goats located within the chromosome 19 QTL interval with high linkage disequilibrium, R2 > 0.8 with the most significant SNP marker rs268292132
Additional file 3 Supplementary 3
Ensembl Variant Effect Predictor (VEP) calculated variant consequences on the 340 variants identified from whole genome sequencing of 302 goats
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, A., Ankersmit-Udy, A., Turner, SA. et al. A Capra hircus chromosome 19 locus linked to milk production influences mammary conformation. J Animal Sci Biotechnol 13, 4 (2022). https://doi.org/10.1186/s40104-021-00667-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-021-00667-y