
Abstract
Mutations in genes that encode RNA-binding proteins (RBPs) have emerged as critical determinants of neurological diseases, especially motor neuron disorders such as amyotrophic lateral sclerosis (ALS). RBPs are involved in all aspects of RNA processing, controlling the life cycle of RNAs from synthesis to degradation. Hallmark features of RBPs in neuron dysfunction include misregulation of RNA processing, mislocalization of RBPs to the cytoplasm, and abnormal aggregation of RBPs. Much progress has been made in understanding how ALS-associated mutations in RBPs drive pathogenesis. Here, we focus on several key RBPs involved in ALS—TDP-43, HNRNP A2/B1, HNRNP A1, FUS, EWSR1, and TAF15—and review our current understanding of how mutations in these proteins cause disease.
Similar content being viewed by others

Introduction
RBPs play a central role in mediating post-transcriptional gene expression. They directly bind to RNA and control all aspects of RNA processing, including RNA synthesis, maturation, localization, translation, and decay. A genome-wide survey of RBPs in the developing mouse nervous system revealed that two-thirds of RBPs are expressed in a cell-type specific manner (McKee et al. 2005). This specialized expression of RBPs is thought to maintain a diverse gene expression profile to support the varied and complex functions of neurons. An increasing number of RBPs are being recognized as causal drivers or associated with neurological diseases (Polymenidou et al. 2012; Belzil et al. 2013; Nussbacher et al. 2015), which reinforces the importance of RBPs in maintaining normal physiology in the nervous system.
Mutations in genes that encode RBPs have been observed in patients with motor neuron disorders such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), multisystem proteinopathy (MSP) and frontotemporal lobar degeneration (FTLD). In adults, ALS is the most common motor neuron disorder and is characterized by progressive loss of upper and lower motor neurons, which leads to fatal paralysis (Eykens and Robberecht 2015). The causes of ALS remain largely unknown, with 90% of cases being sporadic and the remaining 10% having a hereditary component (Pasinelli and Brown 2016). Model organisms (mouse, yeast, and drosophila) and human cells [patient tissue or neurons generated from patient-derived induced pluripotent stem cells (iPSCs)] have enabled significant progress in dissecting the normal and pathological functions of ALS-linked RBPs.
There are more than one hundred genes associated with ALS, a handful of which encode proteins that control RNA processing (Wroe et al. 2008). Here, we focus on several RBPs that are known to be mutated in patients with ALS: TAR DNA-binding protein 43 (TDP-43), heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP A2/B1), fused in sarcoma/translocated in liposarcoma (FUS/TLS, herein referred to as FUS), Ewing’s sarcoma breakpoint region 1 (EWSR1), and TAF15 (TATA-box binding protein associated factor 15) (Fig. 1). There are many commonalities between these proteins. TDP-43, hnRNP A1, and hnRNP A2/B1 are structurally similar in that they contain two RNA recognition motifs (RRMs) and a Gly-rich C-terminal domain (Fig. 1) (He and Smith 2009). FUS, EWSR1 and TAF15, which form the FET family, also share a similar structure: a zinc finger domain and RRM that facilitates DNA and RNA binding, respectively, an N-terminal low complexity, prion-like domain that mediates protein interactions and self-assembly (Han et al. 2012; Kato et al. 2012; Kwon et al. 2013; Schwartz et al. 2012), multiple C-terminal Arg-Gly-Gly (RGG) domains that facilitate non-specific RNA binding and protein interactions, and an atypical Pro-Tyr nuclear localization signal (PY-NLS) that is recognized by transportin to control nuclear–cytoplasmic shuttling (Lee et al. 2006) (Fig. 1). They are predominantly nuclear, multifunctional proteins that are widely expressed in most cell and tissue types and are implicated in a broad range of cellular processes. The importance of these proteins is exemplified by the observations that compete loss of TDP-43 or hnRNP A1 in mice is embryonic lethal (Kraemer et al. 2010; Liu et al. 2017) while complete loss of FUS or EWSR1 in mice is postnatal lethal (Hicks et al. 2000; Li et al. 2007). Disease-related mutations in these RBP have been reported to alter the normal functions of the protein in various steps of RNA processing (Fig. 2). In this review, we discuss their normal functions in RNA processing, their connection to ALS, and speculate how ALS-associated mutations in the genes that encode these proteins may contribute to pathogenesis.

Mutation and RNA interaction maps for ALS-associated RNA-binding proteins (RBPs). The locations of mutations identified in familial and sporadic ALS patients are mapped against the domain structure of the RBP. Mutations that cause a change in the protein sequence—missense mutations, frame shifts, and deletions—are reported. Points of contact between RBPs and mRNA are shown (TDP-43, Buratti and Baralle 2001; all others, Castello et al. 2016)
TDP-43
Roles in RNA processing
Initially described as a transcription factor (Ou et al. 1995), TDP-43 is now better known for its role in RNA processing. Several groups have used crosslinking and immunoprecipitation followed by sequencing (CLIP-seq) to identify the global RNA targets of TDP-43 and showed that TDP-43 binds to thousands of RNAs (Tollervey et al. 2011; Xiao et al. 2011; Polymenidou et al. 2011; Sephton et al. 2011). Two RRMs in TDP-43 mediate the protein’s interaction with RNA (Fig. 1) with the first RRM (RRM1) being necessary and sufficient to bind RNA (Buratti and Baralle 2001). TDP-43 shows a strong preference for UG-repeat motifs, but also binds to non-UG sequences. A critical function of TDP-43 is controlling alternative splicing. Indeed, TDP-43 interacts with proteins involved in splicing (Freibaum et al. 2010) and a loss of TDP-43 causes widespread changes in alternative splicing (Polymenidou et al. 2011; Tollervey et al. 2011). The majority of TDP-43 binding occurs in introns (Polymenidou et al. 2011; Xiao et al. 2011; Tollervey et al. 2011), but when localized to the cytoplasm TDP-43 is mostly bound within 3′UTR of mRNAs (Colombrita et al. 2012). A concentration of TDP-43 binding sites near 5′ or 3′ splice sites of several exons was associated with both splicing repression (Buratti et al. 2001; Passoni et al. 2012; Mercado et al. 2005) and splicing activation (Fiesel et al. 2012).
Under physiological conditions, TDP-43 is predominantly nuclear but shuttles to and from the cytoplasm (Ayala et al. 2008b; Colombrita et al. 2012). Both Tollervey et al. (2011) and Colombrita et al. (2012) observed that the majority of cytoplasmic TDP-43 bound to the 3′UTRs of target mRNAs, suggesting a role in RNA transport, translation, and/or stability. Indeed, a large fraction of proteins that co-purify with TDP-43 are involved in RNA transport and translation (Freibaum et al. 2010). In neurons, the pathways that control RNA trafficking and translation are intimately linked to control the transport of select mRNAs from the soma to distal axonal and dentritic compartments, including neuromuscular junctions (Bramham and Wells 2007). While in transit, mRNAs are translationally repressed, and only upon reaching their synaptic destination are signals received that activate translation (Liu-Yesucevitz et al. 2011). TDP-43 is present in RNA granules and promotes anterograde axonal transport (from soma to axonal compartments) and translational repression for a subset of target mRNAs (Wang et al. 2008a; Alami et al. 2014). Translation is also tightly regulated during cellular stress. Stress granules are cytoplasmic protein–RNA assemblies that form in response to cellular stress and contain non-translating mRNA and various proteins, including TDP-43 (Li et al. 2013). TDP-43 associates with mRNAs that are bound by stalled ribosomes during cellular stress (Higashi et al. 2013) and this association may be mediated by the ribosomal scaffold protein RACK1 (Receptor Activated C Kinase 1) (Russo et al. 2017). TDP-43 is also reported to control RNA stability through several mechanisms. TDP-43 negatively regulates its own transcript through a mechanism that requires binding to the 3′UTR of TDP43 mRNA to promote degradation, possibly by the exosome (Ayala et al. 2008a; Ayala et al. 2011). TDP-43 has also been shown to regulate its own expression by mediating alternative splicing of an exon in the TDP43 mRNA 3′UTR, making the transcript a substrate for nonsense-mediated decay (Polymenidou et al. 2011).
Disease association
TDP-43 was first associated with autosomal dominant ALS (and FTLD) in 2006. It was identified as a major component of ubiquitinated cytoplasmic inclusions in spinal motor neurons from ALS patients (Neumann et al. 2006; Arai et al. 2006). The clinical and pathological features of ALS associated with TDP-43 are highly variable, presenting as a spectrum of disorders collectively referred to as TDP-43 proteinopathies (Geser et al. 2010). Mutations in TARDBP were later identified in sporadic and familial ALS cases (Sreedharan et al. 2008). More than 50 missense mutations have been identified in TARDBP in ALS, accounting for 1–2% of total ALS cases (Van Deerlin et al. 2008; Gitcho et al. 2008; Sreedharan et al. 2008; Kabashi et al. 2008; Yokoseki et al. 2008; Millecamps et al. 2010; Corrado et al. 2009; Xiong et al. 2010; Kirby et al. 2010; Nozaki et al. 2010; Chiang et al. 2012; Orrù et al. 2012; Lemmens et al. 2009; Del Bo et al. 2009; Kamada et al. 2009; Zou et al. 2012; Iida et al. 2012; Huey et al. 2012; Rutherford et al. 2008; Kühnlein et al. 2008; Fujita et al. 2011; Ticozzi et al. 2011b; Tsai et al. 2011; Origone et al. 2010; Daoud et al. 2009; Chio et al. 2012). The majority of TARDBP mutations cluster in the Gly-rich domain (Fig. 1), a region that mediates protein–protein interactions (Buratti et al. 2005). The common features of TDP-43 proteinopathies are an accumulation of TDP-43 in insoluble, cytoplasmic inclusions, concomitant with a loss of nuclear TDP-43, the presence of truncated 20–25 kDa and 35 TDP-43 C-terminal fragments, and abnormal phosphorylation and ubiquitination of TDP-43 in the upper and lower motor neurons and in other regions of the central nervous system (Geser et al. 2010; Mackenzie et al. 2010).
Mechanisms of pathogenicity
Many studies have been performed to determine how mutations in TARDBP cause neuron dysfunction, in particular testing the possibility of a loss of nuclear function, a gain of cytoplasmic function, or both (reviewed in Lee et al. 2012; Buratti 2015). TDP-43 has multiple roles in RNA processing that influence global changes in gene expression (Ratti and Buratti 2016), and mutations in TARDBP can alter gene expression, for example by affecting alternative splicing (Arnold et al. 2013; Highley et al. 2014). It is unclear whether mutant forms of TDP-43 have different affinities for target RNAs. RNA binding is required for TDP-43-mediated toxicity since mutations in TDP-43 that disrupt RNA binding (F147L and F149L) can prevent TDP-43-mediated toxicity (Voigt et al. 2010). Interestingly, ALS-associated variants of TDP-43 caused neural defects in Drosophila to varying degrees but did not exacerbate the neural defects caused by ectopic expression wild-type TDP-43 alone. This may reflect a dose-dependent effect of TDP-43-mediated toxicity (Voigt et al. 2010). TDP-43 variants can increase the half-life of the protein (Ling et al. 2010; Barmada et al. 2010; Watanabe et al. 2013). The exact mechanism by which mutant forms of TDP-43 affect its degradation is not clear, but there is evidence to suggest that ALS-associated mutations in TARDBP, most of which reside in the disordered prion-like domain, promote protein misfolding and stabilize TDP-43 within cytoplasmic inclusions (Kabashi et al. 2010; Johnson et al. 2009). TDP-43 variants affect RNA processing to varying extents and it is unclear how much of this functional variation is attributed to dosage. Several mutant forms of TDP-43 showed impaired axonal transport of RNA (Alami et al. 2014) and TDP-43 Q331K preferentially enhanced exon exclusion for select target pre-mRNAs (Arnold et al. 2013). Mutant TDP-43 protein may also influence gene expression by altering its association with other proteins. Ling et al. (2010) showed that TDP-43 Q331K and M337V mutants had enhanced binding to FUS, another RBP implicated in ALS (discussed below). TDP-43 and FUS co-regulate HDAC6 mRNA in a manner that requires the mutation-prone Gly-rich domain of TDP-43 (Kim et al. 2010). While this is but one example of co-regulated gene expression by TDP-43 and FUS, both proteins have thousands of shared RNA targets (Lagier-Tourenne et al. 2012), and more examples of TDP-43/FUS co-regulation are likely to emerge.
Cytoplasmic mislocalization of TDP-43 is a well-established feature of neurological diseases but not an absolute requirement for cell toxicity. Some studies have shown that mutations in TDP-43 promote mislocalization to various subcellular locations in the cytoplasm (Johnson et al. 2009; Barmada et al. 2010), while other studies have shown that TDP-43 mutants are retained in the nucleus (Arnold et al. 2013). Clearance of TDP-43 from the nucleus can cause numerous downstream effects that lead to neurotoxicity. Wang et al. (2016a) showed that TDP-43 localized to mitochondria and repressed the expression of mitochondrial mRNAs. Interestingly, mutant forms of TDP-43 had increased mislocalization to the mitochondria, suggesting that mutant TDP-43 can cause greater mitochondria dysfunction (Wang et al. 2016a). In rat hippocampal neurons, cytoplasmic TDP-43 resides within RNA granules that travel to dendritic arbors upon depolarization (Wang et al. 2008a); however, ALS-associated mutant forms of TDP-43 (A315T and Q343R) severely reduced the movement of the granules to dendrites (Liu-Yesucevitz et al. 2014; Alami et al. 2014), likely preventing the transport and therefore local translation of mRNAs encoding proteins that are required for proper synaptic function. Nuclear clearance of TDP-43 into the cytoplasm is often accompanied by the formation of protein aggregates. Aggregation of TDP-43 protein is dependent on the C-terminal domain where the majority of ALS-associated mutations occur. Interestingly, Johnson et al. (2009) observed that mutant TDP-43 proteins accelerated TDP-43 aggregation in yeast; however, it is unclear if insoluble aggregates in the cytoplasm confer toxicity or are secondary effects. The accumulation of TDP-43 into insoluble cytoplasmic foci may be a consequence of nuclear clearance since abolishing one or both nuclear localization signals in TDP-43 are sufficient to cause cytoplasmic aggregation (Winton et al. 2008; Igaz et al. 2011). It is important to note that TDP-43 aggregation in the cytoplasm is not necessary to confer cellular toxicity (Barmada et al. 2010).
TDP-43 was recently reported to undergo phase transition and form liquid droplets: membrane-less organelles that contain a morphologically, physically, and functionally distinct sub-structure (Molliex et al. 2015; discussed in more detail below). Membrane-less organelles include structures such as stress granules to which TDP-43 and cellular toxicity are strongly linked (Bentmann et al. 2013; Li et al. 2013). The ability of TD-43 to form liquid droplets, at least in the nucleus, was independent of RNA binding, but rather required the prion-like QGSY-rich domain and low-complexity Gly-rich domain (Schmidt and Rohatgi 2016). Interestingly, TDP-43 variants M337V, N345K, or A382T altered the fluid characteristics of the TDP-43-containing liquid droplets. Schmidt and Rohatgi (2016) found that the exchange of TDP-43 N345K and A382T monomers between droplets and the nucleoplasm was impaired. In contrast, TDP-43 M337V monomers did not rearrange within the droplet to the same extent as wild-type TDP-43. M337V resides within a region of the low-complexity domain that forms an α-helix structure. This α-helix structure was reported to be necessary to form liquid droplets (Conicella et al. 2016) and it is possible that M337V changes the α-helix structure to alter the liquid droplet properties of TDP-43. M337V, N345V, and A382T TDP-43 variants had different effects on the phase transition properties of TDP-43 and it will be of interest to determine how other ALS-associated mutations affect the phase transition properties of TDP-43.
HNRNP A1
Roles in RNA processing
Heterogeneous nuclear ribonucleoprotein (hnRNP) A1 is a member of the hnRNP family, a group of related proteins known to associate with nascent mRNA (Dreyfuss et al. 1993; Jean-Philippe et al. 2013; Piñol-Roma et al. 1988). HnRNPs generally participate in many stages of the mRNA life cycle and hnRNP A1 is no exception. Previous reports have implicated hnRNP A1 in constitutive (Jurica et al. 2002; Tavanez et al. 2012; Zhou et al. 2002) and alternative splicing (Mayeda and Krainer 1992; Fisette et al. 2010). HnRNP A1 associates directly with the spliceosome as well as with a variety of exonic and intronic sequences (Han et al. 2005). Binding is typically associated with repression of nearby exons (Blanchette and Chabot 1999; Nasim et al. 2002). In certain cases, hnRNP A1 and other hnRNPs compete with serine-rich proteins to regulate alternative splicing in an opposing fashion (Kashima and Manley 2003; Cartegni and Krainer 2002; Dirksen et al. 2000; Sun et al. 1993). Synergistic control of alternative splicing has been demonstrated through direct interaction between hnRNP A1 and hnRNP H via their Gly-rich domains (Fisette et al. 2010). Similarly, functional genomic studies have revealed a broad collaboration between hnRNP A1 and other hnRNPs to control alternative splicing throughout the transcriptome (Huelga et al. 2012). Besides pre-mRNA splicing, hnRNP A1 has been linked to other mRNA processing events. HnRNP A1 is primarily nuclear, but nuclear–cytoplasmic shuttling is enabled through the M9 sequence (Mili et al. 2001; Nakielny and Dreyfuss 1999). This behavior suggests a role in nuclear export of mRNA (Gallouzi and Steitz 2001; Izaurralde et al. 1997). HnRNP A1 also plays a role in the regulation of mature mRNAs. HnRNP A1 has been reported to bind AU-rich sequences to control the stability of a number of transcripts (Henics et al. 1994; Hamilton et al. 1997). Internal Ribosomal Entry Sequence (IRES) mediated and cap-dependent translation is also regulated by hnRNP A1 (Cammas et al. 2007; Bonnal et al. 2005; Svitkin et al. 1996). Finally, hnRNP A1 regulates the processing of noncoding RNAs, especially miRNAs (Michlewski et al. 2008). HnRNP A1 was reported to specifically bind pri-miR-18a and the pri-let7a loop to either facilitate or inhibit pri-miRNA processing in a target-specific manner (Michlewski and Cáceres 2010; Guil and Cáceres 2007).
Disease association
To date, there are three reported cases of HNRNPA1 mutations linked to ALS or multisystem proteinopathy (MSP), a group of pleiotropic neurodegenerative disorders that includes ALS. Kim et al. (2013) described two families, one with MSP and another with ALS with the phenotype segregating according to an autosomal dominant mode of inheritance. The affected family members harbored damaging mutations in the Gly-rich domain of HNRNPA1. In one family affected by MSP, the HNRNPA1 (p. D262V) mutation was identified (Fig. 1). Strikingly, this aspartic residue is homologous to the D290 residue identified in hnRNP A2/B1 that is associated with ALS (discussed below). Recently, Liu et al. (2016) sequenced a Chinese family with “Flail arm ALS” and discovered a P288S mutation (P340S in the long isoform) in HNRNPA1 that segregated with the disease, according to an autosomal dominant mode of inheritance. In general, mutations in hnRNP A1 are not common and studies in European and other populations have failed to identify additional HNRNPA1 variants linked to ALS. In the case of ALS, the mechanisms by which HNRNPA1 mutations cause disease are not well understood (see below). However, in the case of Alzheimer’s disease, a simple link between hnRNP protein levels and disease has been established. Along with hnRNP A2/B1, hnRNP A1 was shown to be depleted in Alzheimer’s patient brains (Berson et al. 2012). These data linking hnRNP mutations and protein levels to multiple neurodegenerative diseases imply a strong connection between hnRNP homeostasis and neurological health.
Mechanisms of pathogenicity
The mechanisms of pathogenicity of hnRNP A1 variants are not well studied. It is unclear if damaging mutations represent gain or loss of function or both. Mutations in the region of HNRNPA1 that encode the Gly-rich domain are shown to strengthen “steric zippers” already present in the wild-type protein. This leads to increased fibrillization of the protein in vitro and an accumulation in stress granules in cell-based models (Kato et al. 2012; Kim et al. 2013; Molliex et al. 2015; Liu et al. 2016). HnRNP A1 has been reported to interact with other ALS-linked proteins, for example TDP-43, through its C-terminus. Honda et al. (2015) examined spinal cord motor neurons from ALS patients and found that cytoplasmic relocalization of TDP-43 was associated with loss of nuclear hnRNP A1. In another study, Gilpin et al. (2015) characterized a direct interaction between hnRNP A1 and ubiquilin-2 by yeast two-hybrid studies and other methods, showing that this interaction depended on the Gly-rich domain of hnRNP A1. Mutations in ubiquilin-2 as well as the hnRNP A1 D262V mutation reduced or eliminated hnRNP A1-ubiquilin-2 interactions. Finally, the authors demonstrated that ubiquilin-2-hnRNP A1 interactions may stabilize hnRNP A1 and increase its steady-state protein levels (Gilpin et al. 2015). Importantly, mutations in UBQLN2 are also responsible for X-linked ALS and FTLD (Deng et al. 2011). This implies that seemingly disparate ALS proteins may in fact regulate each other through the critical Gly-rich domain. These data are compelling considering a recent study that indicated that hnRNP A1 autoregulates its own alternative splicing. HnRNP A1 protein was shown to negatively regulate HNRNPA1 mRNA levels by inhibiting splicing of intron 10 (Suzuki and Matsuoka 2017). This mode of regulation was essential for maintaining the protein at a non-cytotoxic level. Although a definitive consensus for how mutant hnRNP A1 causes neurological dysfunction has not emerged, a significant amount of evidence has accumulated linking hnRNP A1 misfolding and fibrilization to disease. However, other studies indicating that ALS pathogenesis may simply be due to changes in hnRNP A1 levels should not be discounted. These hypotheses are not exclusive. Pathogenesis could very well result from some combination of toxic misfolded protein, reduced normal function due to accumulation in stress granules, and alterations in overall protein levels.
HNRNP A2/B1
Roles in RNA processing
A close relative of hnRNP A1, hnRNP A2/B1 exists in two distinct isoforms, A2 (341 amino acids) and B1 (353 amino acids), both transcribed from the HNRNPA2B1 gene. The putative functions of hnRNP A2/B1 include pre-mRNA splicing (Clower et al. 2010; Hutchison et al. 2002), mRNA trafficking (Gao et al. 2008; Raju et al. 2011; Shan et al. 2003; Fahling et al. 2006; Goodarzi et al. 2012), and translational control (Kosturko et al. 2006). Similar to its relatives TDP-43 and hnRNP A1, hnRNP A2/B1 contains two RRMs and has a Gly-rich domain near its C-terminal end (Fig. 1). HnRNP A2/B1 is primarily localized in the nucleus; however, nuclear–cytoplasmic trafficking does occur and is controlled in part by the M9 nuclear localization signal near the Gly-rich domain (Siomi et al. 1997). A previous report has also implicated hnRNP A2/B1 in miRNA processing and biogenesis (Alarcón et al. 2015). However, subsequent studies have found only minimal effects on mature miRNA expression (Martinez et al. 2016). The role of hnRNP A2/B1 in miRNA processing remains unclear and its effects may be context or cell-type specific. MiRNAs can be sorted into exosomes and then transported to the processes of oligodendrocytes in a manner that is mediated by hnRNP A2/B1 (Villarroya-Beltri et al. 2013). Putative targets of these exosomal miRNAs included BDNF and MBP mRNAs. There is a well-characterized role for hnRNP A2/B1 in trafficking mRNAs to neuronal dendrites, where hnRNP A2/B1 recognizes a 21-nucleotide A2 response element (A2RE) in target transcripts (Raju et al. 2011; Munro et al. 1999; Shan et al. 2000, 2003). In one report, hnRNP A2/B1 binding to an A2RE in the VEGFA 3′UTR promoted translational read-through and consequently increased the production of VEGF-Ax, an anti-angiogenic form of VEGFA (Eswarappa et al. 2014).
Disease association
To date, only a single mutation in HNRNPA2B1 (p. D290V) has been reported in a family with MSP, but not specifically ALS (Kim et al. 2013). The authors described a constellation of symptoms affecting the brain, motor neurons, muscle, and bone. These symptoms were previously characterized by a disorder called inclusion body myositis, Paget’s disease of blood and bone, and frontotemporal dementia (IBMPFD), now commonly referred to as MSP. Family members were affected according to an autosomal dominant mode of inheritance. It is notable that the pathogenic D290V mutation occurred within the Gly-rich domain of hnRNP A2/B1 (Fig. 1). This is akin to TDP-43 where most ALS-causing mutations also occur in the Gly-rich domain (Lagier-Tourenne et al. 2010). Drosophila models expressing hnRNP A2 D290V recapitulated the myopathy and protein inclusion pathology seen in patients (Kim et al. 2013). Other groups have attempted but were unable to identify other hnRNP A2/B1 mutations in European ALS and MSP populations (Seelen et al. 2014; Le Ber et al. 2014). A large study involving exome sequencing of 2869 ALS patients also failed to find additional ALS cases with mutations in HNRNPA2B1. This indicates that mutations in HNRNPA2B1 associated with ALS and MSP are exceedingly rare. Loss of hnRNP A2/B1 has been implicated in the pathogenicity of Alzheimer’s disease, although no associated genetic variants have been reported to date. Berson et al. (2012) found that depletion of hnRNP A2/B1 in mouse cortex resulted in impaired cognitive function and aberrant alternative splicing. The same study observed decreased levels of hnRNP A2/B1 and hnRNP A1 in the entorhinal cortex of patients with sporadic Alzheimer’s disease. In an in vivo experiment, ablation of cholinergic input to the entorhinal cortex resulted in reduced hnRNP A2/B1 expression and mis-splicing events consistent with those observed in post-mortem Alzheimer’s brain (Berson et al. 2012). These data link cholinergic neuron loss, a hallmark of Alzheimer’s disease, to reduced hnRNP levels and mis-splicing, which may in turn explain some of the cognitive deficits observed in Alzheimer’s disease.
Mechanisms of pathogenicity
Investigations on the mechanisms of hnRNP A2/B1 D290V toxicity are still at an early stage. Martinez et al. (2016) showed that hnRNP A2/B1 D290V is not equivalent to loss of hnRNP A2/B1 protein, at least with regard to alternative splicing. Several studies have reported increased aggregation and localization of hnRNP A2/B1 D290V to stress granules in cell-based systems (Kim et al. 2013; Martinez et al. 2016). Martinez et al. (2016) also showed that iPSC-derived motor neurons expressing hnRNP A2/B1 D290V had increased cell death and an exacerbated stress response. These data imply that hnRNP A2/B1 D290V may be a gain-of-function mutation with toxic properties. Biochemical experiments exploring fibrillization have confirmed that hnRNP A2/B1 D290V is more prone to self-seeding compared to its wild-type counterpart (Kato et al. 2012; Kim et al. 2013; Shorter and Taylor 2013). The pathogenicity of hnRNP A2/B1 D290V may lie in the protein’s ability to form fibrils and aggregates through interactions involving its Gly-rich domain. Pathogenic aggregates of proteins typically occur in the cytoplasm; however, an increase in nuclear-insoluble hnRNP A2/B1 was observed in a subset of ALS and MSP patients (Martinez et al. 2016). This implies that aggregation and fibrillization may also occur in the nucleus and could affect alternative splicing through the assembly of aberrant ribonucleoprotein complexes. It is unclear if and how nuclear aggregation of hnRNP A2/B1 D290V accounts for the alternative splicing changes caused by the mutant protein. In the CGG expanded repeat RNA disorder Fragile X-associated tremor/ataxia syndrome (FXTAS), pathogenicity may emerge through functional sequestration of hnRNP A2/B1 by toxic RNA repeats (Muslimov et al. 2011). CGG repeats were found to directly bind hnRNP A2/B1 and lead to mis-splicing (He et al. 2014; Blanchette et al. 2009). This effect was abrogated by ectopic expression of TDP-43 and was dependent on an intact C-terminal Gly-rich domain (He et al. 2014). These data raise interesting questions regarding the relevance of RBP protein–protein interactions and RBP-repeat interactions in ALS. Indeed, the intronic GGGGCC repeat expansion in C9orf72 is responsible for a large fraction of ALS cases (DeJesus-Hernandez et al. 2011; Renton et al. 2011). This hexanucleotide repeat is known to form G-quadruplexes that can bind hnRNP A2/B1 and hnRNP A1 (Mori et al. 2013; Sareen et al. 2013) and cause TDP-43 pathology when expressed in transgenic mice (Chew et al. 2015). Going forward, it will be important to determine how TDP-43, hnRNP A2/B1, and GGGGCC repeats interact to modulate the toxicity observed in C9orf72 linked ALS.
In Alzheimer’s disease, loss of hnRNP A2/B1 might generate pathogenicity independently of any aggregation or fibrilization. Extending the work of Berson et al. (2012) mentioned above, Kolisnyk et al. (2016) described cholinergic regulation of hnRNP A2/B1 through the M1 muscarinic receptor. The mechanism of protein reduction was identified as differential translation efficiency due to an autoregulatory splicing event in the HNRNPA2B1 3′UTR. Loss of muscarinic input biased the HNRNPA2B1 transcript toward an NMD-sensitive isoform with reduced translation efficiency (Kolisnyk et al. 2016; McGlincy et al. 2010). Importantly, the authors did not observe cytoplasmic aggregates or increased detergent insoluble fractions when they examined hnRNP A2/B1 in Vacht-deficient mice, indicating a pathogenic mechanism distinct from what is observed in ALS.
FUS
Role in RNA processing
FUS has been implicated in several RNA processing events, particularly transcription, alternative splicing, and mRNA trafficking (Lagier-Tourenne et al. 2010; Fujii and Takumi 2005). FUS carries out these functions by directly binding to RNA in a RRM-dependent manner (Daigle et al. 2013), but the Gly-rich domain may also interact with RNA (Fig. 1) (Castello et al. 2012). Using CLIP-based methods, several groups have identified the RNA targets of FUS in various contexts including human cell lines (Hoell et al. 2011), mouse cell lines (Ishigaki et al. 2012; Colombrita et al. 2012), and human or mouse brain tissue (Rogelj et al. 2012; Lagier-Tourenne et al. 2012). In two studies, FUS-binding sites were enriched for GGU motifs (Rogelj et al. 2012) or GUGGU motifs (Lagier-Tourenne et al. 2012), and a sequence preference for GGUG was been previously identified in vitro (Lerga et al. 2001). FUS appears to have limited sequence specificity, since both iCLIP and CLIP-seq studies identified a 2- to 3-fold enrichment for the GGU motif, which contrasts with a 15-fold enrichment for the GU-rich motif in TDP-43 iCLIP studies (Rogelj et al. 2012). This may explain why other studies did not identify a preferred FUS biding motif, but instead found that FUS preferred to bind RNA regions with AU-rich stem-loop structures (Hoell et al. 2011; Ishigaki et al. 2012).
The repertoire of FUS RNA targets is large with thousands of RNA targets and the RNA-binding patterns of FUS have been used to inform our understanding of its cellular functions. It was observed that FUS-binding sites frequently occurred in introns indicating a role in splicing. Indeed, a reduction in FUS caused global changes in alternative splicing (Lagier-Tourenne et al. 2012; Rogelj et al. 2012). Also, FUS interacts with several factors involved in splicing such as the U1–snRNP splicing complex, the SMN complex, SC35, SRSF1, SRSF3, and SRSF10 (Sun et al. 2015; Shang and Huang 2016). FUS-binding sites were also enriched in the 3′UTRs of target transcripts (Hoell et al. 2011; Lagier-Tourenne et al. 2012), which may allow FUS to mediate RNA localization and stability (Fujii and Takumi 2005; Kapeli et al. 2016).
FUS has additional roles in promoting transcription either by activating transcription of genes regulated by RNA polymerase II, or by blocking transcriptional repression of cyclin D1 and RNA polymerase III (Bertolotti et al. 1996; Hallier et al. 1998; Uranishi et al. 2001; Wang et al. 2015; Tan and Manley 2009; Schwartz et al. 2012; Kwon et al. 2013). FUS also has a role in maintaining genomic integrity through its interactions with PGC-1α (Sánchez-Ramos et al. 2011) and controlling the DNA damage response pathway through its interactions with CBP/p300 and HDAC1 (Wang et al. 2008b, 2013).
Disease associations
FUS was initially identified as an oncofusion protein in human liposarcomas and later in other cancers where chromosomal rearrangements paired the FUS transactivation domain with transcription factors such as CHOP (Rabbitts et al. 1993; Crozat et al. 1993), CREB3L2 (Panagopoulos et al. 2004), and ERG (Shimizu et al. 1993). Mutations in FUS were later reported in patients with ALS (Kwiatkowski et al. 2009; Vance et al. 2009). FUS pathology has also been observed in other neurological diseases, namely FTLD (Neumann et al. 2009a; Van Langenhove et al. 2010), essential tremor (Tio et al. 2016), and Huntington’s disease (Doi et al. 2008). Over 70 mutations in FUS have been identified in ALS patients, some of which are verified as disease causing (Fig. 1) (Deng et al. 2014). Interestingly, no genetic alterations in FUS have been reported in FTLD-FUS, a subtype of FTLD where FUS-positive inclusions are present (Neumann et al. 2009b; Rohrer et al. 2011; Urwin et al. 2010; Snowden et al. 2011). The pathological phenotypes caused by mutant forms of FUS vary widely in ALS patients with different mutations and even in ALS patients with the same mutation. A feature of FUS-related neuropathies, including ALS, is mislocalization of FUS to the cytoplasm either within intracellular protein aggregates or diffused throughout the region. FUS can be present in various types of protein aggregates including stress granules, ubiquitin-positive inclusions, and p62-positive inclusions (Kwiatkowski et al. 2009; Vance et al. 2009). The presence of FUS aggregates stratifies patients into a FUS-positive ALS (ALS-FUS) subtype (Woulfe et al. 2010; Deng et al. 2010).
Mechanisms of pathogenicity
It is unclear how ALS-associated mutations in FUS lead to neuron dysfunction. Numerous loss- and gain-of-function studies have been performed in mice, zebrafish, rats, worms, and fly to understand how disruption of FUS activity affects normal cell physiology (reviewed in Shang and Huang 2016). Data from transgenic mouse studies generally agree that expressing mutant FUS proteins is sufficient to cause motor neuron degeneration (Nolan et al. 2016). While loss of FUS in mice causes perinatal lethality, conditional loss of FUS in the nervous system does not cause motor system dysfunction (Sharma et al. 2016). This suggests that mutant FUS protein is not equivalent to a loss of FUS function. It is likely that mutant FUS proteins instead have acquired abnormal functions perhaps by some combination of disrupting RNA and protein homeostasis, altering subcellular localization, or promoting toxic aggregates.
The pathological activity of mutant FUS depends on its ability to bind RNA. ALS-associated FUS mutants in which the RRM domain was deleted could no longer cause neurological defects in Drosophila (Daigle et al. 2013). Although ALS-associated mutations in FUS do not reside within the regions that make direct contact with RNA, the mutations could affect the ability of FUS to interact with RNA. FUS R521C formed more stable complexes with Bdnf mRNA compared to wild-type FUS (Qiu et al. 2014). Since mutant forms of FUS are prone to mislocalize to the cytoplasm (Dormann et al. 2010), they are exposed to different sets of RNA substrates, particularly pre-mRNA versus mature RNA. Indeed, Hoell et al. (2011) found that the majority of binding sites for wild-type FUS resided in introns, whereas binding sites for ALS-mutant forms of FUS were located in 3′UTRs. Furthermore, FUS downregulates its own expression by binding to exon 7 of FUS mRNA and causing exon skipping (Zhou et al. 2013). The alternatively spliced product is a FUS transcript that undergoes nonsense-mediated decay. FUS variants, on the other hand, were unable to induce exon 7 skipping in FUS and therefore caused an upregulation in FUS protein; this misregulation led to an accumulation of the protein and may be a pathogenic mechanism.
FUS interacts with numerous proteins, many of which are involved in RNA metabolism (Sun et al. 2015; Kamelgarn et al. 2016). Kamelgarn et al. (2016) performed a proteomics study to determine whether FUS R521G exhibited different protein interactions from wild-type FUS and found little difference. These studies were performed in HEK293 cells at steady state. In contrast, mutant FUS proteins were reported to have stronger interactions with Survival of Motor Neuron (SMN) than wild-type FUS (Groen et al. 2013; Sun et al. 2015). The interaction between FUS and SMN is of note since mutations in SMN1 cause a reduction in SMN protein and leads to SMA, a degenerative disease of the lower motor neurons (Chari et al. 2009). This enhanced mutant FUS–SMN interaction was reported to alter global changes in alternative splicing by reducing the number of SMN-positive intranuclear bodies, called Gems, where spliceosome complexes are assembled (Sun et al. 2015) and impair the transport of SMN to axons (Groen et al. 2013).
The majority of FUS mutations associated with ALS cluster in the NLS and the N-terminal QGSY-rich and RGG1 prion-like domains (Fig. 1). These prion-like domains make FUS inherently prone to aggregation (Cushman et al. 2010). Some of these FUS variants, like R521C and P525L, appear to further exacerbate FUS aggregation, which is consistent with the notion that redistribution of FUS to the cytoplasm is a pathological event (Dormann et al. 2010; Bosco et al. 2010; Mackenzie et al. 2011; Gal et al. 2011; Ito et al. 2011; Kino et al. 2011; Niu et al. 2012; Suzuki et al. 2012; Zhang and Chook 2012). Mutations in FUS have so far only been observed in ALS, but not FTLD patients with FUS pathology (ALS-FUS and FTLD-FUS, respectively) (Neumann et al. 2009b; Rohrer et al. 2011; Urwin et al. 2010; Snowden et al. 2011). Neumann et al. (2011) showed that FUS-positive inclusions in ALS-FUS do not contain the other FET family members EWSR1 and TAF15. In contrast, FUS-positive inclusions in FTLD-FUS also contain EWSR1 and TAF15. These differences in pathological features indicate that there is a general breakdown of the nuclear import pathway in FTLD-FUS, whereas in ALS-FUS there is a defect specifically in FUS. Mutations in the NLS of FUS may impair nuclear import by weakening the interaction between FUS and transportin (Trn1) (Dormann et al. 2010; Niu et al. 2012). Trn1 interacts with the NLS of FUS to mediate its import into the nucleus. Although previous studies reported that nuclear FUS does not incorporate into stress granules, mutant FUS can bind and sequester nuclear localized FUS to cytoplasmic stress granules (Vance et al. 2013).
EWSR1
Role in RNA processing
EWSR1 is essential for diverse cellular processes including meiosis and mitosis (Li et al. 2007; Azuma et al. 2007), hematopoiesis and B lymphocyte development (Cho et al. 2011; Li et al. 2007), and adipogenesis (Park et al. 2013; Park and Lee 2015). Loss of EWSR1 in mice causes cellular senescence of hematopoietic stem cells (Cho et al. 2011) and genomic instability in cell types that undergo physiological DNA breaks, such as B cells and meiotic germ cells (Li et al. 2007).
Compared to FUS, less is known about the role of EWSR1 in RNA processing. The zinc finger and RRM domains of EWSR1 make contact with RNA (Fig. 1) (Castello et al. 2016). To begin understanding the role of EWSR1 in RNA processing, two groups independently mapped the RNA-binding sites of EWSR1 using PAR-CLIP in human cell lines. EWSR1 bound broadly across pre-mRNA transcripts, especially within introns, rather than at distinct sites (Mittal et al. 2015; Hoell et al. 2011). This binding profile is reminiscent of FUS, which also binds RNA broadly and is enriched in introns (Lagier-Tourenne et al. 2012; Rogelj et al. 2012). More specifically, Mittal et al. (2015) observed that EWSR1 preferred to bind G-rich RNA motifs in mRNA, consistent with previous in vitro binding studies. In contrast, Hoell et al. (2011) found that EWSR1 tended to interact with stem-loop structures. These discrepancies may be attributed to differences in how the CLIP libraries were prepared and could introduce biases in EWSR1 RNA-binding site identification.
The majority of proteins that interact with EWSR1 appear to be other RBPs that are involved in transcription, splicing, and translation (Pahlich et al. 2009). Some of these EWSR1 interactions are direct and robust, for example with FUS, TAF15, and EWSR1 itself (Thomsen et al. 2013), while other interactions are dependent on RNA (i.e., indirect interaction), like hnRNP M, hnRNP U, RNA helicases p68 and p72, the hsRPB7 subunit of RNAPII, and splicing factor YB-1 (Petermann et al. 1998; Chansky et al. 2001; Pahlich et al. 2009). EWSR1 is required for homologous recombination during the DNA damage response (Li et al. 2007) and regulates mitosis, in part through interactions with α-tubulin (Wang et al. 2016b). Further investigation is needed to determine how EWSR1 is involved in these processes and whether EWSR1 somehow connects the DNA damage response with RNA processing.
Disease association
A role for EWSR1 in human disease is well established in the context of cancer, particularly in Ewing’s sarcoma from which the gene derives its name. The common oncogenic event involves a chromosomal rearrangement that positions the DNA transactivation/low-complexity domain of EWSR1 upstream of DNA-binding domains of numerous genes, including FLI-1, ATF-1, and ERG, to form fusion oncogenes (Fisher 2014). The emergence of pathogenic mutations in FUS and TDP43 and the propensity of FUS and TDP-43 proteins to form pathological aggregates in patients with ALS suggest that defects in RNA regulation may contribute to ALS pathogenesis. This motivated a search for proteins with similar features to FUS and TDP-43, namely the presence of RRMs and prion-like domains, to identify novel ALS candidate genes (Couthouis et al. 2011). EWSR1 emerged as a top candidate. Ticozzi et al. (2011a) screened the entire coding region of EWSR1 in ALS patient and healthy control samples, but did not find any mutations that altered the protein sequence of EWSR1. Couthouis et al. (2012) also scanned the EWSR1 gene for mutations in a different cohort of ALS patients, this time focusing only on the exons that encoded the C-terminal RGG- and PY-motif regions, as the equivalent regions in FUS and TDP43 are hotspots for ALS-associated mutations. They identified two missense mutations—G511A and P552L—that were present in two unrelated sporadic ALS patients and not in control samples (Couthouis et al. 2012).
Mechanisms of pathogenicity
G511A and P552L variants in EWSR1 reside in the RGG domains (Fig. 1). Interestingly, these mutant EWSR1 proteins accumulated in the cytoplasm of ESC-derived neurons and in the neurites of cultured mouse spinal cord neurons, while the wild-type form of EWSR1 remained in the nucleus. The relocation of pathological forms of RBPs from the nucleus to the cytoplasm is a common feature in ALS patients. Indeed, EWSR1 protein was localized diffusely or within punctate granular structures in the cytoplasm of motor neurons from sporadic ALS patient samples but not healthy control samples. Furthermore, Couthouis et al. (2012) evaluated whether EWSR1 mutants were prone to aggregation and if they caused a neurodegeneration phenotype in the drosophila nervous system. The prion-like domain in EWSR1 (amino acids 1–280) makes the protein intrinsically prone to aggregation, but EWSR1 G511A and P552L mutants formed aggregates in vitro more rapidly than wild-type EWSR1. Wild-type EWSR1 alone was sufficient to cause neurodegeneration phenotypes when overexpressed in the fly retina and shortened the lifespan of Drosophila when expressed throughout the nervous system. One way in which mutations in EWSR1 may confer toxicity is by increasing the total abundance of EWSR1 since overexpression of the wild-type protein alone is toxic. Overexpression of EWSR1 G511A or P552L mutants did not exacerbate these phenotypes. These findings imply that the wild-type version of EWSR1 has intrinsic pathogenic properties, but this does not discount the possibility that the two ALS-associated mutant forms of EWSR1 may confer additional pathogenic properties in humans.
Taf15
Role in RNA processing
Like its family members FUS and EWSR1, TAF15 is involved in several steps of RNA processing. TAF15 was initially discovered as a component of the TFIID complex (Bertolotti et al. 1996) and later Kwon et al. (2013) showed that TAF15 interacts specifically with the unphosphorylated form of RNA polymerase II via its low-complexity domain. The unphosphorylated form of RNA polymerase II is associated with the pre-initiation complex. A prevailing hypothesis is that TAF15, likely in complex with TFIID, recruits RNA polymerase II to sites of active transcription. TAF15 also interacts with components of the spliceosome and auxiliary proteins involved in splicing. TAF15 was reported to co-immunoprecipitate with components of the U1–snRNP complex and splicing-associated hnRNP M. These interactions required the N-terminal, low-complexity domain, but not the C-terminal domain, of TAF15 (Leichter et al. 2011; Marko et al. 2014). CLIP-seq binding studies have revealed that TAF15 binds to long introns of a subset of pre-mRNAs in a “saw tooth-like” pattern (Kapeli et al. 2016) similar to FUS (Lagier-Tourenne et al. 2012). This binding profile suggests that TAF15 is deposited onto nascent transcripts as they emerge from the RNA polymerase II complex. TAF15 also binds to the 3′UTR of select mRNAs. In fact, TAF15-binding sites were most highly enriched in the 3′UTRs of its target RNAs when the length of the different genic regions was accounted for. This implies a role in RNA stability (Kapeli et al. 2016).
Interestingly, TAF15 and FUS bind to a large number of similar RNA targets (Ibrahim et al. 2013; Kapeli et al. 2016), indicating that they regulate common networks. For a large number of these common RNA targets, TAF15 and FUS-binding sites are in close proximity (Kapeli et al. 2016). Since TAF15 and FUS proteins strongly interact with each other in a manner that is not dependent on RNA (Thomsen et al. 2013; Sun et al. 2015), it is conceivable that TAF15 and FUS can bind RNA simultaneously.
Disease associations
TAF15 was initially associated with cancer, specifically extraskeletal myxoid chondrosarcoma and acute leukemia, in which the N-terminal transactivation domain of TAF15 was translocated and fused to the DNA-binding domain of NR4A3 or CIZ/MNP4, respectively (Sjögren et al. 1999; Martini et al. 2002). After connections between TDP-43 and FUS with ALS were established, TAF15 became a prime ALS gene candidate (Ticozzi et al. 2011a; Couthouis et al. 2011). Ticozzi and colleagues scanned the entire coding region of TAF15 in patients with sporadic and familial ALS and discovered two missense mutations—A31T and R395Q—that were present in familial ALS but not healthy control patient samples (Ticozzi et al. 2011a). At the same time, Couthouis et al. (2011) had searched specifically within the C-terminal zinc finger and RGG domains of TAF15 (not the entire gene) in 735 individuals diagnosed with ALS and discovered several mutations in patients with familial ALS but not in healthy individuals: M368T, G391E, R408C, and G473E (Fig. 1).
Mechanisms of pathogenicity
Wild-type TAF15 is pathogenic when present at high levels. Overexpression of wild-type TAF15 is toxic in a Drosophila model of neurodegeneration (Couthouis et al. 2011). Furthermore, wild-type TAF15 was mislocalized to the cytoplasm in motor neurons isolated from tissues of deceased sporadic ALS individuals. TAF15 is inherently prone to aggregation as a result of having low-complexity, prion-like domains, and several studies point to this feature as mediating the protein’s pathogenic activity (Couthouis et al. 2011; Kwon et al. 2013). However, ALS-mutant forms of TAF15 can enhance the pathogenic phenotypes of TAF15 over that of wild-type protein. The presence of ALS-linked mutations in TAF15 (G391E, R408C, and G473E) increased the number of TAF15-positive cytoplasmic foci in the dendrites and axons of embryonic rat cultured neurons above wild-type levels (Couthouis et al. 2011). Furthermore, overexpression of TAF15 G391E and R408C in Drosophila decreased lifespan compared to wild-type TAF15. How these mutations promote a degenerative phenotype is still unknown.
Mutant forms of TAF15 may alter the normal activities of TAF15 by disrupting RNA or protein interactions. Mutations in TAF15 do not reside in the RRM of TAF15, the domain that facilitates RNA binding, and so may not perturb TAF15–RNA interactions. Instead, ALS-mutant forms of TAF15 may alter the subcellular localization of TAF15, and as a result prevent nuclear functions of TAF15, promote toxic cytoplasmic functions, or both. Indeed, ALS-mutant forms of TAF15, namely G391E, R408C, and G473E, had a higher propensity to localize to cytoplasmic foci (Couthouis et al. 2011). One mechanism that controls TAF15 subcellular localization involves methylation by PRMT1 (protein arginine methyltransferase 1). When TAF15 methylation was blocked by a chemical inhibitor, a fraction of TAF15 accumulated into TIA1-positive stress granules in the cytoplasm (Jobert et al. 2009). This suggests that methylation of TAF15, presumably by PRMT1, retains the protein in the nucleus. TAF15 contains many arginine residues that are potential substrates for methylation, but R408 does not appear to be a substrate; however, the arginine residue adjacent to G473 is methylated. Further studies are needed to determine whether mutations in TAF15 alter PRMT1-mediated localization of TAF15.
Structural and functional commonalities: implications for unifying mechanisms of pathogenicity
Converging properties in function through protein and RNA interactions
The structural and functional commonalities between ALS-associated RBPs might suggest that they cause disease in a similar manner. As we perform more protein–protein interaction studies, we find that ALS-associated RBPs are often physically connected indicating that they operate in the same pathways. For example, hnRNP A1 and hnRNP A2/B1 interact with TDP-43 to control many of the splicing inhibitory functions of TDP-43 (Buratti et al. 2005; D’Ambrogio et al. 2009). In a proteomics study, FUS and TAF15 were identified as TDP-43 binding partners (Ling et al. 2010) and the FUS–TDP-43 interaction is required to mediate HDAC6 expression (Kim et al. 2010). Several studies have described strong interactions between FUS, EWSR1, and TAF15 (Thomsen et al. 2013; Sun et al. 2015; Kapeli et al. 2016), but the consequences of these interactions on RNA processing is unknown. FET proteins associate with a common set of proteins. For example, all FET proteins associated with TFIID and RNA Pol II to promote transcriptional activation (Bertolotti et al. 1996; Kwon et al. 2013). FUS and EWSR1 interact with a common set of splicing factors such as YB-1, U1C, SR, and SF1 (Chansky et al. 2001; Kovar 2011).
ALS-associated RBPs can also indirectly interact with each other by binding to the same RNA molecule. As RBPs typically have hundreds to thousands of RNA targets, they are likely to have overlapping RNA targets. CLIP studies that are performed under the same conditions (e.g., model system and CLIP protocol) can offer insight into common RNA targets. PAR-CLIP studies performed for FET proteins in human cell lines showed that the family members bind many of the same mRNAs: the RNA targets of FUS, EWSR1, and TAF15 overlapped with ~32, 69, and 48%, respectively, of RNA targets of the other two members (Hoell et al. 2011). In a separate CLIP-seq study performed in the mouse brain, FUS, TAF15, and TDP-43 shared a large percentage (>80%) of RNA targets (Kapeli et al. 2016). Interestingly, in one-third of TAF15 RNA targets there was at least one occurrence of a TAF15 and FUS-binding site within 100 bp of each other (Kapeli et al. 2016). Whether TAF15 and FUS bind the same RNA target in a mutually inclusive or exclusive fashion remains unknown. Genome-wide CLIP studies that define the spatiotemporal binding properties for ALS-associated RBPs with respect to each other will help to reveal new mechanisms of co-regulation between these RBPs (Fig. 2).

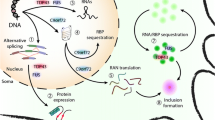
ALS-associated mutations in RBPs may disrupt RNA processing by several mechanisms. a Wild-type RBPs have roles in transcription. Mutant forms of the proteins may have abnormal interactions with transcription factors like TFIID or RNA polymerase II that disrupt transcription. b RBPs bind to introns of pre-mRNAs and splicing factors to regulate constitutive and alternative splicing. ALS-mutant proteins alter global splicing through events like exon skipping. c ALS-associated RBPs predominantly reside in the nucleus. Mutations in the RBPs can cause mislocalization to the cytoplasm where they bind and regulate different sets of RNAs. Some mutant RBPs were found to promote mislocalization of wild-type RBPs to the cytoplasm. d RBPs are involved in RNA trafficking, particularly to distant axonal and dendritic sites in neurons. Mutations in RBPs, such as FUS and TDP-43, disrupt the protein’s ability to transport mRNAs to their proper destinations. e TDP-43, hnRNP A2/B1, and hnRNP A1 are implicated in translation and mutant forms of these RBPs may disrupt this function. For example, mutant forms of TDP-43 have a greater propensity to mislocalize to the mitochondria and block translation of specific mitochondria-transcribed mRNAs (Wang et al. 2016a). f Mutations in ALS-associated RBPs, like FUS and TDP-43, cause the protein to be more resistant to proteasome-mediated degradation. The longer half-lives of mutant proteins results in their accumulation, which may confer toxicity. g Wild-type RBPs naturally form membrane-less organelles through phase transitions into liquid droplets. These reversible interactions are mediated by RNA and the low-complexity Gly-rich domain of the RBP. The presence of ALS-associated mutant RBPs, dipeptide repeats, or RNA repeats alter the biophysical properties of phase transitions and droplet formations. In these cases, droplets may evolve into insoluble structures through fibrilization that disrupt membrane-less organelles and kill neurons
Converging properties in low-complexity domains and liquid droplets
Recent trends in biology have highlighted the importance of membrane-less organelles in diverse cellular processes (reviewed in Banani et al. 2017). These molecular assemblies form dynamic micron-scale environments that mediate many biochemical reactions including those critical for RNA metabolism. The specialized RNP granules are dense collections of RNA and RBPs that exist in a variety of different subtypes including nucleoli, processing bodies (P-bodies), Cajal bodies, germ bodies, and stress granules. Much effort has focused on determining how these granules are formed and stabilized. Recent data indicate a mechanism based on the phase separation of liquid droplets containing high concentrations of RNA and RBPs (Brangwynne et al. 2009, 2011; Kato et al. 2012; Hyman et al. 2014). Indeed many, membrane-less organelles seem to be liquid-like in their behavior: dispersing and reforming, fragmenting, merging, and deforming in response to mechanical stress (Elbaum-Garfinkle et al. 2015; Weber and Brangwynne 2015). Many of the RBPs discussed in this review have been shown to undergo phase transitions and form liquid-like droplets in vitro and in vivo (Molliex et al. 2015; Patel et al. 2015; Kato et al. 2012; Schmidt and Rohatgi 2016; Paul et al. 2017). Importantly, for some RBPs, the presence of ALS-associated mutations seems to alter their phase transitions, stabilize and accelerate the formation of irreversible gel-like states, and in some cases, alter the dynamics of stress granules in cells (Lin et al. 2015; Molliex et al. 2015; Patel et al. 2015). For instance, the ALS-associated D290V mutation in hnRNP A2/B1 was shown to increase the formation of amyloid-like fibrils in vitro (Paul et al. 2017). ALS-associated mutations in hnRNP A1 were similarly shown to increase fibrilization (Molliex et al. 2015). FUS was also shown to form liquid-like droplets in vitro and in vivo and disease-causing mutations led to the formation of an irreversible solid state, not unlike the dense granules observed in post-mortem tissue obtained from ALS patients (Patel et al. 2015). In another study, mutant FUS was shown to accumulate in and disrupt cytoplasmic RNP granules and reduce new protein translation (Murakami et al. 2015). In nearly all cases, the authors demonstrated the requirement of the Gly-rich portion of RBPs for generating liquid droplets and fibrils (Lin et al. 2015; Molliex et al. 2015; Patel et al. 2015). Similarly, individual RBPs may recruit different proteins with Gly-rich domains to liquid droplets in a concentration-dependent manner (Lin et al. 2015; Molliex et al. 2015). Interestingly, the presence of RNA greatly diminishes the threshold necessary for phase transition and droplet formation (Lin et al. 2015).
A handful of recent studies have now established the importance of phase transitions and liquid droplet formation in ALS cases where mutant forms of RBPs were not involved. Dipeptide repeats generated from expanded GGGGCC repeats in C9orf72 ALS were shown to undergo phase transitions and form liquid-like droplets in vitro (Boeynaems et al. 2017; Lee et al. 2016). Dipeptide repeats were also shown to accumulate in stress granules, Cajal bodies, nuclear speckles, and the nucleolus of cells. These peptides reduced protein translation and disrupted stress granule dynamics (Boeynaems et al. 2017; Lee et al. 2016). In both studies, the authors also showed that dipeptide repeats can interact with the Gly-rich domain of several RBPs (hnRNP A1, FUS, TIA-1) and alter their phase transitions, making them more likely to remain in liquid droplets, or form fibrils (Boeynaems et al. 2017; Lee et al. 2016). Very recently, one study demonstrated that GGGGCC repeat containing RNA as well as other RNA repeats can undergo phase transitions and form liquid droplets even when only purified RNA is present (i.e., no RBPs) (Jain and Vale 2017). How the presence of Gly-rich domain containing RBPs might affect this process has not yet been reported.
In summary, the discovery of phase transitions in ALS-associated RBPs is an important step forward in understanding the pathogenic mechanisms of ALS. The observation that GGGGCC-containing RNA can form similar liquid-like droplets further strengthens the hypothesis of a common disease mechanism underlying the varied genetic causes of ALS. The disruption of endogenous membrane-less organelles by ALS proteins or RNA repeats presents a compelling biochemical mechanism for toxicity and a potential strategy for treatment.
Conclusion
It is clear that misregulation of RNA processing caused by defects in RBPs in the nervous system contributes to neurodegeneration. The field continues to accumulate evidence that uncovers how ALS-associated mutant forms of RBPs lead to a loss or gain of function and, importantly, to discern which of these functions are causes or consequences of pathogenesis. The normal and disease forms of these RBPs are often studied in isolation. For the disease-associated RBPs that we have discussed and several that we did not discuss (e.g., TIA-1 and MATRIN 3), there are clear overlaps in physiological and pathological functions. Considering how the functions of these proteins relate to the more global properties of the nerves highlights one of the more perplexing conundrums in the field of ALS: why the neuron? Indeed, all of the proteins discussed here are ubiquitously expressed, and yet patients typically exhibit symptoms in only a few specific tissues, usually the central nervous system. By considering the properties of ALS-linked RBPs, we can speculate on why this may be the case.
We propose two non-exclusive hypotheses. First, nearly every protein discussed above is involved in RNA transport. The upper and lower motor neurons are the longest cells in the body and proper function requires the transport of RNP granules over nearly a meter of distance. Perhaps, due to their unique physiology, motor neurons are most susceptible to subtle disturbances in the RNA transport machinery. Second, most of the proteins discussed in this review are directly or indirectly involved in RNA splicing. RNA splicing is most prevalent in cells of the nervous system than in any other cell types (Yeo et al. 2004; Castle et al. 2008), resulting in a greater diversity of gene expression through alternatively spliced isoforms. If proper function of the central nervous system requires fine-tuning of alternative splicing, then cells of the brain and spinal cord may be extremely sensitive to alterations in RNA splicing, or other RNA processing events, that go unnoticed in other tissues.
References
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E et al (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81(3):536–543. doi:10.1016/j.neuron.2013.12.018
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie SSF (2015) HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162(6):1299–1308. doi:10.1016/j.cell.2015.08.011 (NIH Public Access)
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611. doi:10.1016/j.bbrc.2006.10.093
Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB et al (2013) ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci 110(8):E736–E745. doi:10.1073/pnas.1222809110 (National Academy of Sciences)
Ayala YM, Misteli T, Baralle FE (2008a) TDP-43 Regulates Retinoblastoma Protein Phosphorylation through the Repression of Cyclin-Dependent Kinase 6 Expression. Proc Natl Acad Sci 105(10):3785–3789. doi:10.1073/pnas.0800546105
Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008b) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121(22):3778–3785. doi:10.1242/jcs.038950
Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J et al (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30(2):277–288. doi:10.1038/emboj.2010.310
Azuma M, Embree LJ, Sabaawy H, Hickstein DD (2007) Ewing sarcoma protein ewsr1 maintains mitotic integrity and proneural cell survival in the zebrafish embryo. PLoS One. doi:10.1371/journal.pone.0000979
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18(5):285–298. doi:10.1038/nrm.2017.7
Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S (2010) Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30(2):639–649. doi:10.1523/JNEUROSCI.4988-09.2010 (NIH Public Access)
Belzil VV, Gendron TF, Petrucelli L (2013) RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci 56:406–419. doi:10.1016/j.mcn.2012.12.006
Bentmann E, Haass C, Dormann D (2013) Stress granules in neurodegeneration—lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J 280(18):4348–4370. doi:10.1111/febs.12287
Berson A, Barbash S, Shaltiel G, Goll Y, Hanin G, Greenberg DS, Ketzef M, Becker AJ, Friedman A, Soreq H (2012) Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EMBO Mol Med 4(8):730–742. doi:10.1002/emmm.201100995 (Wiley-Blackwell)
Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L (1996) hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J 15(18):5022–5031. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=452240&tool=pmcentrez&rendertype=abstract
Blanchette M, Chabot B (1999) Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J 18(7):1939–1952. doi:10.1093/emboj/18.7.1939
Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, Eisen MB, Rio DC (2009) Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell 33(4):438–449. doi:10.1016/j.molcel.2009.01.022
Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, Guharoy M et al (2017) Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 65(6):1044–1055.e5. doi:10.1016/j.molcel.2017.02.013
Bonnal S, Pileur F, Orsini C, Parker F, Pujol F, Prats A-C, Vagner S (2005) heterogeneous nuclear ribonucleoprotein A1 is a novel internal ribosome entry site trans-acting factor that modulates alternative initiation of translation of the fibroblast growth factor 2 mRNA. J Biol Chem 280(6):4144–4153. doi:10.1074/jbc.M411492200
Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Sapp P, Mckenna-Yasek D, Brown RH, Hayward LJ (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19(21):4160–4175. doi:10.1093/hmg/ddq335
Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8(10):776–789. doi:10.1038/nrn2150
Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science (New York, NY) 324(5935):1729–1732. doi:10.1126/science.1172046
Brangwynne CP, Mitchison TJ, Hyman AA (2011) Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci USA 108(11):4334–4339. doi:10.1073/pnas.1017150108
Buratti E (2015) Functional significance of TDP-43 mutations in disease. Adv Genet. doi:10.1016/bs.adgen.2015.07.001
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276(39):36337–36343. doi:10.1074/jbc.M104236200
Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20(7):1774–1784. doi:10.1093/emboj/20.7.1774
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280(45):37572–37584. doi:10.1074/jbc.M505557200 (American Society for Biochemistry and Molecular Biology)
Cammas A, Pileur F, Bonnal S, Lewis SM, Lévêque N, Holcik M, Vagner S (2007) Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol Biol Cell 18(12):5048–5059. doi:10.1091/mbc.E07-06-0603 (American Society for Cell Biology)
Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30(4):377–384. doi:10.1038/ng854
Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE et al (2012) Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149(6):1393–1406. doi:10.1016/j.cell.2012.04.031
Castello A, Fischer B, Frese CK, Horos R, Alleaume A-M, Foehr S, Curk T, Krijgsveld J, Hentze MW (2016) Comprehensive identification of RNA-binding domains in human cells. Mol Cell 63:1–15. doi:10.1016/j.molcel.2016.06.029
Castle JC, Zhang C, Shah JK, Kulkarni AV, Kalsotra A, Cooper TA, Johnson JM (2008) Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat Genet 40(12):1416–1425. doi:10.1038/ng.264
Chansky HA, Hu M, Hickstein DD, Yang L (2001) Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res 61(9):3586–3590. http://www.ncbi.nlm.nih.gov/pubmed/11325824
Chari A, Paknia E, Fischer U (2009) The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol. doi:10.1016/j.ceb.2009.02.004
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y-J, Castanedes-Casey M, Lee CW et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science (New York, NY) 348(6239):1151–1154. doi:10.1126/science.aaa9344
Chiang H, Andersen PM, Tysnes OB, Gredal O, Christensen PB, Graff C (2012) Novel TARDBP mutations in nordic ALS patients. J Hum Genet 57(5):316–319. doi:10.1038/jhg.2012.24
Chio A, Calvo A, Mazzini L, Cantello R, Mora G, Moglia C, Corrado L et al (2012) Extensive genetics of ALS: a population-based study in Italy. Neurology 79(19):1983–1989. doi:10.1212/WNL.0b013e3182735d36
Cho J, Shen H, Yu H, Li H, Cheng T, Lee SB, Lee BC (2011) Ewing sarcoma gene Ews regulates hematopoietic stem cell senescence. Blood 117(4):1156–1166. doi:10.1182/blood-2010-04-279349
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR (2010) The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci 107(5):1894–1899. doi:10.1073/pnas.0914845107
Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A (2012) TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem 287(19):15635–15647. doi:10.1074/jbc.M111.333450
Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24(9):1537–1549. doi:10.1016/j.str.2016.07.007
Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N et al (2009) High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat 30(4):688–694. doi:10.1002/humu.20950
Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX et al (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA 108(52):20881–20890. doi:10.1073/pnas.1109434108
Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F et al (2012) Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21(13):2899–2911. doi:10.1093/hmg/dds116
Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363(6430):640–644. doi:10.1038/363640a0
Cushman M, Johnson BS, King OD, Gitler AD, Shorter J (2010) Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 123(Pt 8):1191–1201. doi:10.1242/jcs.051672
D’Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, Baralle FE (2009) Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res 37(12):4116–4126. doi:10.1093/nar/gkp342
Daigle GG, Lanson A, Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, Pandey UB (2013) RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet 22(6):1193–1205. doi:10.1093/hmg/dds526
Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupré N, Camu W, Meininger V et al (2009) Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet 46(2):112–114. doi:10.1136/jmg.2008.062463
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. doi:10.1016/j.neuron.2011.09.011
Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, Ghione I et al (2009) TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol 16(6):727–732. doi:10.1111/j.1468-1331.2009.02574.x
Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M et al (2010) FUS-immunoreactive inclusions are a common feature in Sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67(6):739–748. doi:10.1002/ana.22051
Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, Yang Y et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363):211–215. doi:10.1038/nature10353
Deng H, Gao K, Jankovic J (2014) The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10(6):337–348. doi:10.1038/nrneurol.2014.78
Dirksen WP, Li X, Mayeda A, Krainer AR, Rottman FM (2000) Mapping the SF2/ASF binding sites in the bovine growth hormone exonic splicing enhancer. J Biol Chem 275(37):29170–29177. doi:10.1074/jbc.M001126200
Doi H, Okamura K, Bauer PO, Furukawa Y, Shimizu H, Kurosawa M, Machida Y et al (2008) RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem 283(10):6489–6500. doi:10.1074/jbc.M705306200
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME et al (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J 29(16):2841–2857. doi:10.1038/emboj.2010.143
Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG (1993) hnrnp proteins and the biogenesis of mRNA. Annu Rev Biochem 62(1):289–321. doi:10.1146/annurev.bi.62.070193.001445
Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CC-H, Eckmann CR, Myong S, Brangwynne CP (2015) The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci. doi:10.1073/pnas.1504822112
Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, Fox PL (2014) Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell 157(7):1605–1618. doi:10.1016/j.cell.2014.04.033
Eykens C, Robberecht W (2015) The Genetic basis of amyotrophic lateral sclerosis: recent breakthroughs. Adv Genom Genet 2015(5):327–345. doi:10.2147/AGG.S57397
Fahling M, Mrowka R, Steege A, Martinka P, Persson PB, Thiele BJ (2006) Heterogeneous nuclear ribonucleoprotein-A2/B1 modulate collagen prolyl 4-hydroxylase, (I) mRNA stability. J Biol Chem 281(14):9279–9286. doi:10.1074/jbc.M510925200
Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ (2012) TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res 40(6):2668–2682. doi:10.1093/nar/gkr1082
Fisette J-F, Toutant J, Dugré-Brisson S, Desgroseillers L, Chabot B (2010) hnRNP A1 and hnRNP H can collaborate to modulate 5′ splice site selection. RNA (New York, NY) 16(1):228–238. doi:10.1261/rna.1890310
Fisher C (2014) The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology. doi:10.1111/his.12269
Freibaum BD, Chitta RK, High AA, Paul Taylor J (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9(2):1104–1120. doi:10.1021/pr901076y
Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118(24):5755–5765. doi:10.1242/jcs.02692
Fujita Y, Ikeda M, Yanagisawa T, Senoo Y, Okamoto K (2011) Different clinical and neuropathologic phenotypes of familial ALS with A315E TARDBP mutation. Neurology. doi:10.1212/WNL.0b013e318232ab87
Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H (2011) Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2010.06.010
Gallouzi IE, Steitz JA (2001) Delineation of mRNA export pathways by the use of cell-permeable peptides. Science (New York, NY) 294(5548):1895–1901. doi:10.1126/science.1064693
Gao Y, Tatavarty V, Korza G, Levin MK, Carson JH (2008) Multiplexed dendritic targeting of calcium calmodulin-dependent protein kinase II, neurogranin, and activity-regulated cytoskeleton-associated protein RNAs by the A2 pathway. Mol Biol Cell 19(5):2311–2327. doi:10.1091/mbc.E07-09-0914
Geser F, Lee VMY, Trojanowski JQ (2010) Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. doi:10.1111/j.1440-1789.2009.01091.x
Gilpin KM, Chang L, Monteiro MJ (2015) ALS-linked mutations in ubiquilin-2 or hnRNPA1 reduce interaction between ubiquilin-2 and hnRNPA1. Hum Mol Genet 24(9):2565–2577. doi:10.1093/hmg/ddv020
Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ et al (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63(4):535–538. doi:10.1002/ana.21344
Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S (2012) Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 485(7397):264–268. doi:10.1038/nature11013
Groen EJ, Fumoto K, Blokhuis AM, Engelen-Lee JYY, Zhou Y, van den Heuvel DMA, Koppers M et al (2013) ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 22(18):3690–3704. doi:10.1093/hmg/ddt222
Guil S, Cáceres JF (2007) The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol 14(7):591–596. doi:10.1038/nsmb1250
Hallier M, Lerga A, Barnache S, Tavitian A, Moreau-Gachelin F (1998) The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J Biol Chem 273(9):4838–4842. doi:10.1074/jbc.273.9.4838
Hamilton BJ, Burns CM, Nichols RC, Rigby WF (1997) Modulation of AUUUA response element binding by heterogeneous nuclear ribonucleoprotein A1 in human T lymphocytes. The roles of cytoplasmic location, transcription, and phosphorylation. J Biol Chem 272(45):28732–28741. http://www.ncbi.nlm.nih.gov/pubmed/9353343
Han K, Yeo G, An P, Burge CB, Grabowski PJ (2005) A combinatorial code for splicing silencing: UAGG and GGGG motifs. PLoS Biol 3(5):e158. doi:10.1371/journal.pbio.0030158 (Public Library of Science)
Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149(4):768–779. doi:10.1016/j.cell.2012.04.016
He Y, Smith R (2009) Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci 66(7):1239–1256. doi:10.1007/s00018-008-8532-1
He F, Krans A, Freibaum BD, Paul Taylor J, Todd PK (2014) TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet 23(19):5036–5051. doi:10.1093/hmg/ddu216
Henics T, Sanfridson A, Hamilton BJ, Nagy E, Rigby WF (1994) Enhanced stability of interleukin-2 mRNA in MLA 144 cells. Possible role of cytoplasmic AU-rich sequence-binding proteins. J Biol Chem 269(7):5377–5383. http://www.ncbi.nlm.nih.gov/pubmed/8106521
Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D et al (2000) Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 24(2):175–179. doi:10.1038/72842
Higashi S, Kabuta T, Nagai Y, Tsuchiya Y, Akiyama H, Wada K (2013) TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J Neurochem 126(2):288–300. doi:10.1111/jnc.12194
Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A et al (2014) Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol 40(6):670–685. doi:10.1111/nan.12148
Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T (2011) RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18(12):1428–1431. doi:10.1038/nsmb.2163
Honda H, Hamasaki H, Wakamiya T, Koyama S, Suzuki SO, Fujii N, Iwaki T (2015) Loss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusions. Neuropathology 35(1):37–43. doi:10.1111/neup.12153
Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP et al (2012) Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1(2):167–178. doi:10.1016/j.celrep.2012.02.001
Huey ED, Ferrari R, Moreno JH, Jensen C, Morris CM, Potocnik F, Kalaria RN et al (2012) FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2011.08.004
Hutchison S, LeBel C, Blanchette M, Chabot B (2002) Distinct sets of adjacent heterogeneous nuclear ribonucleoprotein (hnRNP) A1/A2 binding sites control 5′ splice site selection in the hnRNP A1 mRNA precursor. J Biol Chem 277(33):29745–29752. doi:10.1074/jbc.M203633200
Hyman AA, Weber CA, Jülicher F (2014) Liquid–liquid phase separation in biology. Annu Rev Cell Dev Biol 30(1):39–58. doi:10.1146/annurev-cellbio-100913-013325
Ibrahim F, Maragkakis M, Alexiou P, Maronski MA, Dichter MA, Mourelatos Z (2013) Identification of in vivo, conserved, TAF15 RNA binding sites reveals the impact of TAF15 on the neuronal transcriptome. Cell Rep 3(2):301–308. doi:10.1016/j.celrep.2013.01.021
Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J et al (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Investig 121(2):726–738. doi:10.1172/JCI44867
Iida A, Kamei T, Sano M, Oshima S, Tokuda T, Nakamura Y, Ikegawa S (2012) Large-scale screening of TARDBP mutation in amyotrophic lateral sclerosis in Japanese. Neurobiol Aging 33(4):786–790. doi:10.1016/j.neurobiolaging.2010.06.017
Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K (2012) Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2(January):529. doi:10.1038/srep00529
Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N (2011) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69(1):152–162. doi:10.1002/ana.22246
Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G, Fischer U (1997) A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol 137(1):27–35. http://www.ncbi.nlm.nih.gov/pubmed/9105034
Jain A, Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546(7657):243–247. doi:10.1038/nature22386
Jean-Philippe J, Paz S, Caputi M (2013) hnRNP A1: the swiss army knife of gene expression. Int J Mol Sci 14(9):18999–19024. doi:10.3390/ijms140918999
Jobert L, Argentini M, Tora L (2009) PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res 315(7):1273–1286. doi:10.1016/j.yexcr.2008.12.008
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284(30):20329–20339. doi:10.1074/jbc.M109.010264 (American Society for Biochemistry and Molecular Biology)
Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ (2002) Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA (New York, NY) 8(4):426–439. http://www.ncbi.nlm.nih.gov/pubmed/11991638
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard J-P et al (2008) TARDBP Mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40(5):572–574. doi:10.1038/ng.132
Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D et al (2010) Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet 19(4):671–683. doi:10.1093/hmg/ddp534
Kamada M, Maruyama H, Tanaka E, Morino H, Wate R, Ito H, Kusaka H et al (2009) Screening for TARDBP mutations in Japanese familial amyotrophic lateral sclerosis. J Neurol Sci 284(1–2):69–71. doi:10.1016/j.jns.2009.04.017
Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, Gal J (2016) Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta Mol Basis Dis 1862(10):2004–2014. doi:10.1016/j.bbadis.2016.07.015
Kapeli K, Pratt GA, Vu AQ, Hutt KR, Martinez FJ, Sundararaman B, Batra R et al (2016) Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat Commun 7:12143. doi:10.1038/ncomms12143
Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34(4):460–463. doi:10.1038/ng1207
Kato M, Han TW, Xie S, Shi K, Xinlin D, Wu LC, Mirzaei H et al (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149(4):753–767. doi:10.1016/j.cell.2012.04.017
Kim SH, Shanware NP, Bowler MJ, Tibbetts RS (2010) Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J Biol Chem 285(44):34097–34105. doi:10.1074/jbc.M110.154831
Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, MacLea KS et al (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495(7442):467–473. doi:10.1038/nature11922 (Nature Research)
Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N (2011) Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res 39(7):2781–2798. doi:10.1093/nar/gkq1162
Kirby J, Goodall EF, Smith W, Highley JR, Masanzu R, Hartley JA, Hibberd R et al (2010) Broad clinical phenotypes associated with TAR-DNA binding protein (TARDBP) mutations in amyotrophic lateral sclerosis. Neurogenetics 11(2):217–225. doi:10.1007/s10048-009-0218-9
Kolisnyk B, Al-Onaizi MA, Xu J, Parfitt GM, Ostapchenko VG, Hanin G, Soreq H, Prado MA, Prado VF (2016) Cholinergic regulation of hnRNPA2/B1 translation by M1 muscarinic receptors. J Neurosci Off J Soc Neurosci 36(23):6287–6296. doi:10.1523/JNEUROSCI.4614-15.2016
Kosturko LD, Maggipinto MJ, Korza G, Lee JW, Carson JH, Barbarese E (2006) Heterogeneous nuclear ribonucleoprotein (hnRNP) E1 binds to hnRNP A2 and inhibits translation of A2 response element mRNAs. Mol Biol Cell 17(8):3521–3533. doi:10.1091/mbc.E05-10-0946 (American Society for Cell Biology)
Kovar H (2011) Dr. Jekyll and Mr. Hyde: the two faces of the FUS/EWS/TAF15 protein family. Sarcoma, vol. 2011. Hindawi Publishing Corporation. doi:10.1155/2011/837474
Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VMY, Schellenberg GD (2010) Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 119(4):409–419. doi:10.1007/s00401-010-0659-0 (NIH Public Access)
Kühnlein P, Sperfeld A-D, Vanmassenhove B, Van Deerlin V, Lee VM-Y, Trojanowski JQ, Kretzschmar HA, Ludolph AC, Neumann M (2008) Two german kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol 65(9):1185–1189. doi:10.1001/archneur.65.9.1185
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY) 323(5918):1205–1208. doi:10.1126/science.1166066
Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155(1–2):374. doi:10.1016/j.cell.2013.10.033 (Elsevier)
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19(R1):R46–R64. doi:10.1093/hmg/ddq137
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15(11):1488–1497. doi:10.1038/nn.3230
Le Ber I, Van Bortel I, Nicolas G, Bouya-Ahmed K, Camuzat A, Wallon D, De Septenville A et al (2014) hnRNPA2B1 and hnRNPA1 mutations are rare in patients with ‘multisystem proteinopathy’ and frontotemporal lobar degeneration phenotypes. Neurobiol Aging 35(4):934.e5–934.e6. doi:10.1016/j.neurobiolaging.2013.09.016
Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM (2006) Rules for nuclear localization sequence recognition by karyopherinβ2. Cell 126(3):543–558. doi:10.1016/j.cell.2006.05.049
Lee EB, Lee VM-Y, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13(1):38–50. doi:10.1038/nrn3121
Lee K-H, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(3):774–788.e17. doi:10.1016/j.cell.2016.10.002
Leichter M, Marko M, Ganou V, Patrinou-Georgoula M, Tora L, Guialis A (2011) A fraction of the transcription factor TAF15 participates in interactions with a subset of the spliceosomal U1 snRNP complex. Biochim Biophys Acta Proteins Proteom 1814(12):1812–1824. doi:10.1016/j.bbapap.2011.09.008
Lemmens R, Race V, Hersmus N, Matthijs G, Van Den Bosch L, Van Damme P, Dubois B, Boonen S, Goris A, Robberecht W (2009) TDP-43 M311V mutation in familial amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 80(3):354–355. doi:10.1136/jnnp.2008.157677
Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F (2001) Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem 276(9):6807–6816. doi:10.1074/jbc.M008304200
Li H, Watford W, Li C, Parmelee A, Bryant MA, Deng C, O’Shea J, Sean BL (2007) Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J Clin Investig 117(5):1314–1323. doi:10.1172/JCI31222
Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201(3):361–372. doi:10.1083/jcb.201302044
Lin Y, Protter DS, Rosen MK, Parker R (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell 60(2):208–219. doi:10.1016/j.molcel.2015.08.018
Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S et al (2010) ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA 107(30):13318–13323. doi:10.1073/pnas.1008227107
Liu Q, Shu S, Wang RR, Liu F, Cui B, Guo XN, Lu CX et al (2016) Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology 87(17):1763–1769. doi:10.1212/WNL.0000000000003256
Liu T-Y, Chen Y-C, Jong Y-J, Tsai H-J, Lee C-C, Chang Y-S, Chang J-G, Chang Y-F (2017) Muscle developmental defects in heterogeneous nuclear ribonucleoprotein A1 knockout mice. Open Biol. doi:10.1098/rsob.160303 (The Royal Society)
Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B (2011) Local RNA translation at the synapse and in disease. J Neurosci 31(45):16086–16093. doi:10.1523/JNEUROSCI.4105-11.2011
Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu Y-F, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B (2014) ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci Off J Soc Neurosci 34(12):4167–4174. doi:10.1523/JNEUROSCI.2350-13.2014 (Society for Neuroscience)
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. doi:10.1016/S1474-4422(10)70195-2
Mackenzie IR, Ansorge O, Strong M, Bilbao J, Zinman L, Ang LC, Baker M et al (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol 122(1):87–98. doi:10.1007/s00401-011-0838-7
Marko M, Leichter M, Patrinou-Georgoula M, Guialis A (2014) Selective interactions of hnRNP M isoforms with the TET proteins TAF15 and TLS/FUS. Mol Biol Rep 41(4):2687–2695. doi:10.1007/s11033-014-3128-3
Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P et al (2016) Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron 92(4):780–795. doi:10.1016/j.neuron.2016.09.050
Martini A, La Starza R, Janssen H, Bilhou-Nabera C, Corveleyn A, Somers R, Aventin A et al (2002) Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res 62(19):5408–5412
Mayeda A, Krainer AR (1992) Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 68(2):365–375. http://www.ncbi.nlm.nih.gov/pubmed/1531115
McGlincy NJ, Tan L-Y, Paul N, Zavolan M, Lilley KS, Smith CWJ (2010) Expression proteomics of UPF1 knockdown in HeLa cells reveals autoregulation of hnRNP A2/B1 mediated by alternative splicing resulting in nonsense-mediated mRNA decay. BMC Genom 11(1):565. doi:10.1186/1471-2164-11-565
McKee AE, Minet E, Stern C, Riahi S, Stiles CD, Silver PA (2005) A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev Biol 5:14. doi:10.1186/1471-213X-5-14
Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE (2005) Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res 33(18):6000–6010. doi:10.1093/nar/gki897
Michlewski G, Cáceres JF (2010) Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat Struct Mol Biol 17(8):1011–1018. doi:10.1038/nsmb.1874
Michlewski G, Guil S, Semple CA, Cáceres JF (2008) Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell 32(3):383–393. doi:10.1016/j.molcel.2008.10.013
Mili S, Shu HJ, Zhao Y, Pinol-Roma S (2001) Distinct RNP complexes of shuttling hnrnp proteins with pre-mRNA and mRNA: candidate intermediates in formation and export of mRNA. Mol Cell Biol 21(21):7307–7319. doi:10.1128/MCB.21.21.7307-7319.2001
Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, Guillot-Noël L et al (2010) SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype–phenotype correlations. J Med Genet 47(8):554–560. doi:10.1136/jmg.2010.077180
Mittal N, Kunz C, Gypas F, Kishore S, Martin G, Wenzel F, van Nimwegen E, Schaer P, Zavolan M (2015) Ewing sarcoma breakpoint region 1 prevents transcription-associated genome instability. bioRxiv. doi:10.1101/034215
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163(1):123–133. doi:10.1016/j.cell.2015.09.015 (Howard Hughes Medical Institute)
Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, Edbauer Dr et al (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125(3):413–423. doi:10.1007/s00401-013-1088-7
Munro TP, Magee RJ, Kidd GJ, Carson JH, Barbarese E, Smith LM, Smith R (1999) Mutational analysis of a heterogeneous nuclear ribonucleoprotein A2 response element for RNA trafficking. J Biol Chem 274(48):34389–34395. http://www.ncbi.nlm.nih.gov/pubmed/10567417
Murakami T, Qamar S, Lin JQ, Kaminski GS, Schierle ER, Miyashita A, Costa AR et al (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88(4):678–690. doi:10.1016/j.neuron.2015.10.030
Muslimov IA, Patel MV, Rose A, Tiedge H (2011) Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. J Cell Biol 194(3):441–457. doi:10.1083/jcb.201010027
Nakielny S, Dreyfuss G (1999) Transport of proteins and RNAs in and out of the nucleus. Cell 99(7):677–690. http://www.ncbi.nlm.nih.gov/pubmed/10619422
Nasim FU, Hutchison S, Cordeau M, Chabot B (2002) High-affinity hnRNP A1 binding sites and duplex-forming inverted repeats have similar effects on 5′ splice site selection in support of a common looping out and repression mechanism. RNA (New York, NY) 8(8):1078–1089. http://www.ncbi.nlm.nih.gov/pubmed/12212851
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314(5796):130–133. doi:10.1126/science.1134108
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, MacKenzie IRA (2009a) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132(11):2922–2931. doi:10.1093/brain/awp214
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IRA (2009b) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118(5):605–616. doi:10.1007/s00401-009-0581-5
Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, Roeber S et al (2011) FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain J Neurol 134(Pt 9):2595–2609. doi:10.1093/brain/awr201
Niu C, Zhang J, Gao F, Yang L, Jia M, Zhu H, Gong W (2012) FUS-NLS/transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS One. doi:10.1371/journal.pone.0047056
Nolan M, Talbot K, Ansorge O (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4(1):99. doi:10.1186/s40478-016-0358-8
Nozaki I, Arai M, Takahashi K, Hamaguchi T, Yoshikawa H, Muroishi T, Noguchi-Shinohara M et al (2010) Familial ALS with G298S mutation in TARDBP: a comparison of CSF tau protein levels with those in sporadic ALS. Intern Med 49(12):1209–1212. doi:10.2169/internalmedicine.49.3300
Nussbacher JK, Batra R, Lagier-Tourenne C, Yeo GW (2015) RNA-binding proteins in neurodegeneration: seq and you shall receive. Trends Neurosci. doi:10.1016/j.tins.2015.02.003
Origone P, Caponnetto C, Di Poggio MB, Ghiglione E, Bellone E, Ferrandes G, Mancardi GL, Mandich P (2010) Enlarging clinical spectrum of FALS with TARDBP gene mutations: S393L variant in an Italian family showing phenotypic variability and relevance for genetic counselling. Amyotroph Later Scler 11(1–2):223–227. doi:10.3109/17482960903165039
Orrù S, Manolakos E, Orrù N, Kokotas H, Mascia V, Carcassi C, Petersen MB (2012) High frequency of the TARDBP p.Ala382Thr mutation in sardinian patients with amyotrophic lateral sclerosis. Clin Genet 81(2):172–178. doi:10.1111/j.1399-0004.2011.01668.x
Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69(6):3584–3596. http://www.ncbi.nlm.nih.gov/pubmed/7745706
Pahlich S, Quero L, Roschitzki B, Leemann-Zakaryan RP, Gehring H (2009) Analysis of ewing sarcoma (EWS)-binding proteins: interaction with hnRNP M, U, and RNA-helicases p68/72 within protein-RNA complexes. J Proteome Res 8(10):4455–4465. doi:10.1021/pr900235t
Panagopoulos I, Storlazzi CT, Fletcher CDM, Fletcher JA, Nascimento A, Domanski HA, Wejde J et al (2004) The chimeric FUS/CREB3L2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosom Cancer 40(3):218–228. doi:10.1002/gcc.20037
Park JH, Lee SB (2015) An essential role for Ewing sarcoma gene (EWS) in early white adipogenesis. Obesity 23(1):138–144. doi:10.1002/oby.20934
Park JH, Kang HJ, Kang SI, Lee JE, Hur J, Ge K, Mueller E, Li H, Lee BC, Lee SB (2013) A multifunctional protein, EWS, is essential for early brown fat lineage determination. Dev Cell 26(4):393–404. doi:10.1016/j.devcel.2013.07.002
Pasinelli P, Brown RH (2016) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723. doi:10.1038/nrn1971 (Nature Publishing Group)
Passoni M, De Conti L, Baralle M, Buratti E (2012) UG repeats/TDP-43 interactions near 5′ splice sites exert unpredictable effects on splicing modulation. J Mol Biol 415(1):46–60. doi:10.1016/j.jmb.2011.11.003
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162(5):1066–1077. doi:10.1016/j.cell.2015.07.047
Paul KR, Molliex A, Cascarina S, Boncella AE, Paul Taylor J, Ross ED (2017) Effects of mutations on the aggregation propensity of the human prion-like protein hnRNPA2B1. Mol Cell Biol 37(8):e00652-16. doi:10.1128/MCB.00652-16
Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H (1998) Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 17(5):603–610. doi:10.1038/sj.onc.1201964
Piñol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2(2):215–227. http://www.ncbi.nlm.nih.gov/pubmed/3129338
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S-C et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14(4):459–468. doi:10.1038/nn.2779
Polymenidou M, Lagier-Tourenne C, Hutt KR, Frank Bennett C, Cleveland DW, Yeo GW (2012) Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res 1462(June):3–15. doi:10.1016/j.brainres.2012.02.059
Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ et al (2014) ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Investig 124(3):981–999. doi:10.1172/JCI72723
Rabbitts TH, Forster A, Larson R, Nathan P (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4(2):175–180. doi:10.1038/ng0693-175
Raju CS, Fukuda N, López-Iglesias C, Göritz C, Visa N, Percipalle P (2011) In neurons, activity-dependent association of dendritically transported mRNA transcripts with the transacting factor CBF-A is mediated by A2RE/RTS elements. Mol Biol Cell. doi:10.1091/mbc.E10-11-0904 (American Society for Cell Biology)
Ratti A, Buratti E (2016) Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem 138(August):95–111. doi:10.1111/jnc.13625
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Raphael Gibbs J, Schymick JC et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 72(2):257–268. doi:10.1016/j.neuron.2011.09.010
Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B et al (2012) Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2(January):603. doi:10.1038/srep00603 (Nature Publishing Group)
Rohrer JD, Lashley T, Holton J, Revesz T, Urwin H, Isaacs AM, Fox NC, Rossor MN, Warren J (2011) The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 82(12):1405–1407. doi:10.1136/jnnp.2010.214437
Russo A, Scardigli R, La Regina F, Murray ME, Romano N, Dickson DW, Wolozin B, Cattaneo A, Ceci M (2017) Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet 26(8):1407–1418. doi:10.1093/hmg/ddx035
Rutherford NJ, Zhang Y-J, Baker M, Gass JM, Finch NA, Xu Y-F, Stewart H et al (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. Edited by Gregory A. Cox. PLoS Genet 4(9):e1000193. doi:10.1371/journal.pgen.1000193 (Public Library of Science)
Sánchez-Ramos C, Tierrez A, Fabregat-Andrés O, Wild B, Sánchez-Cabo F, Arduini A, Dopazo A, Monsalve M (2011) PGC-1α regulates translocated in liposarcoma activity: role in oxidative stress gene expression. Antioxid Redox Signal 15(2):325–337. doi:10.1089/ars.2010.3643
Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S et al (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5(208):208ra149. doi:10.1126/scitranslmed.3007529
Schmidt HB, Rohatgi R (2016) In vivo formation of vacuolated multi-phase compartments lacking membranes cell reports report in vivo formation of vacuolated multi-phase compartments lacking membranes. Cell Rep 16:1228–1236. doi:10.1016/j.celrep.2016.06.088
Schwartz JC, Ebmeier CC, Podell ER, Heimiller Joseph, Taatjes DJ, Cech TR (2012) FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev 26(24):2690–2695. doi:10.1101/gad.204602.112
Seelen M, Visser AE, Overste DJ, Kim HJ, Palud A, Wong TH, van Swieten JC et al (2014) No mutations in hnRNPA1 and hnRNPA2B1 in Dutch patients with amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy. Neurobiol Aging 35(8):1956.e9–1956.e11. doi:10.1016/j.neurobiolaging.2014.01.152
Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM et al (2011) Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 286(2):1204–1215. doi:10.1074/jbc.M110.190884
Shan J, Moran-Jones K, Munro TP, Kidd GJ, Winzor DJ, Hoek KS, Smith R (2000) Binding of an RNA trafficking response element to heterogeneous nuclear ribonucleoproteins A1 and A2. J Biol Chem 275(49):38286–38295. doi:10.1074/jbc.M007642200
Shan J, Munro TP, Barbarese E, Carson JH, Smith R (2003) A molecular mechanism for mRNA trafficking in neuronal dendrites. J Neurosci Off J Soc Neurosci 23(26):8859–8866. http://www.ncbi.nlm.nih.gov/pubmed/14523087
Shang Y, Huang EJ (2016) Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. doi:10.1016/j.brainres.2016.03.036
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA (2016) ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7(January):10465. doi:10.1038/ncomms10465
Shimizu K, Ichikawa H, Tojo A, Kaneko Y, Maseki N, Hayashi Y, Ohira M, Asano S, Ohki M (1993) An ets-related gene, ERG, is rearranged in human myeloid leukemia with t(16;21) chromosomal translocation. Proc Natl Acad Sci USA 90(21):10280–10284. doi:10.1073/pnas.90.21.10280
Shorter J, Taylor JP (2013) Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis 1:e25200. doi:10.4161/rdis.25200 (Taylor & Francis)
Siomi MC, Eder PS, Kataoka N, Wan L, Liu Q, Dreyfuss G (1997) Transportin-mediated nuclear import of heterogeneous nuclear RNP proteins. J Cell Biol 138(6):1181–1192. http://www.ncbi.nlm.nih.gov/pubmed/9298975
Sjögren H, Meis-Kindblom J, Kindblom LG, Åman P, Stenman G (1999) Fusion of the EWS-related gene TAF2N to TEC in extraskeletal myxoid chondrosarcoma. Can Res 59(20):5064–5067
Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, Momeni Parastoo et al (2011) The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 122(1):99–110. doi:10.1007/s00401-011-0816-0
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (New York, NY) 319(5870):1668–1672. doi:10.1126/science.1154584
Sun Q, Hampson RK, Rottman FM (1993) In vitro analysis of bovine growth hormone pre-mRNA alternative splicing. involvement of exon sequences and trans-acting factor(s). J Biol Chem 268 (21): 15659–15666. http://www.ncbi.nlm.nih.gov/pubmed/8340391
Sun S, Ling S-C, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H et al (2015) ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6(January):6171. doi:10.1038/ncomms7171 (Nature Publishing Group)
Suzuki H, Matsuoka M (2017) hnRNPA1 autoregulates its own mRNA expression to remain non-cytotoxic. Mol Cell Biochem 427(1–2):123–131. doi:10.1007/s11010-016-2904-x
Suzuki N, Kato S, Kato M, Warita H, Mizuno H, Kato M, Shimakura N et al (2012) FUS/TLS-immunoreactive neuronal and glial cell inclusions increase with disease duration in familial amyotrophic lateral sclerosis with an R521C FUS/TLS mutation. J Neuropathol Exp Neurol 71(9):779–788. doi:10.1097/NEN.0b013e318264f164
Svitkin YV, Ovchinnikov LP, Dreyfuss G, Sonenberg N (1996) General RNA binding proteins render translation cap dependent. EMBO J 15(24):7147–7155. http://www.ncbi.nlm.nih.gov/pubmed/9003790 (European Molecular Biology Organization)
Tan AY, Manley JL (2009) The TET family of proteins: functions and roles in disease. J Mol Cell Biol. doi:10.1093/jmcb/mjp025
Tavanez JP, Madl T, Kooshapur H, Sattler M, Valcárcel J (2012) hnRNP A1 proofreads 3′ splice site recognition by U2AF. Mol Cell 45(3):314–329. doi:10.1016/j.molcel.2011.11.033
Thomsen C, Grundevik P, Elias P, Stahlberg A, Aman P, Ståhlberg A, Aman P (2013) A conserved N-terminal motif is required for complex formation between FUS, EWSR1, TAF15 and their oncogenic fusion proteins. FASEB J 27(12):4965–4974. doi:10.1096/fj.13-234435
Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D, Sapp PC et al (2011a) Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet 156B(3):285–290. doi:10.1002/ajmg.b.31158
Ticozzi N, LeClerc AL, van Blitterswijk M, Keagle P, McKenna-Yasek DM, Sapp PC, Silani V, Wills AM, Brown RH, Landers JE (2011b) Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol Aging 32(11):2096–2099. doi:10.1016/j.neurobiolaging.2009.11.018
Tio M, Wen R, Lim YL, Wang H, Ling SC, Zhao Y, Tan EK (2016) FUS-linked essential tremor associated with motor dysfunction in Drosophila. Hum Genet 135(11):1223–1232. doi:10.1007/s00439-016-1709-z
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosc 14(4):452–458. doi:10.1038/nn.2778 (Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved)
Tsai CP, Soong BW, Lin KP, Tu PH, Lin JL, Lee YC (2011) FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2010.04.009
Uranishi H, Tetsuka T, Yamashita M, Asamitsu K, Shimizu M, Itoh M, Okamoto T (2001) Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kB p65-mediated transcription as a coactivator. J Biol Chem 276(16):13395–13401. doi:10.1074/jbc.M011176200
Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Seelaar H et al (2010) FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120(1):33–41. doi:10.1007/s00401-010-0698-6
Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7(5):409–416. doi:10.1016/S1474-4422(08)70071-1
Van Langenhove T, Van Der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van Den Broeck M et al (2010) Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74(5):366–371. doi:10.1212/WNL.0b013e3181ccc732
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. doi:10.1126/science.1165942
Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H et al (2013) ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet 22(13):2676–2688. doi:10.1093/hmg/ddt117
Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, Sánchez-Madrid F (2013) Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4(December):2980. doi:10.1038/ncomms3980
Voigt A, Herholz D, Fiesel FC, Kaur K, Müller D, Karsten P, Weber SS, Kahle PJ, Marquardt T, Schulz JB (2010) TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One 5(8):e12247. doi:10.1371/journal.pone.0012247
Wang IF, Wu LS, Chang HY, Shen CK (2008a) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105(3):797–806. doi:10.1111/j.1471-4159.2007.05190.x
Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R (2008b) Induced ncRNAs allosterically modify rNA-binding proteins in cis to inhibit transcription. Nature 454(7200):126–130. doi:10.1038/nature06992
Wang W-Y, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IRA, Huang EJ, Tsai L-H (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16(10):1383–1391. doi:10.1038/nn.3514
Wang X, Schwartz JC, Cech TR (2015) Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res 43(15):7535–7543. doi:10.1093/nar/gkv679
Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S et al (2016a) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22(8):869–878. doi:10.1038/nm.4130
Wang YL, Chen H, Zhan YQ, Yin RH, Li CY, Ge CH, Yu M, Yang XM (2016b) EWSR1 regulates mitosis by dynamically influencing microtubule acetylation. Cell Cycle 15(16):2202–2215. doi:10.1080/15384101.2016.1200774
Watanabe S, Kaneko K, Yamanaka K (2013) Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J Biol Chem 288(5):3641–3654. doi:10.1074/jbc.M112.433615 (American Society for Biochemistry and Molecular Biology)
Weber SC, Brangwynne CP (2015) Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr Biol CB 25(5):641–646. doi:10.1016/j.cub.2015.01.012
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VMY (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283(19):13302–13309. doi:10.1074/jbc.M800342200
Woulfe J, Gray DA, MacKenzie IRA (2010) FUS-immunoreactive intranuclear inclusions in neurodegenerative disease. Brain Pathol 20(3):589–597. doi:10.1111/j.1750-3639.2009.00337.x
Wroe R, Wai-Ling Butler A, Andersen PM, Powell JF, Al-Chalabi A (2008) ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Later Scler 9(4):249–250. doi:10.1080/17482960802146106
Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L, Rogaeva E, Robertson J (2011) RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci 47(3):167–180. doi:10.1016/j.mcn.2011.02.013
Xiong H-L, Wang J-Y, Sun Y-M, Wu J-J, Chen Y, Qiao K, Zheng Q-J, Zhao G-X, WU Z-Y (2010) Association between novel TARDBP mutations and chinese patients with amyotrophic lateral sclerosis. BMC Med Genet 11(1):8. doi:10.1186/1471-2350-11-8
Yeo G, Holste D, Kreiman G, Burge CB (2004) Variation in alternative splicing across human tissues. Genome Biol 5(10):R74. doi:10.1186/gb-2004-5-10-r74
Yokoseki A, Shiga A, Tan C-F, Tagawa A, Kaneko H, Koyama A, Eguchi H et al (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63:538–542. doi:10.1002/ana.21392
Zhang ZC, Chook YM (2012) Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the fused in sarcoma protein (FUS). Proc Natl Acad Sci 109(30):12017–12021. doi:10.1073/pnas.1207247109
Zhou Z, Licklider LJ, Gygi SP, Reed R (2002) Comprehensive proteomic analysis of the human spliceosome. Nature 419(6903):182–185. doi:10.1038/nature01031
Zhou Y, Liu S, Liu G, Öztürk A, Hicks GG (2013) ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. doi:10.1371/journal.pgen.1003895
Zou Z-Y, Peng Y, Wang X-N, Liu M-S, Li X-G, Cui L-Y (2012) Screening of the TARDBP gene in familial and sporadic amyotrophic lateral sclerosis patients of chinese origin. Neurobiol Aging 33(9):2229.e11–2229.e18. doi:10.1016/j.neurobiolaging.2012.03.014
Acknowledgements
The authors thank Julia Nussbacher of the Yeo lab for critical reading of this manuscript. KK is supported by research grants from the National University of Singapore. FMJ was supported by the National Institute of General Medical Sciences of the NIH under award number T32GM008666. GWY is supported by grants from the National Institute of Health (HG004659, NS075449, HG007005) and the ALS Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kapeli, K., Martinez, F.J. & Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet 136, 1193–1214 (2017). https://doi.org/10.1007/s00439-017-1830-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1830-7