
Abstract
In order to dissect amyotrophic lateral sclerosis (ALS), a multigenic, multifactorial, and progressive neurodegenerative disease with heterogeneous clinical presentations, researchers have generated numerous animal models to mimic the genetic defects. Concurrent and comparative analysis of these various models allows identification of the causes and mechanisms of ALS in order to finally obtain effective therapeutics. However, most genetically modified rodent models lack overt pathological features, imposing challenges and limitations in utilizing them to rigorously test the potential mechanisms. Recent studies using large animals, including pigs and non-human primates, have uncovered important events that resemble neurodegeneration in patients’ brains but could not be produced in small animals. Here we describe common features as well as discrepancies among these models, highlighting new insights from these models. Furthermore, we will discuss how to make rodent models more capable of recapitulating important pathological features based on the important pathogenic insights from large animal models.
Similar content being viewed by others
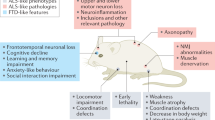

Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a classical neurodegenerative disease characterized by progressive degeneration of both upper and lower motor neurons in the brain and spinal cord [1]. ALS is divided into sporadic (sALS) and familial (fALS) forms based on heredity, with approximately 5%–10% of cases being fALS [2]. The onset of ALS usually occurs at late middle ages, and the clinical manifestations are progressive muscle atrophy and weakness. Most patients will die of respiratory failure within 2–5 years [3]. Whole-exome and whole-genome sequencing has identified fALS-associated mutations in approximately 50 genes, and more than 30 are considered causative genes [4]. The most commonly mutated genes are superoxide dismutase-1 (SOD1) with a mutation frequency of 25% in fALS cases (the wild-type [WT] or misfolded SOD1 has also been implicated in a significant fraction of sALS cases) [5], chromosome 9 open reading frame 72 (C9ORF72) with a mutation frequency of 39% in fALS cases and 7% in sALS cases [6,7,8], fused in sarcoma (FUS) with a mutation frequency of 2.8% in fALS cases and 0.3% in sALS cases [9,10,11], and TAR DNA-binding protein (TARDBP) with a mutation frequency of 4.2% in fALS cases and 0.8% in sALS cases [11, 12]. Most of the current ALS models are based on these four genes, including both vertebrate and invertebrate models such as yeast, elegans, fruit flies, zebrafish, mice, rats, dogs, pigs, and more recently, non-human primates. These models have different characteristics and are complementary in the dissection of pathological mechanisms underlying motor neuron degeneration and ALS progression. However, most transgenic small animal models lack significant neurodegenerative features compared to large animal models, presenting challenges and limitations in their use [13]. In this review, the four genes currently most associated with ALS prevalence are discussed, with a focus on evidence derived from ALS patients. Then we describe common features as well as discrepancies among these models, highlighting new insights and emerging roles of experimental organisms in ALS research.
ALS genes in ALS patients
SOD1
The SOD1 gene encodes a ubiquitous Cu/Zn superoxide dismutase that catalyzes the dismutation of superoxide radicals into hydrogen peroxide and dioxygen. Rosen et al. [14] first reported the genetic link between fALS and the SOD1 gene in 1993. Teepu Siddique et al. [15] subsequently reported a breakthrough finding that dominant, gain-of-function mutations in SOD1 contribute to the pathogenesis of fALS. In the ensuing two decades, more than 185 disease-associated SOD1 variants were identified, distributed throughout the gene [16]. Genotype–phenotype correlations are evident in SOD1-ALS, with distinct clinical features in patients harboring specific variants [17]. Globally, the most common mutation in the SOD1 gene is D90A, and carriers of this mutation typically present with a slowly progressive paralysis that begins in the legs and spreads upwards, with atypical features such as bladder dysfunction [18]. Mutations in SOD1 are found in approximately 20% of fALS patients, but the mechanism by which mutant SOD1 triggers motor neuron damage remains controversial. Currently, more evidence supports that the mutation-induced SOD1 conformational and functional changes confer toxicity through interactions with many proteins and cause a series of consequences, including excitotoxicity, endoplasmic reticulum stress, oxidative stress through upregulation of reactive oxygen species, mitochondrial dysfunction, and prion-like proliferation [19]. The fact that SOD1-knockout mice also develop muscle denervation and mitochondrial oxidative stress over time suggests that chronic loss of SOD1 activity may contribute to disease [20, 21].
TARDBP
TDP-43, encoded by the TARDBP gene, is a DNA/RNA-binding protein consisting of 414 amino acids. TDP-43 contains nuclear localization signals and nuclear export signals, allowing it to shuttle between the nucleus and the cytoplasm, but typically it resides in the nucleus to exert important functions such as gene regulation [22]. More than 50 missense mutations in the TARDBP gene have been identified in ALS patients, accounting for 1%–2% of the total cases [23]. TDP-43 has been reported to be involved in several RNA processing steps, including pre-mRNA splicing, mRNA transport, regulation of mRNA stability, translation, and regulation of non-coding RNAs [24]. In 2006, TDP-43 was identified as a key component of neuronal and glial cytoplasmic inclusions in patients with ALS and frontotemporal lobar degeneration (FTLD or FTLD-TDP) [25, 26]. These inclusion bodies are mainly aggregates of pathological TDP-43 proteins that are hyperphosphorylated, ubiquitinated, and cleaved at the C-terminus. Pathological TDP-43 protein aggregation is often accompanied by loss of nuclear TDP-43, suggesting a loss of normal function in the nucleus, increased abnormal cytoplasmic function, or both [27]. The accumulation of TDP-43 in ubiquitin-positive cytoplasmic neuronal inclusions in the brain and spinal cord has been recognized as a pathological hallmark of ALS [28].
FUS
In 2009, pathogenic variants in the gene encoding FUS, another TDP-43-like RNA-binding protein, were reported in patients with ALS [9]. FUS is a ubiquitously expressed 526-amino-acid protein, which shares many physiological roles with TDP-43 in various aspects of gene expression and is involved in several RNA processing events, in particular transcription, alternative splicing, and mRNA trafficking. FUS is predominantly localized to the nucleus under normal physiological conditions, but it crosses the nuclear membrane to play a role in nucleoplasmic transport, similar to TDP-43 [29]. More than 50 autosomal-dominant FUS variants have been identified in ALS patients [30]. Many of these mutations affect the nuclear localization of FUS, leading to loss of function as a regulator of transcription and RNA maturation, and formation of toxic FUS aggregates in the cytoplasm [31,32,33,34]. Therefore, the pathogenic mechanism of FUS is similar as TDP-43, involving combined loss of function and toxic aggregation. In addition, FUS is also involved in DNA repair mechanisms, including strand-break repair, non-homologous end joining, and homologous recombination during DNA binuclear recombination [35].
C9ORF72
The C9ORF72 gene contains 12 exons and encodes a small protein that plays a key role in the regulation of autophagy. Crystal structures and biochemical analysis of purified recombinant proteins support a role for the C9ORF72 complex as a GTPase-activating protein [36,37,38]. In 2011, expansion of a hexanucleotide repeat (GGGGCC) in the noncoding region of C9ORF72 was reported to be the most common genetic cause of ALS in European populations [39]. There are 5–10 copies of the hexanucleotide repeat in the normal C9ORF72 gene, but the number of repeats may increase to hundreds to thousands in ALS patients. This hexanucleotide repeat expansion is found in approximately 34% of fALS and 5% of sALS cases in European populations but occurs less frequently in Asian populations [11]. The pathogenic role of C9ORF72 is still controversial, but increasing evidence indicates that the pathogenic mechanism of C9ORF72-ALS involves a cascade of reactions, including multiple cellular mechanisms: (1) G4C2 hexanucleotide repeat expansion causes RNA toxicity [40, 41]; (2) aggregation of toxic dipeptide repeat proteins (DPRs) translated from the hexanucleotide repeats through repeat-associated non-ATG translation [42, 43]; and (3) decreased levels of normally functioning C9ORF72 protein, leading to loss-of-function mechanisms [44]. There is no consensus on the extent to which each of these mechanisms contributes to disease progression, but all of them could explain the pathogenic role of the hexanucleotide expansion in C9ORF72.
The specific pathogenic mechanism of the four genes are illustrated in Fig. 1. Apart from these genes, other less common genetic mutations have also been reported to be associated with ALS, such as mutations in OPTN (Optineurin), VABP (VAMP-associated protein B), UBQLN2 (Ubiquilin-2), VCP (Valosin containing protein), MATR3 (Matrin 3), TBK1 (TANK-binding kinase-1), NEK1 (NIMA-related kinase-1), and C21orf2 [45]. In addition, the interactions between environmental factors and genetic mutations must also be considered.

Pathogenesis of TARDBP-, FUS-, SOD1- and C9OR72-associated ALS. a TARDBP mutations act through both loss-of-function and gain-of-function mechanisms. Mutant (Mt) TDP-43 proteins inhibit normal TDP-43 binding to pre-mRNA and generate TDP-43 inclusions in the cytoplasm. b Like TDP-43, FUS mutations act through both loss-of-function and gain-of-function mechanisms. Mutant FUS proteins cause loss-of-function by inhibiting normal FUS binding to pre-mRNA. c SOD1 mutations act through a gain-of-function mechanism. Mutant SOD1 dimer accumulation in the cytoplasm leads to prune-like toxin production and Golgi apparatus stress, and generation of mitochondrial reactive oxygen species (ROS), leading to mitochondrial destruction. d The C9ORF72 mutation acts through a gain-of-function mechanism. GGGGCC[G4C2] translocates to the cytosol, translates to form aggregates of poly (GP) DPRs, and misfolds to form aggregates of ubiquitinated RNA aggregates associated with TDP-43 or FUS proteins. Both proteins mediate neuronal toxicity. Cytosol vacuolization (vac) is caused by all the above-mentioned mutations
Models for ALS
Caenorhabditis elegans and Zebrafish (Danio rerio)
The anatomical transparency of C. elegans and Zebrafish enables visualization of neurons and monitoring of neuronal activity over time using co-expressed fluorescent proteins. In addition, the well-defined and genetically controllable nervous system of C. elegans and Zebrafish is also an advantage for exploring the pathological mechanisms of neurodegenerative diseases and provides a good tool for screening new potential drugs [46,47,48,49].
Caenorhabditis elegans and Zebrafish models of SOD1 mutation
In a C. elegans SOD1 model, overexpression of human SOD1 (G85R) in neurons results in cytoplasmic aggregates, reduced number and diameter of cellular processes, reduced number of mitochondria and vesicles, and motility deficits. Caenorhabditis elegans overexpressing human SOD1 (H46R or H48Q) also exhibits motor deficits, but to a lesser extent than those overexpressing SOD1 (G85R). Overexpression of human SOD1 (G93A) in motor neurons leads to age-dependent paralysis due to axonal defects, but no overt neuronal death was observed [50, 51]. Injection of human SOD1 (G93A, G37R or A4V) mRNA into zebrafish embryos resulted in abnormal axonal branching and shortened axon length, and these phenotypes were aggravated when the models expressed higher levels of mutant SOD1 [52, 53]. Furthermore, in zebrafish expressing mutant SOD1, the motor impairment, protein misfolding, and ER-Golgi transport dysfunction were rescued by protein disulfide isomerase, suggesting that redox regulation is essential for maintaining cellular homeostasis [54].
Caenorhabditis elegans and Zebrafish models of FUS mutation
Expression of FUS variants in C. elegans induces protein aggregation in GABAergic neurons, leading to neurodegeneration, synaptic dysfunction, and paralysis, together with cytoplasmic mislocalization of FUS, whereas expression of WT FUS does not cause significant changes [55, 56]. Furthermore, electron microscopy and electrophysiological analysis showed that transgenic expression of mutant FUS in C. elegans resulted in reduced synaptic transmission from motor neurons to muscles and impaired synaptic vesicle docking at the neuromuscular junction (NMJ) [57]. In zebrafish models, knockdown of endogenous FUS or overexpression of human defective FUS alleles leads to defective presynaptic function at the NMJ, producing pathological motor phenotypes that can be rescued by co-expression of WT human FUS [58]. Additional studies indicate that FUS mutations in zebrafish also induce protein aggregation, oxidative stress, NMJ damage, and motor dysfunction in motor neurons (MN) and other cells [59,60,61,62].
Caenorhabditis elegans and Zebrafish models of TDP-43 mutation
Ash et al. developed the first TDP-43 overexpression model in C. elegans and showed that pan-neuronal expression of human WT TDP-43 in transgenic C. elegans resulted in movement disorders, as well as fasciculations of motor neurons [63, 64]. Furthermore, expression of mutant TDP-43 in nematode GABAergic motor neurons induces oxidative stress and aberrant expression of endogenous TDP-1 (an ortholog of TARDBP), leading to age-dependent progressive paralysis, neurodegeneration and synaptic damage [56, 65]. Expression of mutant TARDBP in zebrafish embryos induced motor neuron degeneration and movement deficits, which were rescued by co-expression of the WT human TARDBP gene, suggesting the functional importance of TDP-43 and the potential for pathogenic mutations to cause loss-of-function and toxicity function [66].
Caenorhabditis elegans and Zebrafish models of C9ORF72 mutation
The ALS/FTD-related gene homolog (alfa-1) is a C. elegans ortholog of C9ORF72. Loss of alfa-1 leads to motor deficits, motor neuron degeneration, and paralysis in C. elegans [67]. In addition, alfa-1 deletion also leads to dysfunction of lysosomal reorganization and degradation of endocytic elements in nematodes, which disrupts lysosomal homeostasis [68]. Knockdown of a zebrafish ortholog of C9ORF72 results in axonopathy in MNs, cytoplasmic accumulation of TDP-43, and swimming abnormalities [49]. Recently, a stable C9ORF72 transgenic zebrafish model was constructed, characterized by accumulation of RNA foci and DPR in muscles and the central nervous system (CNS), increased apoptosis, abnormal motor axons, motor deficits, muscle atrophy, loss of MNs, cognitive impairment, and reduced survival [69, 70].
Studies of neurodegeneration and toxicity associated with ALS may benefit from the C. elegans and Zebrafish models. However, it should be recognized that these are very simple organisms with several limitations, including large anatomical differences from the human brain, lack of obvious tissues and organs, wide use of embryonic stages, and inability to perform more informative behavioral analyses. Simulating some pathological features and phenotypes of human ALS disease is difficult.
Drosophila melanogaster
Drosophila melanogaster is an easy handling and cost-effective animal model with short lifespan and complete genome sequenced. They are widely used for studying neurodegenerative diseases including ALS [71, 72]. Most of the mutated genes known to be associated with ALS have been modeled in Drosophila, and transgenic flies expressing ALS genes can reliably reproduce some ALS phenotypes such as movement disorders, cellular inclusions, and mitochondrial dysfunction [73].
Drosophila melanogaster model of SOD1 mutation
Several studies have shown that transgenic flies expressing the human SOD1 gene carrying point mutations (G85R, A4V, G37R, or G41D) exhibit motor neuron dysfunction, dyskinesia, mitochondrial damage, oxidative stress, and pathological SOD1 aggregation [74,75,76]. One of the important reasons why Drosophila melanogaster is a powerful genetic model is the UAS/Gal4 system, which is widely used to overexpress disease-associated human genes in Drosophila [77]. The UAS/Gal4-driven expression of WT or ALS-associated forms of SOD1 (A4V or G85R) in MNs does not alter the lifespan of flies but results in progressive motor function deterioration [74]. Besides, expression of zinc-deficient human SOD1 in Drosophila neurons also produces a locomotor defect linked to mitochondrial dysfunction [76]. Human SOD1 mutations in a Drosophila knock-in model also cause neuronal metamorphosis, muscle contraction and decreased survival rates [78].
Drosophila melanogaster model of FUS mutation
The only FUS ortholog in Drosophila melanogaster is cabeza [70]. Drosophila expressing mutant human FUS or Cabeza have phenotypes such as neurodegeneration, impaired photoreceptors, and neuronal complications [79,80,81,82]. It has been reported that these phenotypes can be rescued by introducing a WT human FUS transgene in fly mutants of cabeza [83]. Investigations in FUS transgenic flies have shown that human FUS-induced neurotoxicity can be attenuated by inhibiting nuclear export, confirming that nucleoplasmic transport is involved in the pathogenesis of ALS [84, 85]. Several studies on transgenic Drosophila have also confirmed that the FUS-induced neurodegeneration is associated with cellular processes such as transcriptional and translational regulation [86], piRNA biogenesis [87], stress granule assembly [88], and Hippo-signaling pathways [89, 90], and further elucidated the complex pathogenesis of ALS.
Drosophila melanogaster model of TDP-43 mutation
Several studies have reported that overexpression of the TBPH gene (fly ortholog of TARDBP) in Drosophila and overexpression of mutant or WT human TDP-43 affects motility, axonal transport, pupal eclosion, and lifespan [81, 91,92,93,94]. Loss of TBPH in Drosophila also results in motor impairment and shortened lifespan [95]. Interestingly, several potential therapeutic approaches have been identified using the Drosophila TDP-43 transgenic model. Modulations of autophagy [96], mitophagy [97], mitochondrial dynamics [98], glucose and lipid metabolism [99], and stress granule dynamics [100, 101] have been reported as beneficial for fly motor behavior and longevity.
Drosophila melanogaster model of C9ORF72 mutation
Drosophila melanogaster has no C9ORF72 ortholog, so the consequences of C9ORF72 deletion in Drosophila cannot be determined. A Drosophila model of C9ORF72-associated ALS has been developed by overexpressing the G4C2 repeat RNA to mimick DPR proteotoxicity, and has revealed some important insights into the pathogenesis of C9ORF72-ALS. Ectopic expression of expanded G4C2 or toxic dipeptide repeats in Drosophila tissues results in motor deficits, abnormalities of the NMJ, and disorganized microphthalmia [102,103,104,105]. Recent studies in several C9ORF72 transgenic Drosophila models have shown that different cellular processes contribute to C9ORF72-ALS pathogenesis, such as transcription [104, 106], nucleocytoplasmic transport [107, 108], translation [109], and protein degradation [110].
Despite the many advantages of the Drosophila model, the major limitations of this organism are the large anatomical differences from human brains and the impossibility of performing more informative behavioral analyses.
Rodents (mouse and rat)
Transgenic rodents are the most used animal models and provide important insights into pathogenesis. Rodent models have been widely used in ALS research since the first SOD1 (A4V and G93A) ALS mouse models were developed in 1994 [15]. Although rats are used much less than mice, they also have physiological characteristics like those in humans and the possibilities for genetic manipulation are historically more recent in rats than in mice. Rodent ALS models with ALS-associated mutations are listed in Table 1.
Rodent models of SOD1 mutation
Several SOD1 transgenic rodent models (G93A, D83G, D85G, D86G, D90A, and G37R, among others) have been constructed based on variants found in ALS patients over the past 28 years. Most are established by overexpressing missense, mutated, or truncated human SOD1 [15, 68, 140,141,142]. Among them, the SOD1 G93A model is the most widely used. It reproduces some pathological mechanisms in ALS patients such as abundant cytoplasmic inclusions, NMJ injury and extensive inflammation in the spinal cord with reactive gliosis, and exhibits gender difference in disease progression [143, 144]. However, a major drawback of the SOD1 G93A mouse model is the absence of motor neuron degeneration in the cerebral cortex, which is one of the main hallmarks in human patients [145]. SOD1 (D83G) transgenic mice show some motor neuron degeneration in the cerebral cortex, but with no paralytic phenotype in the adulthood [146]. Mice homozygous for the SOD1 D90A mutation accumulate SOD1 aggregates in the ventral horn of the spinal cord and develop a fatal motor neuron disease that progresses slowly, similar to bladder disturbances observed in human ALS patients [147]. Several other models (D85G, D86G, and G37R) all express high levels of SOD1 aggregates, and share common features such as neuroinflammation, glutamate excitotoxicity, mitochondrial alterations, and defective axonal transport in neurons [148]. Most mutations in SOD1 associated with ALS are generally thought to cause ALS through a gain-of-function mechanism. However, the SOD1-knockout mice also develop muscle denervation and mitochondrial oxidative stress over time, suggesting that chronic loss of SOD1 activity may contribute to the disease [20, 21]. These SOD1 mouse models have been used for preclinical evaluation of potential treatments for ALS. While some potential treatments have been able to show benefits in the SOD1 mouse models, translation into clinical trials has been poor. For example, minocycline was able to slow disease in SOD1 (G37R) mice, but it accelerated disease in human clinical trials in a diverse ALS patient group [149].
Rat models of SOD1 mutation have also been developed, among which the more studied are SOD1 G93A and H46R models. They show similar pathological features derived from genetic alterations as described in mice, such as upper and lower motor neuron degeneration [150, 151]. Notably, the SOD1 G93A mutation causes more aggressive disease than the H46R mutation [152]. Recently, in the SOD1 G93A rat model, Maggot et al. showed that disrupting the blood-spinal cord barrier directly leads to motor neuron degeneration. Intravenous infusion of healthy mesenchymal stem cells into these rats delayed disease progression, preserved barrier function, increased expression of the neurotrophic factor neurturin, and protected motor neurons [153].
Rodent models of TDP-43 mutation
Based on the known variants of TDP-43 in patients with fALS, approximately 20 TDP-43 mouse models have been established [154]. The earliest transgenic TDP-43 mouse models were generated by overexpression of WT or mutant (A315T and M337V) TDP-43 cDNAs under the prion protein gene promoter [120, 155, 156]. Subsequently, researchers generated transgenic mouse models overexpressing exogenous human TDP-43 based on promoters such as Thy1.2 and Camkllα. These transgenic mice display accumulation of pathological aggregates of ubiquitinated proteins in specific neuronal populations, resulting in abnormalities of early neuronal morphology, gliosis, varying degrees of spinal cord pathology, and progressive paralysis and death [124, 157]. Studies have shown that the phenotypic severity correlates with the expression level of mutant TDP-43, and in those animals with high expression, death usually occurs within a week. To understand whether there is a loss-of-function mechanism in TDP-43-related ALS, researchers generated a knockout mouse model. They found that homozygous knockout led to impaired embryogenesis, while heterozygous knockout did not induce symptoms of neuromuscular disease and preserved normal protein expression, suggesting that TDP-43 has an important function during development [28, 157, 158]. Furthermore, in patients with TDP-43 mutation or certain pathological conditions such as FTLD, nuclear TDP-43 redistributes in the cytoplasm and forms cytoplasmic inclusions [25, 159]. While some mouse models can have minimal levels of cytoplasmic TDP-43 [120, 160, 161], most TDP-43 mutant mice show predominantly nuclear localization of TDP-43 and do not reproduce the critical cytoplasmic mislocalization of TDP-43 [162, 163]. These findings raise concerns about the reliability of these mouse models in studies of ALS pathogenesis.
Overexpression of human WT and mutant M337V TDP-43 has been studied in rats, with early phenotypes of immobility, paralysis, and presexual death. In addition, the rats expressing comparable levels of TDP-43 M337V show a more severe phenotype than those expressing WT TDP-43 after 6 months, suggesting that the mutant TDP-43 protein is more toxic than WT TDP-43 [164].
Rodent models of FUS mutation
Like TDP-43, the nuclear depletion of FUS proteins and the formation of toxic aggregates in the cytoplasm are important events leading to ALS pathogenesis [165]. Since the discovery of FUS association with ALS [9, 166], several mouse models with FUS knockdown or overexpression of WT and mutant FUS have been developed (R521C, R521G, P525L, FUSΔNLS, etc.) [129, 131,132,133]. All these models have varying degrees of phenotypes such as early neuronal loss, motor deficits, and mild behavioral impairments. Two mutants, FUS P525L and R521C, have been reported to cause early-onset and late-onset disease in humans, respectively, and induce NMJ deficits and progressive neurodegeneration in mice at 2 and 1 month of age, respectively. Furthermore, the FUS P525L mice exhibit more cytoplasmic FUS than FUS R521C mice, suggesting that the frequency of FUS accumulation is directly related to the severity of ALS [167]. The FUS R521G mice develop hindlimb clenching, NMJ denervation, muscle atrophy, and mild behavioral disturbances before locomotion loss, and eventually die before age of 1 month due to the loss of locomotor function [130, 167]. Partial cytoplasmic mislocalization of FUS in FUSΔNLS mice is sufficient to cause several behavioral abnormalities, including locomotor hyperactivity and altered social interaction, which precede motor neuron degeneration [131,132,133].
In the rat FUS R512C model, protein aggregation is observed in the brain and the spinal cord, leading to early neuronal loss, gliosis, and motor dysfunction, and eventually typical FTD phenotypic behaviors such as social restriction and hyperactivity [168, 169]. Transgenic rats can also be generated by intravenous administration of adeno-associated virus vector serotype 9 (AAV9) in adult rats. These rats exhibit progressive motor changes and respiratory dysfunction [170].
Rodent models of C9ORF72 mutation
In 2013, researchers developed the first mouse model carrying a mutation in the C9ORF72 gene [171], which exhibits NMJ damage, apoptosis, cognitive impairment, and motor deficits [138, 171]. The researchers then used bacterial artificial chromosomes to generate a cohort of transgenic mice that carry approximately 500 and 1000 repeats of the G4C2 motif and develop extensive RNA foci and DPRs. However, no behavioral changes or neurodegeneration was observed [135, 172,173,174]. This supports the hypothesis that RNA foci and dipeptides are insufficient to drive degeneration, although this finding may be only specific to these models and not representative of what happens in humans. Furthermore, the C9ORF72 transgenic model developed by Liu et al. showed phenotypes such as extensive nuclear and cytoplasmic inclusions in neurons, MN loss, muscle denervation, hindlimb paralysis, and decreased survival [139]. However, the degenerative phenotypes of this model were not reproducible when tested in two independent laboratories [175]. Although repeat expansion has been shown to generate toxic products, mRNAs encoding the C9ORF72 protein are also reduced in affected individuals, and the reduced C9ORF72 has been shown to suppress the repeat-mediated increase in autophagy. These results support a disease mechanism in ALS/FTD resulting from reduced C9ORF72, which may lead to autophagy deficits that synergize with the repeat-dependent increase in toxicity [176]. Repeat expansion reduces C9ORF72 expression and induces neurodegeneration by two mechanisms: accumulation of glutamate receptors leading to excitotoxicity and impaired clearance of neurotoxic DPRs derived from the repeat expansion [177]. Thus, a combined gain-of-function and loss-of-function mechanism leads to neurodegeneration. Currently, conditional knockout mice with neuron-specific deletion of C9ORF72 have been generated, but they do not exhibit MN degeneration, overt motor deficits, or reduced survival [178].
Knockdown of the C9ORF72 gene in rats using CRISPR/cas9 technology has been reported without MN loss and motor deficits. Interestingly, when the rats were treated with low doses of a glutamic acid analog kainic acid, the release of excitatory neurotransmitters was stimulated, leading to susceptibility of motor neurons to excitotoxicity, motor neuron degeneration, and motor deficits [179]. C9ORF72 knock-in rats were generated by knockin of 80 G4C2 repeats with human flanking fragments within exon 1a and exon 1b of the rat C9ORF72 locus. These rat models have reduced C9ORF72 protein expression in several CNS regions and show motor deficits due to motor neuron loss at 4 months of age [180].
Although some rodent models can reproduce protein misfolding and aggregation observed in patient brains, most rodent models cannot fully mimic the symptoms and pathologies of many neurodegenerative diseases, including ALS [120, 181,182,183,184]. This phenomenon could be due to the genomic, molecular, and anatomic differences between rodents and humans.
Canine models
Canine degenerative myelopathy (CDM) is an idiopathic pathology that occurs in specific dog breeds and is thought to be a human SOD1-related ALS model due to clinical and molecular similarities. More precisely, CDM is characterized by progressive axonal degeneration, muscle atrophy, astrocytosis, peripheral demyelination, and SOD1 inclusions leading to adult-onset neurodegenerative myelopathy and progressive motor dysfunction [185, 186]. CDM shares some molecular and clinical features with upper MN-dominant forms of ALS, such as disease progression and distribution of lesions [187, 188]. Interestingly, to date, only two missense mutations in SOD1 dismutase, T18S and E40K, have been identified as causative for CDM. Unlike ALS, in which SOD1 pathogenic variants are inherited as dominant, CDM shows recessive inheritance with reduced penetrance [189, 190]. The T18S and E40K mutations do not disrupt the dismutase domain, but both may induce SOD1 aggregation either by reducing the negative charge repulsion or by forming disulfide-linked enzymatically active dimers, thus supporting the gain-of-function hypothesis for SOD1 toxicity [191]. In addition to the loss of MNs, canine models affected by CDM share some other pathological features with SOD1-ALS rodent models and patients, such as oligodendrocyte damage leading to demyelination [192], an increase of arginase 1-expressing microglia in the vicinity of motor neurons [193], and upregulation of CB2 receptors in glia cells that serve as a marker of major cellular and biological responses to disease [194].
Considering the clinical and molecular similarities between idiopathic CDM and SOD1-ALS, studies of canine idiopathic CDM may help better dissect the pathological mechanisms of ALS. However, it should be considered that dogs with CDM are often euthanized at an early stage of the disease, and therefore the tissues used for investigation can only provide insight into early disease stages.
Swine models
The swine model has been widely used to study human disease pathology because of its anatomical, physiological, and biochemical similarities with humans, including high similarities in the genome [195] and neuropsychiatric disease characteristics [196]. Currently, several neurodegenerative diseases have been recapitulated in pigs [197,198,199,200], including ALS (TDP-43-related and SOD1-related). Chieppa et al. generated the first ALS pig model expressing G93A hSOD1 by in vitro culture transfection combined with somatic cell nuclear transfer (SCNT) [201]. The transgenic SOD1G93A pigs exhibit motor neuron degeneration, hindlimb motor deficits, expression of mutated SOD1 copies, gliosis, and protein aggregation in an age-dependent manner [202, 203]. During early disease stages, the mutant SOD1 does not form cytoplasmic inclusions but instead shows nuclear accumulation and ubiquitinated nuclear aggregates in the SOD1G93A pigs, as in the brains of some ALS patients [202]. At approximately 27 months of age, the transgenic SOD1G93A pigs undergo a prolonged symptomatic phase characterized by increased amounts of total TDP-43 in peripheral blood mononuclear cells. Severe skeletal muscle pathology, including inflammation and necrosis, is observed in the late stages of the disease [203]. Subsequently, Wang et al. also used SCNT to generate the first M337V TDP-43 transgenic pig model exhibiting severe phenotypes and early death. TDP-43 aggregates were detected in the cytoplasm of the spinal cord and brain neurons in the M337V TDP-43 transgenic pigs. The M337V TDP-43 protein alters neuronal RNA splicing by interacting with NeuN-associated protein-associated RNA splicing factors, as reported in ALS patients [204]. Convincingly, with gene silencing approaches such as viral-mediated delivery of shRNAs, large animal models such as pigs could be widely used in drug development and drug safety studies [205, 206].
Although the pig model can recapitulate some pathological characteristics of neurodegenerative diseases, there are still some limitations, such as high cost, long growth cycle, and lack of systematic behavioral and cognitive testing systems.
Non-human primate models
The brains of non-human primates are evolutionarily closest to the human brain with many structural, cognitive, and functional features. The brains of non-human primates have a complex network of brain connections involving the neocortex and prefrontal cortex, thus enabling the development of higher brain functions such as thinking, learning, decision-making, and judgment [207, 208]. At the molecular level, the brains of non-human primates share more similar gene expression patterns with human brains than mouse brains do [209]. These features, along with neuroanatomical and genetic similarities, make non-human primates highly desirable models for neurodegenerative diseases, including ALS. In 2012, Uchida et al. created TDP-43-overexpressing cynomolgus monkeys by injecting adeno-associated virus (AAV)-based human WT TDP-43 coding sequences into the C5-C6 spinal segment of cynomolgus monkeys [210]. After 2–3 weeks, the monkeys exhibited progressive motor weakness and muscle atrophy with fasciculations in the muscles of the distal hand on the injection side; complete paralysis of the ipsilateral hand was observed 2–5 weeks after onset. At the same time, symptoms such as muscle atrophy and weakness also appeared in the contralateral hand. At the cellular level, diffuse mislocalization of TDP-43 in the cytoplasm was evident in α-MNs, but accumulation was infrequent, suggesting that this model does recapitulate some of the clinical features of ALS patients as well as pathological features in the spinal cord. In subsequent years, Borel et al. obtained marmoset and macaque SOD1-ALS models by intrathecal delivery of AAV encoding an artificial SOD1-specific microRNA and determined reduced SOD1 levels in motoneurons and spinal cord slices [211, 212]. Using the same technique, stereotaxic injection of FUS-targeting shRNA in Callithrix jacchus was used to generate a FUS-ALS marmoset model [213]. It is undeniable that gene silencing methods using viral delivery can play an important role in drug development and drug safety research. However, these monkeys were only manipulated with gene silencing to simulate the loss of SOD1 and FUS in the neurons of ALS patients and were not studied for neuropathological characteristics and behavioral phenotypes. Recently, to investigate the subcellular distribution of mutant TDP-43 in the monkey brain, Yin et al. injected a viral vector expressing mutant TDP-43 (M337V) directly into the substantia nigra of rhesus monkeys. Three months after injection, all the monkeys with TDP-43 injection developed significant left upper-extremity weakness at 2–4 weeks post-injection, and the severity increased and stabilized 3–4 months later [214]. Most of the mutant TDP-43 was distributed in the cytoplasm of the monkey brain, which is consistent with the cytoplasmic distribution of TDP-43 in the brains of ALS patients and the spinal cords of monkeys overexpressing WT TDP-43. Notably, non-human primate-specific caspase-4, but not the mouse homolog caspase-11, removes the nuclear localization signal-containing N-terminal domain, leading to accumulation of fragmented TDP-43 in the cytoplasm [210, 214]. The cleavage of TDP-43 mediated by the primate-specific caspase-4 provides additional clues to the cytoplasmic TDP-43-mediated gain-of-toxicity and points to potential therapeutic strategies to prevent or reduce TDP-43-associated neuropathology.
Non-human primate models have anatomical, physiological, and biochemical characteristics relevant to humans, including high genomic similarity. However, similar as pig models, the non-human primate models of neurodegenerative diseases have limitations of high cost and ethical issues. Table 2 lists large animal models of ALS with ALS-associated mutations.
Challenges and future directions
Effective therapeutics for ALS are urgently needed, but for drug applications, translating results from small animal models to human clinical trials remains limited and challenging. This may be due to the genomic, molecular, and anatomical differences between small animals and humans. For instance, in rodents, some genetic mutation models show a milder ALS phenotype than humans, while some models show no signs of neurodegeneration. Rodents typically live for less than 3 years, a short lifespan that may not be long enough for the occurrence of neurodegeneration that typically takes decades to occur in humans. Furthermore, only about a quarter of alternatively spliced exons for a given transcript is conserved between humans and rodents [216]. The importance of this difference in the pathogenesis of ALS remains unexplored. These factors may lead to substantial failures in the translation from preclinical animal studies to effective clinical treatments for ALS.
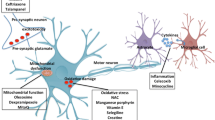
The current large animal models have demonstrated important species-dependent differences in neuropathology. They serve as important tools to study pathogenesis and pathological events that may uniquely occur in humans and thus could serve as new therapeutic targets. For example, the cytoplasmic distribution of mutant TDP-43 (M337V) in rhesus monkey and pig brains highlights the value of large animal models for studying the cytoplasmic toxicity of TDP-43 [204, 210, 214]. Thus, the selective expression of modifiers or targets of disease proteins in large animal models contributes to specific neuropathological events, which may not occur in small animal models. Given these factors, it is expected that more and more large animals will be accepted as models of choice for research on human diseases and translational medicine. In addition, the important information gained from large animal models is invaluable for generating more humanized mouse models. However, there are barriers to generating large animal models, such as the high cost of animals, the current inefficiency of gene targeting in large animals, and the large amount of time required, which have hindered the widespread use of such animal models for research (Fig. 2).

New pathogenic insights from monkey models expressing mutant TDP-43. TDP-43 remains in the nucleus of rodent neurons to elicit nuclear toxicity. In the primate neurons, however, the primate-specific caspase-4 cleaves TDP-43 to produce truncated TDP-43, which redistributes in the cytoplasm, resulting in cytoplasmic toxicity
Conclusions
In this review, we describe several commonly used model organisms (worms, flies, zebrafish, rodents, dogs, pigs, and non-human primates) as ALS models. Undoubtedly, the models summarized here play a pivotal role in uncovering the myriad cellular and molecular determinants involved in ALS and its progression, and in showing the multifactorial and non-cell-autonomous nature of the disease. To understand the causes and mechanisms of ALS, simultaneous and comparative analyses of these animal models are required while keeping in mind the limitations of these models. Overall, this review describes the pros and cons of various ALS models as well as common features and differences, highlights insights unique to large animal models of ALS, and discusses the use of important pathogenic insights from large animal models to advance the use of rodent models to simulate important pathological features of ALS.
Availability of data and materials
Not applicable.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- SOD1:
-
Superoxide dismutase-1
- C9ORF72:
-
Chromosome 9 open reading frame 72
- FUS:
-
Fused in sarcoma
- TARDBP:
-
TAR DNA-binding protein
- FTLD:
-
Frontotemporal lobar degeneration
- DPR:
-
Dipeptide repeat protein
- NMJ:
-
Neuromuscular junction
- CNS:
-
Central nervous system
- AAV:
-
Adeno-associated virus
- CDM:
-
Canine degenerative myelopathy
- SCNT:
-
Somatic cell nuclear transfer
- MN:
-
Motor neuron
References
Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162–72.
Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. ALS. 2009;10(5–6):310–23.
Shatunov A, Al-Chalabi A. The genetic architecture of ALS. Neurobiol Dis. 2021;147:105156.
Paré B, Lehmann M, Beaudin M, Nordström U, Saikali S, Julien JP, et al. Misfolded SOD1 pathology in sporadic amyotrophic lateral sclerosis. Sci Rep. 2018;8(1):14223.
Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet. 2013;92(3):345–53.
Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. The C9ORF72 expansion mutation is a common cause of ALS+/− FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21(1):102–8.
Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(5):510–8.
Kwiatkowski T Jr, Bosco D, Leclerc A, Tamrazian E, Vanderburg C, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8.
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Zou Z-Y, Zhou Z-R, Che C-H, Liu C-Y, He R-L, Huang H-P. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9.
Kapeli K, Martinez FJ, Yeo GW. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 2017;136:1193–214.
Yin P, Li S, Li XJ, Yang W. New pathogenic insights from large animal models of neurodegenerative diseases. Protein Cell. 2022;13(10):707–20.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5.
Yamashita S, Ando Y. Genotype–phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener. 2015;4:13.
Li HF, Wu ZY. Genotype–phenotype correlations of amyotrophic lateral sclerosis. Transl Neurodegener. 2016;5:3.
Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keranen ML, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. 1996;119(Pt 4):1153–72.
Hayashi Y, Homma K, Ichijo H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul. 2016;60:95–104.
Fischer LR, Igoudjil A, Magrané J, Li Y, Hansen JM, Manfredi G, et al. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011;134(1):196–209.
Fischer LR, Li Y, Asress SA, Jones DP, Glass JD. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp Neurol. 2012;233(1):163–71.
Tziortzouda P, Van Den Bosch L, Hirth F. Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat Rev Neurosci. 2021;22(4):197–208.
Kapeli K, Martinez FJ, Yeo GW. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 2017;136(9):1193–214.
Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010;7(4):420–9.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138(Suppl 1):95–111.
de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2020;92(1):86–95.
Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–19.
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110(Pt 15):1741–50.
Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34(6):812–26.
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7:10465.
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun. 2018;9(1):335.
Zhou Y, Liu S, Liu G, Ozturk A, Hicks GG. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9(10):e1003895.
Humphrey J, Birsa N, Milioto C, McLaughlin M, Ule AM, Robaldo D, et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020;48(12):6889–905.
Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol. 2015;210(4):529–39.
Pang W, Hu F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J Neurochem. 2021;157(3):334–50.
Tang D, Sheng J, Xu L, Zhan X, Liu J, Jiang H, et al. Cryo-EM structure of C9ORF72–SMCR8–WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci USA. 2020;117(18):9876–83.
Su M-Y, Fromm SA, Zoncu R, Hurley JH. Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature. 2020;585(7824):251–5.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126(6):829–44.
Gendron TF, Belzil VV, Zhang YJ, Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127(3):359–76.
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108(1):260–5.
Cleary JD, Ranum LP. New developments in RAN translation: insights from multiple diseases. Curr Opin Genet Dev. 2017;44:125–34.
Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):e1775–e1713.
Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol. 2014;262:91–101.
Patten SA, Parker JA, Wen XY, Drapeau P. Simple animal models for amyotrophic lateral sclerosis drug discovery. Expert Opin Drug Discov. 2016;11(8):797–804.
Ma L, Zhao Y, Chen Y, Cheng B, Peng A, Huang K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur J Pharmacol. 2018;819:169–80.
Patten SA, Armstrong GA, Lissouba A, Kabashi E, Parker JA, Drapeau P. Fishing for causes and cures of motor neuron disorders. Dis Model Mech. 2014;7(7):799–809.
Fortier G, Butti Z, Patten SA. Modelling C9orf72-related amyotrophic lateral sclerosis in zebrafish. Biomedicines. 2020;8(10):440.
Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5(1):e1000350.
Baskoylu SN, Yersak J, O’Hern P, Grosser S, Simon J, Kim S, et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018;14(10):e1007682.
Lemmens R, Van Hoecke A, Hersmus N, Geelen V, D’Hollander I, Thijs V, et al. Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum Mol Genet. 2007;16(19):2359–65.
Ramesh T, Lyon AN, Pineda RH, Wang C, Janssen PM, Canan BD, et al. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis Model Mech. 2010;3(9–10):652–62.
Parakh S, Shadfar S, Perri ER, Ragagnin AMG, Piattoni CV, Fogolin MB, et al. The redox activity of protein disulfide isomerase inhibits ALS phenotypes in cellular and zebrafish models. iScience. 2020;23(5):101097.
Murakami T, Yang SP, Xie L, Kawano T, Fu D, Mukai A, et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet. 2012;21(1):1–9.
Vaccaro A, Tauffenberger A, Aggad D, Rouleau G, Drapeau P, Parker JA. Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS ONE. 2012;7(2):e31321.
Markert SM, Skoruppa M, Yu B, Mulcahy B, Zhen M, Gao S, et al. Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol Open. 2020;9(12):bio055129.
Armstrong GA, Drapeau P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet. 2013;22(21):4282–92.
Armstrong GAB, Liao M, You Z, Lissouba A, Chen BE, Drapeau P. Homology directed knockin of point mutations in the zebrafish tardbp and fus genes in ALS using the CRISPR/Cas9 system. PLoS ONE. 2016;11(3):e0150188.
Campanari M-L, Bourefis A-R, Buee-Scherrer V, Kabashi E. Freezing activity brief data from a new FUS mutant zebrafish line. Data Brief. 2020;31:105921.
Bourefis A-R, Campanari M-L, Buée-Scherrer V, Kabashi E. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiol Dis. 2020;142:104935.
Kabashi E, Brustein E, Champagne N, Drapeau P. Zebrafish models for the functional genomics of neurogenetic disorders. Biochim Biophys Acta Mol Basis Dis. 2011;1812(3):335–45.
Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19(16):3206–18.
Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci. 2010;30(48):16208–19.
Vaccaro A, Tauffenberger A, Ash PE, Carlomagno Y, Petrucelli L, Parker JA. TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012;8(7):e1002806.
Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19(4):671–83.
Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE. 2013;8(12):e83450.
Corrionero A, Horvitz HR. A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr Biol. 2018;28(10):1522–35.
Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018;135(3):427–43.
Shaw MP, Higginbottom A, McGown A, Castelli LM, James E, Hautbergue GM, et al. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol Commun. 2018;6(1):125.
Bolus H, Crocker K, Boekhoff-Falk G, Chtarbanova S. Modeling neurodegenerative disorders in Drosophila melanogaster. Int J Mol Sci. 2020;21(9):3055.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–95.
Azuma Y, Mizuta I, Tokuda T, Mizuno T. Amyotrophic lateral sclerosis model. Adv Exp Med Biol. 2018;1076:79–95.
Watson MR, Lagow RD, Xu K, Zhang B, Bonini NM. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283(36):24972–81.
Elia AJ, Parkes TL, Kirby K, St George-Hyslop P, Boulianne GL, Phillips JP, et al. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic Biol Med. 1999;26(9–10):1332–8.
Bahadorani S, Mukai ST, Rabie J, Beckman JS, Phillips JP, Hilliker AJ. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol Aging. 2013;34(10):2322–30.
Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–15.
Sahin A, Held A, Bredvik K, Major P, Achilli TM, Kerson AG, et al. Human SOD1 ALS mutations in a drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics. 2017;205(2):707–23.
Chen Y, Yang M, Deng J, Chen X, Ye Y, Zhu L, et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell. 2011;2(6):477–86.
Sasayama H, Shimamura M, Tokuda T, Azuma Y, Yoshida T, Mizuno T, et al. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE. 2012;7(6):e39483.
Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet. 2016;25(12):2378–92.
Lanson NA Jr, Maltare A, King H, Smith R, Kim JH, Taylor JP, et al. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20(13):2510–23.
Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Investig. 2011;121(10):4118–26.
Steyaert J, Scheveneels W, Vanneste J, Van Damme P, Robberecht W, Callaerts P, et al. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum Mol Genet. 2018;27(23):4103–16.
Fallini C, Khalil B, Smith CL, Rossoll W. Traffic jam at the nuclear pore: all roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol Dis. 2020;140:104835.
Mallik M, Catinozzi M, Hug CB, Zhang L, Wagner M, Bussmann J, et al. Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicity. J Cell Biol. 2018;217(11):3947–64.
Wakisaka KT, Tanaka R, Hirashima T, Muraoka Y, Azuma Y, Yoshida H, et al. Novel roles of Drosophila FUS and Aub responsible for piRNA biogenesis in neuronal disorders. Brain Res. 2019;1708:207–19.
Casci I, Krishnamurthy K, Kour S, Tripathy V, Ramesh N, Anderson EN, et al. Muscleblind acts as a modifier of FUS toxicity by modulating stress granule dynamics and SMN localization. Nat Commun. 2019;10(1):5583.
Azuma Y, Tokuda T, Kushimura Y, Yamamoto I, Mizuta I, Mizuno T, et al. Hippo, Drosophila MST, is a novel modifier of motor neuron degeneration induced by knockdown of Caz, Drosophila FUS. Exp Cell Res. 2018;371(2):311–21.
Gogia N, Sarkar A, Mehta AS, Ramesh N, Deshpande P, Kango-Singh M, et al. Inactivation of Hippo and cJun-N-terminal Kinase (JNK) signaling mitigate FUS mediated neurodegeneration in vivo. Neurobiol Dis. 2020;140:104837.
Estes PS, Boehringer A, Zwick R, Tang JE, Grigsby B, Zarnescu DC. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum Mol Genet. 2011;20(12):2308–21.
Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, et al. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci USA. 2010;107(7):3169–74.
Lin MJ, Cheng CW, Shen CK. Neuronal function and dysfunction of Drosophila dTDP. PLoS ONE. 2011;6(6):e20371.
Langellotti S, Romano V, Romano G, Klima R, Feiguin F, Cragnaz L, et al. A novel Drosophila model of TDP-43 proteinopathies: N-terminal sequences combined with the Q/N domain induce protein functional loss and locomotion defects. Dis Model Mech. 2016;9(6):659–69.
Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583(10):1586–92.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–34.
Sun X, Duan Y, Qin C, Li JC, Duan G, Deng X, et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018;9(10):953.
Khalil B, Cabirol-Pol MJ, Miguel L, Whitworth AJ, Lecourtois M, Lievens JC. Enhancing mitofusin/marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol Aging. 2017;54:71–83.
Manzo E, Lorenzini I, Barrameda D, O’Conner AG, Barrows JM, Starr A, et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife. 2019;8:e45114.
Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, et al. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci. 2014;34(48):15962–74.
Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019;29(3):233–47.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–33.
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–4.
Goodman LD, Prudencio M, Kramer NJ, Martinez-Ramirez LF, Srinivasan AR, Lan M, et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci. 2019;22(6):863–74.
Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science. 2016;353(6300):708–12.
Goodman LD, Bonini NM. New roles for canonical transcription factors in repeat expansion diseases. Trends Genet. 2020;36(2):81–92.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61.
Cunningham KM, Maulding K, Ruan K, Senturk M, Grima JC, Sung H, et al. TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife. 2020;9:e59419.
Goodman LD, Prudencio M, Srinivasan AR, Rifai OM, Lee VM, Petrucelli L, et al. eIF4B and eIF4H mediate GR production from expanded G4C2 in a Drosophila model for C9orf72-associated ALS. Acta Neuropathol Commun. 2019;7(1):62.
Azoulay-Ginsburg S, Di Salvio M, Weitman M, Afri M, Ribeiro S, Ebbinghaus S, et al. Chemical chaperones targeted to the endoplasmic reticulum (ER) and lysosome prevented neurodegeneration in a C9orf72 repeat expansion drosophila amyotrophic lateral sclerosis (ALS) model. Pharmacol Rep. 2021;73(2):536–50.
Canto MD, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu, Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta neuropathol. 1997;93:537–50.
Quarta E, Bravi R, Scambi I, Mariotti R, Minciacchi D. Increased anxiety-like behavior and selective learning impairments are concomitant to loss of hippocampal interneurons in the presymptomatic SOD1 (G93A) ALS mouse model. J Comp Neurol. 2015;523(11):1622–38.
Tafuri F, Ronchi D, Magri F, Comi GP, Corti S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci. 2015;9:336.
Liu Y-C, Chiang P-M, Tsai K-J. Disease animal models of TDP-43 proteinopathy and their pre-clinical applications. Int J Mol Sci. 2013;14(10):20079–111.
Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci USA. 2008;105(10):4022–7.
Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43(1):5–17.
Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, Kusaka H, et al. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph Later Scler Frontotempor Degener. 2000;1(3):163–84.
Jaarsma D, Haasdijk ED, Grashorn J, Hawkins R, van Duijn W, Verspaget HW, et al. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7(6):623–43.
Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(9):2610–26.
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2009;106(44):18809–14.
Wang W, Arakawa H, Wang L, Okolo O, Siedlak SL, Jiang Y, et al. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Ther. 2017;25(1):127–39.
Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C, et al. Overexpression of ALS-associated p. M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol. 2013;48:22–35.
Ditsworth D, Maldonado M, McAlonis-Downes M, Sun S, Seelman A, Drenner K, et al. Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:907–22.
Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110(8):E736–45.
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010;107(8):3858–63.
Corcia P, Valdmanis P, Millecamps S, Lionnet C, Blasco H, Mouzat K, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. 2012;78(19):1519–26.
Efimova A, Ovchinnikov R, Roman AY, Maltsev A, Grigoriev V, Kovrazhkina E, et al. The FUS protein: physiological functions and a role in amyotrophic lateral sclerosis. Mol Biol. 2017;51:341–51.
Corcia P, Danel V, Lacour A, Beltran S, Andres C, Couratier P, et al. A novel mutation of the C-terminal amino acid of FUS (Y526C) strengthens FUS gene as the most frequent genetic factor in aggressive juvenile ALS. Amyotroph Later Scler Frontotempor Degener. 2017;18(3–4):298–301.
Nolan M, Talbot K, Ansorge O. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun. 2016;4(1):99.
Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age-and dose-dependent fashion. Acta Neuropathol. 2013;125(2):273–88.
Scekic-Zahirovic J, Oussini HE, Mersmann S, Drenner K, Wagner M, Sun Y, et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:887–906.
Scekic-Zahirovic J, Sanjuan-Ruiz I, Kan V, Megat S, De Rossi P, Dieterlé S, et al. Cytoplasmic FUS triggers early behavioral alterations linked to cortical neuronal hyperactivity and inhibitory synaptic defects. Nat Commun. 2021;12(1):3028.
Scekic-Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016;35(10):1077–97.
Yanovsky-Dagan S, Mor-Shaked H, Eiges R. Modeling diseases of noncoding unstable repeat expansions using mutant pluripotent stem cells. World J Stem Cells. 2015;7(5):823.
O’Rourke JG, Bogdanik L, Muhammad A, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901.
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y-J, Castanedes-Casey M, et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348(6239):1151–4.
Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14:1–15.
Herranz-Martin S, Chandran J, Lewis K, Mulcahy P, Higginbottom A, Walker C, et al. Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis Model Mech. 2017;10(7):859–68.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90(3):521–34.
Bruijn L, Beal M, Becher M, Schulz J, Wong P, Price D, et al. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997;94(14):7606–11.
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105–16.
Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2012;131(4):535–63.
McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gend Med. 2010;7(6):557–70.
Bame M, Pentiak PA, Needleman R, Brusilow WS. Effect of sex on lifespan, disease progression, and the response to methionine sulfoximine in the SOD1 G93A mouse model for ALS. Gend Med. 2012;9(6):524–35.
Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133(6):863–85.
Joyce PI, Mcgoldrick P, Saccon RA, Weber W, Fratta P, West SJ, et al. A novel SOD1-ALS mutation separates central and peripheral effects of mutant SOD1 toxicity. Hum Mol Genet. 2015;24(7):1883–97.
Jonsson PA, Graffmo KS, Brännström T, Nilsson P, Andersen PM, Marklund SL. Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol. 2006;65(12):1126–36.
Philips T, Rothstein JD. Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol. 2015;69:56721–656761.
Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045–53.
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002;99(3):1604–9.
Sanagi T, Yuasa S, Nakamura Y, Suzuki E, Aoki M, Warita H, et al. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J Neurosci Res. 2010;88(12):2736–46.
Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, et al. Rats expressing human cytosolic copper–zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21(23):9246–54.
Magota H, Sasaki M, Kataoka-Sasaki Y, Oka S, Ukai R, Kiyose R, et al. Intravenous infusion of mesenchymal stem cells delays disease progression in the SOD1G93A transgenic amyotrophic lateral sclerosis rat model. Brain Res. 2021;1757:147296.
Lutz C. Mouse models of ALS: past, present and future. Brain Res. 2018;1693(Pt A):1–10.
Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30(32):10851–9.
Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73.
Alfieri JA, Silva PR, Igaz LM. Early cognitive/social deficits and late motor phenotype in conditional wild-type TDP-43 transgenic mice. Front Aging Neurosci. 2016;8:310.
Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CKJ. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48(1):56–62.
Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):211–20.
Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci USA. 2010;107(37):16325–30.
Mitchell JC, Constable R, So E, Vance C, Scotter E, Glover L, et al. Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol Commun. 2015;3:36.
Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Investig. 2012;122(1):107–18.
Yan S, Wang CE, Wei W, Gaertig MA, Lai L, Li S, et al. TDP-43 causes differential pathology in neuronal versus glial cells in the mouse brain. Hum Mol Genet. 2014;23(10):2678–93.
Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887.
Liu YJ, Tsai PY, Chern Y. Energy homeostasis and abnormal RNA metabolism in amyotrophic lateral sclerosis. Front Cell Neurosci. 2017;11:126.
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Qiu H, Lee S, Shang Y, Wang W-Y, Au KF, Kamiya S, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Investig. 2014;124(3):981–99.
Gois AM, Mendonca DMF, Freire MAM, Santos JR. In vitro and in vivo models of amyotrophic lateral sclerosis: an updated overview. Brain Res Bull. 2020;159:32–43.
Ling SC, Dastidar SG, Tokunaga S, Ho WY, Lim K, Ilieva H, et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife. 2019;8:e40811.
Jackson KL, Dhaibar HA, Dayton RD, Cananzi SG, Mayhan WG, Glasscock E, et al. Severe respiratory changes at end stage in a FUS-induced disease state in adult rats. BMC Neurosci. 2016;17(1):1–11.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110(19):7778–83.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50.
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88(5):902–9.
Batra R, Lee CW. Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/frontotemporal dementia. Front Cell Neurosci. 2017;11:196.
Mordes DA, Morrison BM, Ament XH, Cantrell C, Mok J, Eggan P, et al. Absence of survival and motor deficits in 500 repeat C9ORF72 BAC mice. Neuron. 2020;108(4):775–83.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–24.
Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–25.
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, et al. C 9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78(3):426–38.
Dong W, Ma Y, Guan F, Zhang X, Chen W, Zhang L, et al. Ablation of C9orf72 together with excitotoxicity induces ALS in rats. FEBS J. 2021;288(5):1712–23.
Dong W, Zhang L, Sun C, Gao X, Guan F, Li J, et al. Knock in of a hexanucleotide repeat expansion in the C9orf72 gene induces ALS in rats. Animal Model Exp Med. 2020;3(3):237–44.
Zoghbi HY, Botas J. Mouse and fly models of neurodegeneration. Trends Genet. 2002;18(9):463–71.
Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci. 2018;21(10):1332–40.
Crook ZR, Housman D. Huntington’s disease: Can mice lead the way to treatment? Neuron. 2011;69(3):423–35.
Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111(3):696–702.
Averill DR Jr. Degenerative myelopathy in the aging German Shepherd dog: clinical and pathologic findings. J Am Vet Med Assoc. 1973;162(12):1045–51.
Nardone R, Holler Y, Taylor AC, Lochner P, Tezzon F, Golaszewski S, et al. Canine degenerative myelopathy: a model of human amyotrophic lateral sclerosis. Zoology (Jena). 2016;119(1):64–73.
Engel WK, Kurland LT, Klatzo I. An inherited disease similar to amyotrophic lateral sclerosis with a pattern of posterior column involvement. An intermediate form? Brain. 1959;82:203–20.
Hirano A, Kurland LT, Sayre GP. Familial amyotrophic lateral sclerosis. A subgroup characterized by posterior and spinocerebellar tract involvement and hyaline inclusions in the anterior horn cells. Arch Neurol. 1967;16(3):232–43.
Awano T, Johnson GS, Wade CM, Katz ML, Johnson GC, Taylor JF, et al. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2009;106(8):2794–9.
Wininger FA, Zeng R, Johnson GS, Katz ML, Johnson GC, Bush WW, et al. Degenerative myelopathy in a Bernese Mountain Dog with a novel SOD1 missense mutation. J Vet Intern Med. 2011;25(5):1166–70.
Crisp MJ, Beckett J, Coates JR, Miller TM. Canine degenerative myelopathy: biochemical characterization of superoxide dismutase 1 in the first naturally occurring non-human amyotrophic lateral sclerosis model. Exp Neurol. 2013;248:1–9.
Golubczyk D, Malysz-Cymborska I, Kalkowski L, Janowski M, Coates JR, Wojtkiewicz J, et al. The role of glia in canine degenerative myelopathy: relevance to human amyotrophic lateral sclerosis. Mol Neurobiol. 2019;56(8):5740–8.
Toedebusch CM, Snyder JC, Jones MR, Garcia VB, Johnson GC, Villalon EL, et al. Arginase-1 expressing microglia in close proximity to motor neurons were increased early in disease progression in canine degenerative myelopathy, a model of amyotrophic lateral sclerosis. Mol Cell Neurosci. 2018;88:148–57.
Fernandez-Trapero M, Espejo-Porras F, Rodriguez-Cueto C, Coates JR, Perez-Diaz C, de Lago E, et al. Upregulation of CB(2) receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis Model Mech. 2017;10(5):551–8.
Bendixen E, Danielsen M, Larsen K, Bendixen C. Advances in porcine genomics and proteomics–a toolbox for developing the pig as a model organism for molecular biomedical research. Brief Funct Genom. 2010;9(3):208–19.
Lind NM, Moustgaard A, Jelsing J, Vajta G, Cumming P, Hansen AK. The use of pigs in neuroscience: modeling brain disorders. Neurosci Biobehav Rev. 2007;31(5):728–51.
Bjarkam CR, Nielsen MS, Glud AN, Rosendal F, Mogensen P, Bender D, et al. Neuromodulation in a minipig MPTP model of Parkinson disease. Br J Neurosurg. 2008;22(Suppl 1):S9-12.
Kragh PM, Nielsen AL, Li J, Du Y, Lin L, Schmidt M, et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res. 2009;18(4):545–58.
Uchida M, Shimatsu Y, Onoe K, Matsuyama N, Niki R, Ikeda JE, et al. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res. 2001;10(6):577–82.
Holm IE, Alstrup AK, Luo Y. Genetically modified pig models for neurodegenerative disorders. J Pathol. 2016;238(2):267–87.
Chieppa MN, Perota A, Corona C, Grindatto A, Lagutina I, Vallino Costassa E, et al. Modeling amyotrophic lateral sclerosis in hSOD1 transgenic swine. Neurodegener Dis. 2014;13(4):246–54.
Yang H, Wang G, Sun H, Shu R, Liu T, Wang CE, et al. Species-dependent neuropathology in transgenic SOD1 pigs. Cell Res. 2014;24(4):464–81.
Crociara P, Chieppa MN, Vallino Costassa E, Berrone E, Gallo M, Lo Faro M, et al. Motor neuron degeneration, severe myopathy and TDP-43 increase in a transgenic pig model of SOD1-linked familiar ALS. Neurobiol Dis. 2019;124:263–75.
Wang G, Yang H, Yan S, Wang CE, Liu X, Zhao B, et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol Neurodegener. 2015;10:42.
Dolezalova D, Hruska-Plochan M, Bjarkam CR, Sorensen JC, Cunningham M, Weingarten D, et al. Pig models of neurodegenerative disorders: utilization in cell replacement-based preclinical safety and efficacy studies. J Comp Neurol. 2014;522(12):2784–801.
Bravo-Hernandez M, Tadokoro T, Navarro MR, Platoshyn O, Kobayashi Y, Marsala S, et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med. 2020;26(1):118–30.
Verdier JM, Acquatella I, Lautier C, Devau G, Trouche S, Lasbleiz C, et al. Lessons from the analysis of nonhuman primates for understanding human aging and neurodegenerative diseases. Front Neurosci. 2015;9:64.
Herculano-Houzel S, Mota B, Wong P, Kaas JH. Connectivity-driven white matter scaling and folding in primate cerebral cortex. Proc Natl Acad Sci USA. 2010;107(44):19008–13.
Zhu Y, Sousa AM, Gao T, Skarica M, Li M, Santpere G, et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science. 2018;362(6420):eaat8077.
Uchida A, Sasaguri H, Kimura N, Tajiri M, Ohkubo T, Ono F, et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain. 2012;135(Pt 3):833–46.
Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabrera GC, Song L, et al. Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1(G93A) mice and nonhuman primates. Hum Gene Ther. 2016;27(1):19–31.
Borel F, Gernoux G, Sun H, Stock R, Blackwood M, Brown RH Jr, et al. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci Transl Med. 2018;10(465):eaau6414.
Endo K, Ishigaki S, Masamizu Y, Fujioka Y, Watakabe A, Yamamori T, et al. Silencing of FUS in the common marmoset (Callithrix jacchus) brain via stereotaxic injection of an adeno-associated virus encoding shRNA. Neurosci Res. 2018;130:56–64.
Yin P, Guo X, Yang W, Yan S, Yang S, Zhao T, et al. Caspase-4 mediates cytoplasmic accumulation of TDP-43 in the primate brains. Acta Neuropathol. 2019;137(6):919–37.
Yin P, Bai D, Deng F, Zhang C, Jia Q, Zhu L, et al. SQSTM1-mediated clearance of cytoplasmic mutant TARDBP/TDP-43 in the monkey brain. Autophagy. 2022;18(8):1955–68.
Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34(2):177–80.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (32270564, 81830032, 82071421), Department of Science and Technology of Guangdong Province (2021ZT09Y007, 2018B030337001), Guangzhou Key Research Program on Brain Science (202007030008), Guangdong Basic and Applied Basic Research (2023A1515010811, 2022A1515011205), and the Fundamental Research Funds for the Central Universities (21622113).
Author information
Authors and Affiliations
Contributions
LZ and PY wrote the manuscript. X-JL and SL edited the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, L., Li, S., Li, XJ. et al. Pathological insights from amyotrophic lateral sclerosis animal models: comparisons, limitations, and challenges. Transl Neurodegener 12, 46 (2023). https://doi.org/10.1186/s40035-023-00377-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00377-7