
Abstract
Neurological conditions, including cognitive impairment and Alzheimer’s disease (AD), impose a huge burden on society, affecting millions of people globally. In addition to genetic factors, recent studies indicate that environmental and experiential factors may contribute to the pathogenesis of these diseases. Early life adversity (ELA) has a profound impact on brain function and health later in life. In rodent models, exposure to ELA results in specific cognitive deficits and aggravated AD pathology. Extensive concerns have been raised regarding the higher risk of developing cognitive impairments in people with a history of ELA. In this review, we scrutinize findings from human and animal studies focusing on the connection of ELA with cognitive impairment and AD. These discoveries suggest that ELA, especially at early postnatal stages, increases susceptibility to cognitive impairment and AD later in life. In terms of mechanisms, ELA could lead to dysregulation of the hypothalamus-pituitary-adrenal axis, altered gut microbiome, persistent inflammation, oligodendrocyte dysfunction, hypomyelination, and aberrant adult hippocampal neurogenesis. Crosstalks among these events may synergistically contribute to cognitive impairment later in life. Additionally, we discuss several interventions that may alleviate adverse consequences of ELA. Further investigation into this crucial area will help improve ELA management and reduce the burden of related neurological conditions.
Similar content being viewed by others
Background
Cognitive impairment and Alzheimer’s disease (AD) are highly prevalent, affecting nearly 50 million people worldwide [1]. As a result, these conditions impart an outsized economic and emotional burden on individuals and society [2]. Moreover, as the populations of many nations age, the impact of neurological disorders is anticipated to rise dramatically in the coming decades [3, 4]. Several risk factors have been described to explain the pathogenesis of these diseases, including genetics, inadequate sleep, acquired factors, stress, and other environmental factors [5, 6]. Intriguingly, evidence from both human and animal research suggests that some environmental factors, particularly adverse early life experiences, may serve as key drivers in the pathogenesis of many neurological conditions [7, 8].
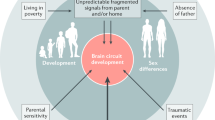
The developing brain is highly vulnerable to environmental factors in the first few years of life. Experiences during this time can permanently alter brain structure and function through epigenetic modifications, consequently increasing susceptibility to mental illness later in life [9,10,11]. Early parental caregiving has been proposed as a modulator for children’s mental development. Adequate early parental caregiving positively affects the brain development of the offspring, while poor parental caregiving could serve as a risk factor for adult mental illness in the offspring, as reviewed previously [12]. Adverse experiences in early life, including neglect and physical and emotional abuse, are known as early life adversity (ELA). Recent discoveries from both human studies and experimental animal models have supported the association between ELA and conduct disorders, impaired cognitive development, and a higher risk of dementia, AD and related neurodegenerative diseases [7, 8]. Although significant efforts have been made to mitigate the public health challenges of ELA in recent years, current understanding of this issue remains limited [13]. Hence, a better understanding of how ELA increases the risk of cognitive impairment and AD, as well as the corresponding management strategies, may help lessen the adverse consequences of ELA and reduce the global burden of these neurological illnesses.
In this review, we discuss the role of ELA in driving cognitive impairment and the onset of AD later in life. Despite the large number of cohort studies focusing on this issue, it is challenging to conduct invasive research on humans, which makes it difficult to establish a causal link. Thus, we will look at animal studies in-depth. In particular, this review will focus on potential mechanisms underlying ELA-related increased susceptibility to cognitive impairment and AD. Understanding these pathogenic processes will facilitate the development of therapeutic approaches. Finally, we summarize the current management strategies for ELA and propose potential interventions.
Main text
Consequences of ELA on later life health
ELA commonly involves exposure to adverse environmental circumstances during early life, such as physical, emotional, or sexual abuse and neglect [14, 15]. Nearly two decades have passed since the first discovery of strong links of adverse childhood experiences to increased lifetime risk of major diseases in a retrospective study[16]. Since then, concerns surrounding the effects of ELA on long-term health outcomes have grown substantially. Although an early report in rodents demonstrated that mild sensory stimulation in early life may favor neural connections and growth of the brain, thus providing benefits for cognitive and emotional development, chronic/extreme stress in early life may have detrimental effects on health [17]. Numerous human studies have demonstrated that adverse experiences during this sensitive period of development can increase the risk of a variety of adult diseases, including psychiatric illnesses, cardiovascular diseases, diabetes mellitus, and neurodegenerative diseases [18, 19].
ELA can also increase the risk of certain diseases in a degree-dependent manner later in life. As demonstrated in a national cross-sectional survey of adults in the US, exposure to multiple or repeated ELAs is associated with even poorer health outcomes [20]. Population-based studies suggest that the prevalence of ELA has increased to epidemic proportions, leading to a public health crisis [13, 21]. This has prompted researchers to consider the broader implications of ELA. Indeed, emerging empirical data from human and animal studies indicate that ELA exposure is connected to an increased risk of cognitive impairment in adulthood, as well as AD and other forms of dementia [7, 22,23,24,25]. Studies in animal models have shown that ELA can modulate neuronal morphology, especially causing dendritic changes [26, 27]. These results highlight the significant effects of ELA on the central nervous system (CNS), although the effects are yet to be fully understood. Moreover, to date, the link between ELA and neurodegenerative conditions has not been thoroughly investigated.
Animal models of ELA
Multiple animal models of ELA have been developed to mimic the long-term consequences of early adverse experiences in humans [28, 29]. With these animal models, research can be conducted under controlled conditions to compensate for limitations in human studies and establish a causal connection between these variables. In most cases, animal models of ELA involve stress exposure and changing the amount and quality of parental care during the early postnatal period, as described below.
Maternal separation/deprivation
Maternal separation or deprivation procedures involve altering mother-pup interactions during the early postnatal period. In the maternal separation procedure, pups are separated from the dam for 2–5 h/day over a set number of days, to induce an acute predictable level of stress [30,31,32,33]. Correspondingly, maternal deprivation involves prolonged separation of dam and pups during the early postnatal period, typically a single 24-h maternal deprivation session [34]. Long-term behavioral abnormalities and impaired cognitive performance have been reported in pups exposed to these paradigms [32].
Limited bedding and nesting (LBN) conditions
LBN conditions are another widely used ELA paradigm to simulate resource scarcity and provoke aberrant maternal care [35,36,37,38]. In this paradigm, most bedding and nesting materials in the home cage are removed, triggering mild, chronic stress in the dam. As a result, maternal care, a crucial source of environmental sensory signals for the developing brain, can become erratic and fragmented [35, 39]. This leads to abnormalities of maternal-derived sensory input and negatively impacts long-term outcomes for pups [35, 39, 40].
Chronic early life stress
Paradigms that cause chronic early life stress involve exposing pups to multiple stressors or repetitive, prolonged stressors. In some paradigms, the pups are exposed to 3 to 4 distinct types of stressors, generally comprising forced swimming, physical restriction, placement on an elevated platform, and foot shock exposure in early postnatal days (PND) [41, 42]. These combined factors can lead to significant physical and psychological stress [41]. Furthermore, an early foot shock paradigm has been developed to imitate early trauma or abuse experiences [43, 44]. Briefly, the pups are put in a closed, dark, electric shock apparatus during early postnatal time windows and receive a continuous electric foot shock, which can cause severe psychological and physiological stress [45, 46].
ELA and cognitive impairment
Studies in animals
Up to now, most studies on the association between ELA and cognitive outcomes have been conducted in rodents (Table 1). These animal models allow studies based on specific hypotheses and enable controlled conditions of experiments to avoid the inherent ethical issues in human studies. Preclinical animal studies have demonstrated that maternal care during early life profoundly impacts many aspects of the cognitive ability of offspring, and these effects can persist into adulthood [47, 48]. Pups that received high levels of maternal caregiving express higher levels of neurotrophic factors and exhibit better spatial learning and memory [47]. Based on these findings, many studies have been conducted in animal models to explore how inadequate maternal care influences the cognitive outcomes of the offspring. Several research groups have reported poor cognitive outcomes in adult rodents with a history of maternal separation/deprivation [22, 49,50,51,52,53,54]. Notably, these deficits are not apparent in PND-21 female animals with maternal deprivation during the early postnatal period, in comparison to age-matched male animals [55]. However, severe cognitive deficits have been observed in female animals on PND 40, indicating that the susceptibility to cognitive impairment might increase with age in female animals [49]. Likewise, using a visual-discrimination task, Yajima and colleagues [22] found that mice exposed to maternal deprivation display more evident cognitive impairment in middle age (1.4 years of age) than at a younger age. Intriguingly, lower levels of brain-derived neurotrophic factor (BDNF) and synapse-related proteins, accompanied by reduced number of mature neurons, were also detected in the hippocampus and prefrontal cortex of animals subjected to maternal separation/deprivation [49, 54]. This further supports poor maternal care as a potential risk factor for abnormal brain development.
Consistent with these findings, abnormal maternal behaviors, such as hypervigilance and abuse, which are induced by the LBN paradigm, profoundly impact the cognitive outcomes of their offspring [56]. Several early studies revealed that animals subjected to sporadic maternal care exhibit progressive cognitive deficits in adulthood. They also display impaired hippocampal long-term potentiation (a molecular basis of learning and memory) and structural changes such as dendritic atrophy and synaptic degeneration [56,57,58]. Furthermore, the survival of newborn neurons in the hippocampus is dramatically reduced in mice exposed to LBN from postnatal day 2 to day 9. These alterations are associated with aberrant cognitive performance. However, no changes in cell proliferation and neuronal differentiation were observed [59].
In light of the evidence that early traumatic experiences increase the vulnerability to mental illness in adulthood, increasing research interest has been focused on the consequences of traumatic experiences in early life. Chronic exposure to unavoidable plantar electroshock in the early post-weaning period leads to impaired spatial memory in adulthood, as evidenced by poor performance in the Y-maze or Morris water maze [44, 60]. Additionally, rats exposed to a single platform and acute swimming stress during adolescence also show inferior cognitive performance in adulthood, indicating that even relatively brief stress experiences early in life might exert profound, long-lasting effects on cognitive health [41, 61]. Overall, these data from animal models provide new insights into the crosstalk between ELA and cognitive impairment later in life.
Studies in humans
The connection between ELA and neurological consequences has been an active area of research since the discovery that adverse childhood experiences are positively linked to poor health outcomes later in life [16]. Numerous clinical studies have identified ELA as a potential risk factor for cognitive impairment (Table 2). These studies primarily focused on child neglect, physical abuse, and parental separation, thus providing an important context for understanding this health concern.
Parent-child coregulation is a dynamic process that involves mutual influence and coordination of emotional, behavioral, and physiological states between parents and their children [62]. Early coregulation is critical for the healthy development of children across multiple domains, including emotional and cognitive functioning [63, 64]. The importance of coregulation has been further highlighted by a longitudinal study which indicates that a secure infant-caregiver attachment during infancy may predict adult competence of children, including educational attainment, occupational success, and social functioning [65]. Intriguingly, a longitudinal study has demonstrated that positive mother–child interactions in kindergarten are associated with an increased likelihood of high school graduation and, for some students, a better academic performance [66].
One study examined the cognitive performances of adolescents with a documented history of ELA. Results showed that individuals who experienced child neglect, maltreatment, and unstable early environments exhibited poorer performance in the change task, a task designed to specifically assess cognitive control [67]. Similarly, a Helsinki birth cohort study investigated the ongoing effects of parental separation. At the age of 20 years, men who were separated from their parents during World War II scored lower in verbal, visuospatial, and general cognitive reasoning, and later at the age of 70, they exhibited lower scores in all tests in comparison to non-separated subjects [68]. Of note, a more extended separation period is associated with poorer overall cognitive performances. Several longitudinal studies have assessed how childhood neglect and maltreatment can influence cognitive outcomes later in life, revealing a remarkable connection between childhood neglect/maltreatment and lower general cognition or poorer working memory [23, 69]. Given that different forms of ELA often co-occur in some individuals, the effect of poly-victimization, which refers to experiencing multiple forms of victimization during a certain period, has been further analyzed. Those exposed to multiple types of ELA including physical/emotional abuse, harsh parenting, and domestic violence, experience more severe detrimental effects [23]. The adverse effects of ELA are further supported by the Romanian orphanage studies performed by many groups, which showed that childhood neglect/deprivation led to long-lasting effects on cognitive and emotional development [70, 71]. Children raised in institutions experienced severe deprivation (lack of individualized care and nurturing environment) early in life, and they exhibited lower executive functioning and increased diagnostic susceptibility to psychopathology compared to their peers who were never institutionalized [70]. Early childhood deprivation is associated with structural changes of the brain in adulthood. Adoptees who experienced early deprivation exhibited smaller total-brain volumes compared to nondeprived adoptees, which may be linked to lower intelligence quotient and higher levels of attention-deficit/hyperactivity disorder symptoms [71].
The effects of other types of ELA on cognition have also been examined. A nationally representative longitudinal cohort study analyzed the cumulative effects of multiple childhood adversities on cognitive decline in later life. The results of this study are profound: poor parent-child relationships, lack of friends, childhood parental mental health issues, and low socioeconomic status all negatively affected cognitive function among the middle-aged and elderly populations [72]. These conclusions are further supported by a longitudinal study demonstrating that low family socioeconomic status and poor social relationships in childhood are positively associated with the risk of mild cognitive impairment [73]. To gain more insights into the relationship between the number of ELA exposures and negative outcome expectations, scales with high internal consistency, validity, and test-retest reliability have been developed and employed in several cross-sectional studies. These studies showed that greater ELA exposure is associated with compromised cognitive flexibility, processing speed, and working memory [74, 75]. Moreover, these negative consequences may be amplified in subjects with depression, as evidenced by smaller orbitofrontal cortex and hippocampal volumes compared to never-depressed individuals [74]. Taken together, these human studies provide moderate evidence that ELA exposure is a risk factor for developing cognitive impairment later in life.
ELA and AD
Studies in animals
Numerous experimental AD models have been developed in rodents, exhibiting multiple key aspects of human AD pathology. The potential involvement of ELA in AD has been studied in rodent models (Table 3). APPswe/PS1dE9 mice exposed to LBN displayed aggravated Aβ plaque load at 10 months of age accompanied by increased glial activation and inflammatory signals in the hippocampus [76, 77]. LBN also increased the hippocampal levels of Aβ40 and Aβ42 and elevated the level of β-site APP-cleaving enzyme 1 (BACE1), a key rate-limiting enzyme for Aβ processing and production, in this mouse model [78]. In addition, synaptic damage and cognitive impairment are exacerbated by LBN exposure [78, 79].
Moreover, using different transgenic mouse models of AD, several groups have investigated the effects of maternal separation on AD disease progression. Hui et al. showed that chronic maternal separation worsened cognitive deficits and led to increased Aβ plaque formation and neural damage in adult APPswe/PS1dE9 mice [80]. Tanaka and colleagues focused on vascular pathological changes following maternal separation [81]. In their study, amyloid precursor protein (APP) wild-type and heterozygous APP mutant (AppNL−G−F/wt) mice displayed narrowed vessels in the prefrontal cortex with decreased capillary pericyte coverage and blood-brain barrier (BBB) disruption. Further analysis revealed that these deficits resulted from microglial activation. In addition, maternally separated AppNL−G−F/wt mice exhibited exacerbated cognitive impairment at four months of age [81]. More importantly, maternal separation has also been reported to induce Aβ formation in animals without an AD background. Martisova et al. exposed Wistar rats to three weeks of maternal separation, and found increases in Aβ40 and Aβ42 levels as well as increased expression of BACE1 in the hippocampus of these animals, along with profound cognitive deficits. These results suggest that ELA increases the risk of AD-like pathology development even in the absence of AD risk genes [82]. Furthermore, the effects of ELA exposure on AD pathology may occur through several common pathogenic mechanisms, perhaps differing by the stressor source.
Studies in humans
Multiple cross-sectional and longitudinal studies have documented the tight association between ELA and increased risk of AD or other dementias. Given that early parental death has profound implications for adult health outcomes, a longitudinal study conducted by Norton and colleagues analyzed the connection between this form of ELA and AD onset [83]. Their study included 4108 subjects, aged 65–105, with a mean follow-up of 18 months. Logistic regression analysis revealed a higher number of confirmed cases of AD within the 18-month follow-up in the group experiencing parental death in childhood [83]. Similarly, a cross-sectional study conducted in the Australian community used the self-reported childhood trauma questionnaire (CTQ) to evaluate the prevalence of ELA in older people. Based on the National Institute of Aging/Alzheimer’s disease Association workgroup criteria for AD diagnosis, the study showed that individuals with higher CTQ scores were more likely to receive a diagnosis of AD [24]. In addition, a longitudinal study investigated the association of childhood stress with late-life dementia and AD in 2682 male subjects. After adjustment via Cox regression analysis, a higher prevalence of AD was revealed among those who lived in custody or an orphanage, experienced an early-life crisis, had problems with teachers, or emigrated because of war during childhood [84].
In light of the increasing prevalence of dementia in Japan, several cohort studies have been conducted in Japan to address the crosstalk between ELA and dementia prevalence [85, 86], using the adverse childhood experience questionnaire, which covers family violence, physical and psychological abuse, neglect, parental death, parental divorce, and parental mental illness. Intriguingly, increased numbers of clinically confirmed dementia cases were reported within a 3-year follow-up period in participants with 3 or more adverse childhood experiences [85]. Further investigations indicated that the individual-level social capital score might be a variable for this vulnerability. After stratification by social capital score, this increased dementia risk was only observed in participants with low social capital [86]. More recently, several cross-sectional studies addressing ELA and cognitive outcomes have been performed in the USA. Among both older individuals or former National Football League players, those who experienced more than 4 ELA events had higher rates of positive dementia screening than those without a history of ELA [87, 88].
Taken together, these results indicate that all types of ELA confer an increased risk of developing dementia/AD, despite the widely varying ELA experiences among individuals. The studies mentioned above are summarized in Table 4.
Potential mechanisms linking ELA to cognitive impairment and AD
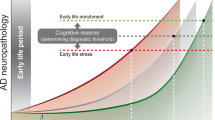
Understanding the mechanisms by which ELA influences the vulnerability to cognitive impairment and AD later in life can facilitate the development of preventive interventions and allow their timely application. Despite different stressors among studies, available evidence indicates that different ELAs can affect cognitive outcomes through shared mechanisms (Fig. 1).

The potential mechanisms underlying increased vulnerability to cognitive impairment and AD following ELA. ELA exposure can induce multiple physiological and pathological processes. Several key mechanisms may be implicated in the increased vulnerability to cognitive impairment and AD in later life, including HPA axis dysregulation, altered gut microbiome, oligodendrocyte dysfunction and hypomyelination, inflammation, and aberrant adult hippocampal neurogenesis. Furthermore, these pathological processes may interact with each other and collectively contribute to adverse neurological outcomes. HPA, Hypothalamus-pituitary-adrenal
Hypothalamus-pituitary-adrenal (HPA) axis dysregulation
The HPA axis is one of the major stress response systems in human bodies, and is responsible for the production of stress hormone glucocorticoids, including cortisol and corticosterone [89]. A well-functioning and dynamic HPA axis is essential for coping with stresses in daily life. Chronic stress, however, can disrupt its negative feedback system, leading to chronic activation of the HPA axis and elevated serum cortisol levels [90]. Dysregulated HPA axis and aberrant circulating cortisol levels have been associated with cognitive impairment and AD [91, 92]. Experiments in laboratory animals have shown that glucocorticoid administration can aggravate Aβ formation by increasing levels of APP and BACE [93]. Furthermore, elevated glucocorticoid also exacerbates the development of neurofibrillary tangles [93]. Conversely, mifepristone, an antagonist for glucocorticoid, has been extensively reported to alleviate AD pathology and cognitive decline [94, 95]. Therefore, an aberrant HPA axis may play a key role in the progression of cognitive impairment.
The HPA axis is highly plastic in early life. Notably, the HPA axis of infants is particularly sensitive to parental care, and absence of maternal care could negatively affect the HPA function of their offspring. Experiments performed on zebra finches revealed that maternally deprived offspring were hyper-responsive to stressors, exhibiting higher corticosterone concentrations and altered corticosteroid receptors in the brain [96]. In addition, prenatal stress can have a long-lasting impact on the reactivity of the HPA axis and the cognitive performance of the offspring [97, 98]. Molenaar et al. [98] found that maternal stress during pregnancy (e.g. long-lasting difficulties, stressful life events, family functioning, and pregnancy-related stress) was associated with higher child hair cortisone levels. Animal studies in rats showed that the offspring of prenatally stressed dams exhibited higher anxiety levels and elevated glucocorticoid levels in response to stressors [99].
The early postnatal environment is another critical factor influencing the HPA axis reactivity and sensitivity to stressors later in life. Mice exposed to LBN display hyperreactivity of the HPA axis and alterations of neuroendocrine stress responsiveness [100]. Moreover, maternal separation in rats induced changes of the HPA axis reactivity and increased stress hormone levels, accompanied by poorer cognitive performances in the Morris water maze and the novel object recognition test [101]. Strikingly, these deficits were entirely reversed by both mifepristone and β-adrenoceptor antagonist propranolol. These findings were supported by another study showing that LBN aggravated AD progression through activation of the HPA axis [78]. Intriguingly, short-term blockade of glucocorticoid receptors at 12 months of age was able to impede the accelerated progression of AD pathology due to ELA [78]. Importantly, a study by Kumsta et al. [102] reported that the effects of early-life institutional deprivation in Romanian orphanages can persist into adulthood. Individuals who experienced early life institutional deprivation showed HPA axis dysregulation in adulthood, including blunted cortisol responses to stress and alterations in cortisol metabolism [102].
Therefore, it is not surprising that ELA can cause aberrant HPA axis reactivity, potentially persisting lifelong and increasing susceptibility to cognitive impairment and AD. In this regard, interventions targeting the HPA axis may hold promise in alleviating the complex sequellae of adverse consequences of ELA.
Altered gut microbiome
A growing body of work over the last several years suggests that the gut microbiota impacts brain function. There are bidirectional interactions between the intestine and the CNS, known as the brain-gut-microbiome axis [103, 104]. To date, several mechanisms have been reported for the interactions between the gut microbiome and the CNS, including the immune system, the vagus nerve, the enteric nervous system, and the microbial-derived intermediates (e.g., short-chain fatty acids, secondary bile acids, and tryptophan metabolites) [103, 105]. Increasing evidence indicates that the gut microbiota plays a crucial role in neuroimmune signaling. Accordingly, absence of normal gut microbiota could directly disrupt CNS neurotransmission [106]. Gut microbiota is highly sensitive to the early environment, and colonization of microbiota in early life is increasingly connected to the host immune system [107]. Therefore, early life experiences may exert lasting effects on gut microbiota and the development of the host immune system through a “window of opportunity”.
Recent studies have reported that abnormalities in the gut microbiome are implicated in AD pathogenesis. The gut microbiota of patients with AD is strikingly different from that of healthy individuals [108, 109]. Moreover, people with mild cognitive impairment show similar alterations of the gut microbiota as AD patients [110, 111]. Experimental animal studies further support these findings. Gut microbiome analysis using 16S rRNA gene sequencing identified and characterized different compositions of the gut microbiota in transgenic AD animals and healthy controls [112,113,114]. Interestingly, cerebral amyloid plaques and neurofibrillary tangle pathology were significantly reduced in germ-free 3×Tg AD mice compared to mice with typical gut microbiota [112]. Microbiota transplantation from healthy animals also alleviated amyloid burden and tau pathology [113]. Conversely, gut microbiota from AD individuals aggravated AD disease progression and impaired cognitive function in normal animals [112, 115]. The reshaping of the gut microbiome using probiotics or nutrients has shown promise for AD treatment in several studies [116,117,118]. Taken together, these findings point toward the role of altered gut microbiota in driving cognitive impairment.
It has been demonstrated that stress induces gut microbiota alterations and disrupts the intestinal barrier integrity [119]. Recently, the effects of ELA on the brain-gut-microbiome axis have gained increasing attention. In young adulthood, maternally separated animals (from PND 2 to 12) showed an altered systemic immune response and increased visceral sensation, along with an alteration of the fecal microbiota [120]. Further studies revealed that alterations in Butyricimonas, Butyricicoccus, and Corynebacterium populations partially mediated the ELA-induced visceral hypersensitivity [121]. Using a multi-hit ELA model, Rincel et al. revealed sex-dependent gut dysbiosis in C3H/HeN mice. Male mice exposed to ELA displayed a significantly lower abundance of Lachnospiraceae and Porphyromonadaceae, accompanied by an increased abundance of Bacteroides, Lactobacillus, Porphyromonas, Alloprevotella, and Firmicutes genera. Female mice, on the other hand, only showed changes in Lactobacillus and Mucispirillum genera [122]. These results resemble gut microbiome alterations observed in early AD [123,124,125]. In addition to these changes, elevated levels of pro-inflammatory cytokines were observed in the colons of ELA-exposed animals [126]. Similarly, early weaning stress in piglets resulted in increased gut barrier permeability and diminished expression of gut epithelial tight junction proteins, along with higher levels of pro-inflammatory cytokines [127]. Notably, significant gut microbiome alterations have been observed in human subjects with a history of ELA [128, 129].
Taken together, these findings suggest that alterations in the brain-gut-microbiome axis play a role in ELA-induced cognitive impairment. However, the mechanisms by which ELA affects the brain-gut-microbiome axis and the exact microbiota that contributes to these adverse neurological consequences remain to be determined.
Oligodendrocyte dysfunction and hypomyelination
Another potential mechanism that links ELA to cognitive impairment and AD is oligodendrocyte dysfunction and hypomyelination. The myelin sheath is a multi-layered structure wrapping around the axon [130]. In the CNS, myelin sheaths are formed by mature oligodendrocytes through a process called myelination [130]. Myelination of axons is essential for normal CNS function in vertebrates. Intact myelin sheaths contribute to the conduction velocity of nerve impulses and provide trophic support for neurons and their axons [130, 131]. Intriguingly, emerging evidence highlights the importance of myelin in retaining cognition [132, 133]. Extensive myelin loss has been reported in AD patients and rodent models of AD, and enhancing myelin renewal rescues cognitive impairment in experimental animals [133, 134]. Moreover, depletion of oligodendrocytes or compromised oligodendrogenesis leads to cognitive dysfunction in normal animals [135, 136], whereas enhanced myelination can rescue age-related cognitive deficits [133]. All these findings imply that dysfunctional oligodendrocytes may directly contribute to cognitive dysfunction.
Stress has been identified as a trigger for oligodendrocyte dysfunction and hypomyelination [137]. Animals exposed to chronic stress show aberrant oligodendrocyte differentiation, hypomyelination, and oligodendroglial apoptosis in the prefrontal cortex and hippocampus, two brain areas involved in cognitive processing [138,139,140]. Furthermore, neuroimaging and postmortem histopathological studies also revealed compromised oligodendrocyte/myelin in individuals who experienced ELA. Postmortem brain samples from individuals with a history of child abuse showed decreased myelin thickness and an aberrant myelin-related transcriptional program [141]. A slower rate of myelin growth was also reported in those who experienced early-life economic disadvantage [142].
Animal studies have provided greater insights into the correlation between ELA, oligodendrocyte dysfunction/hypomyelination, and cognitive impairment. Bordner et al. reported a novel model to mimic early life neglect [143]. They exposed mice to neonatal maternal separation and early weaning, and conducted functional genomic and proteomic analyses after the animals reached adulthood. The results revealed significant dysregulations of myelin-related proteins and genes, including decreased expression of MBP, MAG, MOG, and PLP mRNA [143]. These findings were further supported by studies in rats [144, 145]. Rats separated from their dam during the first three postnatal weeks exhibited pronounced demyelination and aberrant oligodendrocyte differentiation at adulthood, accompanied by notable cognitive deficits [144]. Furthermore, inhibition of the Wnt signaling, which negatively regulates oligodendrocyte development, rescued these deficits [144]. To gain more insights into the abnormal oligodendrocyte processes resulting from ELA, Teissier and colleagues investigated oligodendrocyte changes in both early postnatal and adult stages after ELA induction in mice, and showed different patterns of variations [146]. Specifically, a precocious oligodendrocyte differentiation was observed in the prefrontal cortex immediately after maternal separation. However, depletion of the oligodendrocyte progenitor pool and compromised oligodendrocyte differentiation were observed in adulthood. Further mechanistic studies indicated that ELA decreased neuronal excitability, resulting in premature differentiation of oligodendrocytes and depletion of the oligodendrocyte precursor pool [146]. More importantly, chemogenetic activation of neuronal excitability normalized these alterations in ELA-exposed animals and mitigated memory impairment [146]. These studies suggest that ELA significantly impacts oligodendrocyte physiology, and dysfunctional oligodendrocytes and hypomyelination may contribute to subsequent cognitive deficits. Nevertheless, whether ELA also directly exacerbates myelin loss in the context of AD remains largely unclear. Further research is needed to address this issue.
Inflammation
As an integral part of innate immunity, the inflammatory response is the body’s self-protection mechanism to eliminate pathogens and protect against pathogen invasion. However, sustained or chronic inflammation can contribute to the progression of multiple diseases and neurodegeneration [147, 148]. Inflammation, particularly neuroinflammation, has been increasingly linked with cognitive impairment and neurodegeneration, persisting throughout the course of the diseases. Indeed, inflammation is a prominent feature of mild cognitive impairment and AD [149, 150]; elevated pro-inflammatory cytokines and chemokines have frequently been observed in the cerebrospinal fluid from patients with cognitive impairment or AD and are associated with amyloid and tau pathology [151, 152]. A meta-analysis involving 28 studies also revealed higher levels of inflammatory markers throughout the brain in people with cognitive impairment and confirmed AD cases [150]. In addition, exposure to acute inflammation aggravated the progression of AD in a mouse model of AD, which was accompanied by significant increases in inflammatory cytokines and chemokines in the brain [153]. Notably, loss of the NLR family pyrin domain containing 3 (NLRP3), an inflammasome implicated in multiple neurological diseases, prevented the development of AD-related pathology. In contrast, injection of fibrillar amyloid-beta-containing brain homogenate from AD animals induced AD pathology in an NLRP3-dependent manner [154]. Therefore, the development of inflammation could be a pivotal contributor to cognitive impairment.
Importantly, exposure to stress conditions can also trigger chronic inflammation. Acute electric foot shock has been shown to trigger NLRP3 inflammasome activation in the hippocampus and compromise fear memory and synaptic protein expression [155]. Of note, these deficits were not observed in Nlrp3-/- mice or animals treated with an NLRP3 inhibitor, suggesting the critical involvement of NLRP3 inflammasome in stress-driven inflammation [155]. Similarly, the expression of pro-inflammatory factors was elevated in animals exposed to neonatal maternal separation, which was accompanied by memory impairment [156, 157]. Furthermore, in APP/PS1 mice, LBN exposure led to lasting alterations in inflammation response, including increased hippocampal interleukin-1β expression and microglial activation, coinciding with accelerated disease progression [76].
Stress, including ELA, also leads to significant alterations of the neuroimmune profile. Microglia, the primary resident immune cells in the brain, are involved in phagocytosis and can release cytokines and neurotrophic factors [158]. Typically, microglia polarize into one of two different subtypes: pro-inflammatory (M1 phenotype) and anti-inflammatory (M2 phenotype) [159]. Both acute and chronic stress can trigger M1-phenotype microglial activation, causing the release of pro-inflammatory cytokines [76, 160, 161]. For instance, electric foot shocks and intermittent noise have been reported to induce microglial M1 polarization [136]. At four months of age, APPswe/PS1dE mice exposed to LBN (from PND 2 to 9) showed microglial activation and elevated levels of pro-inflammatory factors, along with an altered microglia response to Aβ neuropathology [76].
The activation of microglia and elevated pro-inflammatory factors were also observed in rats exposed to maternal separation [162]. Furthermore, activation of the stress system usually mediates alterations of the BBB permeability, which may further exacerbate the flow of peripheral inflammatory factors into the brain [163, 164]. Hence, chronic inflammation may be an important player connecting ELA and cognitive impairment/AD. However, further research is warranted to gain an in-depth understanding of the mechanisms by which ELA triggers chronic systemic inflammation and neuroinflammation. Notably, in addition to the pro-inflammatory activation of microglia, ELA has been reported to impair microglial function. Exposure to LBN resulted in compromised microglial process dynamics and deficits in microglial engulfment of synapses, leading to excess excitatory synapses in the hypothalamus and aberrant behavioral stress response [165]. Conversely, selective activation of microglia in early life has been shown to rescue these deficits. Therefore, microglial actions during development may also contribute to the adverse consequences of ELA [165].
Aberrant adult hippocampal neurogenesis (AHN)
The mammalian hippocampus, in particular the dentate gyrus (DG), is one of the few brain regions capable of generating new neurons throughout life. This process is known as AHN [166]. As a well-organized process, AHN involves the proliferation, migration, differentiation, and maturation of adult neural progenitor cells and has been implicated in many cognitive processes [166, 167]. Altered AHN has been reported in AD and may represent an early critical event in disease progression [168, 169]. Using state-of-the-art tissue processing methods, Moreno-Jimenez et al. observed immature neurons with variable degrees of maturation in the DG from neurologically healthy human subjects; however, the number and maturation of these neurons progressively declined alongside AD development [169]. Additionally, AHN depletion exacerbated cognitive deficits in AD and contributed to cognitive impairment in normal individuals [170, 171]. Conversely, pharmacological induction of AHN improved cognitive function in a mouse model of AD, suggesting a critical role of AHN in supporting cognitive function [172].
Importantly, AHN is strongly affected by early life experiences. The effects of ELA on AHN were first reported by Mirescu et al. in 2004. In their study, rat pups were subjected to maternal separation (3 h each day, from PND 1 to 14), and alterations in AHN were detected at adulthood. Strikingly, cell proliferation and immature neuron generation were decreased in the DG of maternally separated animals [173]. In addition, this compromised AHN was further demonstrated to involve a corticosteroid-dependent mechanism, as it could be reversed by lowering the corticosterone level. Similarly, ELA accelerates age-related AHN alterations. Compared to age-matched animals, rats that experienced neonatal maternal separation exhibited diminished dorsal hippocampal neurogenesis in the young adult period. These changes were more profound at 10 months of age, as evidenced by decreased neurogenesis in both the dorsal and the ventral hippocampus [174].
Several studies have further investigated the mechanisms underlying ELA-induced aberrant AHN [59, 175, 176]. Maternally separated animals showed enhanced hippocampal neurogenesis during the postnatal period and young adulthood, whereas compromised AHN was subsequently observed in middle age, compared to age-matched controls [175]. Another study further supported these results, reporting that LBN on PND 2–9 increased the proliferation and differentiation of neurons in the DG at PND 9 [59]. However, DG volume and survival of adult-born neurons were reduced at age of 5 months. Intriguingly, a reduction in neural stem cells was observed in the DG of adult mice that experienced LBN-induced ELA on PND 3–10 [176]. Hence, ELA-induced early progenitor pool depletion may underlie the aberrant AHN, thereby increasing susceptibility to cognitive impairment/AD in later life.
Potential crosstalk between different pathological mechanisms
Although we proposed several mechanisms to explain increased vulnerability to cognitive impairment/AD following ELA exposure, the adverse neurological consequences of ELA may not result from a single mechanism. Indeed, these mechanisms likely interact with each other and collectively contribute to the onset of cognitive impairment/AD later in life. For instance, evidence has shown that acute administration of glucocorticoids prior to lipopolysaccharide injection sensitizes both peripheral and central inflammatory responses to the immune challenge [177], and potentiate the pro-inflammatory response of hippocampal microglia to lipopolysaccharide [177]. These findings raise the possibility that the higher level of stress hormone glucocorticoid resulting from dysfunctional HPA axis may intensify inflammation, which could form a vicious feedback loop and lead to adverse neurological outcomes. There has also been accumulating evidence for the association between inflammation and neurogenesis. Pro-inflammatory cytokines produced peripherally can communicate with the brain and activate resident microglia, which subsequently release additional pro-inflammatory cytokines, ultimately impairing hippocampal neurogenesis [178, 179]. Moreover, the gut microbiome can also affect microglia and myelination in the brain [180, 181]. Disruption of the gut microbiome has been found to worsen the activation of microglia and neuroinflammation in mice, while administration of fecal microbiota transplants from healthy mice mitigates neuroinflammation [182]. Recent investigations have also suggested a connection between gut microbiome and myelination. Compared to conventionally raised mice, germ-free mice exhibited up-regulation of genes linked to myelination and myelin plasticity in the prefrontal cortex, concomitant with increased myelin sheath thickness [181]. Hence, healthy gut microbiomes may have a profound effect on oligodendrocyte development and myelin formation, but the specific intestinal microbes involved in myelination are still poorly understood.
Overall, although there has been a growing interest in the field, further investigations are necessary to ascertain the potential crosstalk between these pathological changes. Such research may provide critical insight into the intervention strategies to prevent the adverse outcomes of ELA.
Management of ELA and potential interventions
ELA affects children worldwide; unfortunately, therapeutic management options remain limited. Currently, ELA management focuses on preventive strategies. Family-targeted anticipatory guidance effectively fosters positive family functioning, improves parent-child relationships, and reduces child abuse and neglect [183]. Accordingly, several policy changes to improve early living conditions of children have been implemented [184]. Nevertheless, a substantial number of adverse consequences of ELA may not be apparent until later in life. Hence, post-hoc interventions aiming to mitigate the negative consequences of ELA are urgently required. In light of the increasing evidence that ELA is a risk factor for cognitive impairment and AD, interventions should be developed to help reduce the burden of these neurological illnesses.
Intriguingly, several interventions have shown the potential to alleviate cognitive impairment in ELA-exposed experimental animal models. Environmental enrichment involves housing animals in stimulating environments to facilitate improvement of cognitive function [185, 186]. Although the programs of environmental enrichment vary among laboratories, they typically involve the introduction of running wheels, toys, and larger cages. Environmental enrichment has shown beneficial effects in preventing ELA-induced cognitive impairment in several studies [187, 188]. Compared to maternally deprived controls, animals treated with early-life environmental enrichment (from PND 21 to 81) exhibited higher hippocampal expression of BDNF and cognitive improvements in adulthood [189]. Early environmental enrichment also preserved normal AHN and hippocampal-dependent functions [188]. More importantly, even short-term early environmental enrichment (from PND 22 to 34) can mitigate the adverse consequences of ELA, as evidenced by the retention of long-term potentiation and improved cognitive performance [190].
Nutritional supplements have also been reported to improve cognitive outcomes following ELA. Omega-3 fatty acids are essential for brain development and function [191, 192]. Recently, Omega-3 fatty acid supplementation has been suggested as a promising approach for AD prevention [193]. In LBN-induced ELA animal models, an early diet with Omega-3 fatty acids prevented cognitive impairments in adulthood and rescued the survival of newborn hippocampal cells [194]. Similarly, early administration of an Omega-3 fatty acid mixture also alleviated cognitive deficits in rats exposed to neonatal maternal separation [195]. Further studies revealed that an Omega-3 fatty acid mixture also altered gut microbiota composition in maternally separated rats, which was associated with a diminished corticosterone response to acute stress [196]. In addition, probiotic supplementation has shown promising results in improving outcomes after ELA [197, 198]. These observations provide promising new therapeutic avenues for ELA.
Exercise represents a promising, cost-effective intervention to prevent adverse consequences of ELA. Our group and others have reported positive effects of exercise in preventing AD and alleviating cognitive impairment in experimental animal models [199, 200]. Likewise, exercise seems to have beneficial effects on ELA-related pathophysiology. In rodent models, early voluntary exercise attenuated ELA-induced behavioral abnormalities in adulthood, accompanied by normalization of corticosterone level and decreased expression of pro-inflammatory factors [201, 202]. In addition, a study by Park and colleagues found that two weeks of treadmill exercise from PND 21 to 35 significantly ameliorated cognitive impairment induced by maternal separation [203]. Furthermore, our recent work demonstrated that photobiomodulation, a non-invasive therapy, alleviated oligodendrocyte dysfunctions induced by early unpredictable electric foot shock and prevented adverse outcomes, indicating the therapeutic potential of this treatment for ELA [42].
Notably, the therapeutic window is an important consideration when establishing ELA management strategies. Most preclinical studies to date have indicated that many interventions are effective in preventing ELA-induced cognitive impairment when administered early. Unfortunately, later interventions may sometimes be less effective. For instance, exercise, when initiated from 8 months of age, significantly promoted AHN; however, this effect was diminished in ELA-exposed animals [45]. Therefore, early intervention might be particularly important. Lastly, despite these encouraging results, the wide varieties of ELA type and degree make it challenging to establish standardized treatments and univocal guidelines from a clinical perspective. Further studies are thus needed to establish optimal treatment strategies, time windows, and dosages for individuals who have experienced different types of ELA.
Conclusion
As an environmental factor, ELA has tremendous impacts on brain development and can increase future susceptibility to neurological illnesses. The present review emphasizes ELA as a risk factor for cognitive impairment and AD in later life. ELA may contribute to the pathogenesis of cognitive impairment/AD through multiple pathophysiologic events, with potential crosstalk between mechanisms. Additionally, several interventions, including environmental enrichment, exercise, and nutritional supplements, have shown potentials in mitigating these adverse neurological consequences. Further studies are warranted to unravel the exact mechanisms underlying increased susceptibility to cognitive impairment and AD following ELA. Developing effective early interventions in parallel would help alleviate the disease burdens on individuals, their families, and the public health.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- AHN:
-
Adult hippocampal neurogenesis
- APP:
-
Amyloid precursor protein
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- CTQ:
-
Childhood trauma questionnaire
- CNS:
-
Central nervous system
- DG:
-
Dentate gyrus
- ELA:
-
Early life adversity
- LBN:
-
Limited bedding and nesting conditions
- BACE1:
-
β-site APP-cleaving enzyme 1
- HPA:
-
Hypothalamus-pituitary-adrenal
- NLRP3:
-
NLR family pyrin domain containing 3
References
World Health Organization. Risk reduction of cognitive decline and dementia: WHO guidelines. Geneva; 2019
Moon W, Han JW, Bae JB, Suh SW, Kim TH, Kwak KP, et al. Disease burdens of Alzheimer’s disease, vascular dementia, and mild cognitive impairment. J Am Med Dir Assoc. 2021;22:2093-2099.e3.
Collaborators GBDD. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18:88–106.
Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48.
Kivimaki M, Singh-Manoux A, Pentti J, Sabia S, Nyberg ST, Alfredsson L, et al. Physical inactivity, cardiometabolic disease, and risk of dementia: an individual-participant meta-analysis. BMJ. 2019;365:l1495.
Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG. Alzheimer’s disease: risk factors and potentially protective measures. J Biomed Sci. 2019;26:33.
Hoeijmakers L, Lesuis SL, Krugers H, Lucassen PJ, Korosi A. A preclinical perspective on the enhanced vulnerability to Alzheimer’s disease after early-life stress. Neurobiol Stress. 2018;8:172–85.
Short AK, Baram TZ. Early-life adversity and neurological disease: age-old questions and novel answers. Nat Rev Neurol. 2019;15:657–69.
Yuan B, Li J, Li K, Chen M. Longstanding health risk across the life course: the influence of early-life experience on health status throughout the life span. J Biosoc Sci. 2022. https://doi.org/10.1017/S002193202200027X.
Kundakovic M, Champagne FA. Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology. 2015;40:141–53.
Song S, Gleeson JG. Early life experience shapes neural genome. Science. 2018;359:1330–1.
Tottenham N. Early adversity and the neotenous human brain. Biol Psychiatry. 2020;87:350–8.
Lopez M, Ruiz MO, Rovnaghi CR, Tam GK, Hiscox J, Gotlib IH, et al. The social ecology of childhood and early life adversity. Pediatr Res. 2021;89:353–67.
McLaughlin KA. Future directions in childhood adversity and youth psychopathology. J Clin Child Adolesc Psychol. 2016;45:361–82.
Jones CM, Merrick MT, Houry DE. Identifying and preventing adverse childhood experiences: implications for clinical practice. JAMA. 2020;323:25–6.
Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, et al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. The adverse childhood experiences (ACE) study. Am J Prev Med. 1998;14:245–58.
Levine S. Stimulation in infancy. Sci Am. 1960;202:81–6.
Loria AS, Ho DH, Pollock JS. A mechanistic look at the effects of adversity early in life on cardiovascular disease risk during adulthood. Acta Physiol (Oxf). 2014;210:277–87.
Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, et al. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Public Health. 2017;2:e356–e66.
Gilbert LK, Breiding MJ, Merrick MT, Thompson WW, Ford DC, Dhingra SS, et al. Childhood adversity and adult chronic disease: an update from ten states and the District of Columbia, 2010. Am J Prev Med. 2015;48:345–9.
McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, Kessler RC. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication II: associations with persistence of DSM-IV disorders. Arch Gen Psychiatry. 2010;67:124–32.
Yajima H, Haijima A, Khairinisa MA, Shimokawa N, Amano I, Takatsuru Y. Early-life stress induces cognitive disorder in middle-aged mice. Neurobiol Aging. 2018;64:139–46.
Holland JF, Khandaker GM, Dauvermann MR, Morris D, Zammit S, Donohoe G. Effects of early life adversity on immune function and cognitive performance: results from the ALSPAC cohort. Soc Psychiatry Psychiatr Epidemiol. 2020;55:723–33.
Radford K, Delbaere K, Draper B, Mack HA, Daylight G, Cumming R, et al. Childhood stress and adversity is associated with late-life dementia in Aboriginal Australians. Am J Geriatr Psychiatry. 2017;25:1097–106.
Hedges DW, Woon FL. Early-life stress and cognitive outcome. Psychopharmacology. 2011;214:121–30.
Muhammad A, Kolb B. Maternal separation altered behavior and neuronal spine density without influencing amphetamine sensitization. Behav Brain Res. 2011;223:7–16.
McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29.
Benmhammed H, El Hayek S, Berkik I, Elmostafi H, Bousalham R, Mesfioui A, et al. Animal models of early-life adversity. Methods Mol Biol. 2019;2011:143–61.
Waters RC, Gould E. Early life adversity and neuropsychiatric disease: differential outcomes and translational relevance of rodent models. Front Syst Neurosci. 2022;16:860847.
Levine S. Infantile experience and resistance to physiological stress. Science. 1957;126:405.
Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM. Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci. 2007;25:3091–8.
Levis SC, Baram TZ, Mahler SV. Neurodevelopmental origins of substance use disorders: evidence from animal models of early-life adversity and addiction. Eur J Neurosci. 2022;55:2170–95.
Marco EM, Llorente R, Lopez-Gallardo M, Mela V, Llorente-Berzal A, Prada C, et al. The maternal deprivation animal model revisited. Neurosci Biobehav Rev. 2015;51:151–63.
Ellenbroek BA, Derks N, Park HJ. Early maternal deprivation retards neurodevelopment in Wistar rats. Stress. 2005;8:247–57.
Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–900.
Hsiao YM, Tsai TC, Lin YT, Chen CC, Huang CC, Hsu KS. Early life stress dampens stress responsiveness in adolescence: evaluation of neuroendocrine reactivity and coping behavior. Psychoneuroendocrinology. 2016;67:86–99.
Bolton JL, Schulmann A, Garcia-Curran MM, Regev L, Chen Y, Kamei N, et al. Unexpected transcriptional programs contribute to hippocampal memory deficits and neuronal stunting after early-life adversity. Cell Rep. 2020;33:108511.
Bonapersona V, Damsteegt R, Adams ML, van Weert L, Meijer OC, Joels M, et al. Sex-dependent modulation of acute stress reactivity after early life stress in mice: relevance of mineralocorticoid receptor expression. Front Behav Neurosci. 2019;13:181.
Molet J, Heins K, Zhuo X, Mei YT, Regev L, Baram TZ, et al. Fragmentation and high entropy of neonatal experience predict adolescent emotional outcome. Transl Psychiatry. 2016;6:e702.
Gallo M, Shleifer DG, Godoy LD, Ofray D, Olaniyan A, Campbell T, et al. Limited bedding and nesting induces maternal behavior resembling both hypervigilance and abuse. Front Behav Neurosci. 2019;13:167.
Avital A, Richter-Levin G. Exposure to juvenile stress exacerbates the behavioural consequences of exposure to stress in the adult rat. Int J Neuropsychopharmacol. 2005;8:163–73.
Huang Z, Zhang Y, Ma X, Feng Y, Zong X, Jordan JD, et al. Photobiomodulation attenuates oligodendrocyte dysfunction and prevents adverse neurological consequences in a rat model of early life adversity. Theranostics. 2023;13:913–30.
Zardooz H, Sadeghimahalli F, Khodagholi F. Early postnatal stress impairs insulin secretion in response to psychological stress in adult rats. J Endocrinol Invest. 2021;44:277–86.
Lu CY, Liu X, Jiang H, Pan F, Ho CS, Ho RC. Effects of traumatic stress induced in the juvenile period on the expression of gamma-aminobutyric acid receptor type a subunits in adult rat brain. Neural Plast. 2017;2017:5715816.
Abbink MR, Naninck EFG, Lucassen PJ, Korosi A. Early-life stress diminishes the increase in neurogenesis after exercise in adult female mice. Hippocampus. 2017;27:839–44.
Sadeghimahalli F, Karbaschi R, Zardooz H, Khodagholi F, Rostamkhani F. Effect of early life stress on pancreatic isolated islets’ insulin secretion in young adult male rats subjected to chronic stress. Endocrine. 2015;48:493–503.
Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci. 2000;3:799–806.
Feldman R, Rosenthal Z, Eidelman AI. Maternal-preterm skin-to-skin contact enhances child physiologic organization and cognitive control across the first 10 years of life. Biol Psychiatry. 2014;75:56–64.
Marco EM, Valero M, de la Serna O, Aisa B, Borcel E, Ramirez MJ, et al. Maternal deprivation effects on brain plasticity and recognition memory in adolescent male and female rats. Neuropharmacology. 2013;68:223–31.
Grassi-Oliveira R, Honeycutt JA, Holland FH, Ganguly P, Brenhouse HC. Cognitive impairment effects of early life stress in adolescents can be predicted with early biomarkers: impacts of sex, experience, and cytokines. Psychoneuroendocrinology. 2016;71:19–30.
Loi M, Mossink JC, Meerhoff GF, Den Blaauwen JL, Lucassen PJ, Joels M. Effects of early-life stress on cognitive function and hippocampal structure in female rodents. Neuroscience. 2017;342:101–19.
Feifel AJ, Shair HN, Schmauss C. Lasting effects of early life stress in mice: interaction of maternal environment and infant genes. Genes Brain Behav. 2017;16:768–80.
Janetsian-Fritz SS, Timme NM, Timm MM, McCane AM, Baucum Ii AJ, O’Donnell BF, et al. Maternal deprivation induces alterations in cognitive and cortical function in adulthood. Transl Psychiatry. 2018;8:71.
Reshetnikov VV, Kovner AV, Lepeshko AA, Pavlov KS, Grinkevich LN, Bondar NP. Stress early in life leads to cognitive impairments, reduced numbers of CA3 neurons and altered maternal behavior in adult female mice. Genes Brain Behav. 2020;19:e12541.
Xu H, Ye Y, Hao Y, Shi F, Yan Z, Yuan G, et al. Sex differences in associations between maternal deprivation and alterations in hippocampal calcium-binding proteins and cognitive functions in rats. Behav Brain Funct. 2018;14:10.
Wang XD, Rammes G, Kraev I, Wolf M, Liebl C, Scharf SH, et al. Forebrain CRF(1) modulates early-life stress-programmed cognitive deficits. J Neurosci. 2011;31:13625–34.
Brunson KL, Kramar E, Lin B, Chen Y, Colgin LL, Yanagihara TK, et al. Mechanisms of late-onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–38.
Ivy AS, Rex CS, Chen Y, Dube C, Maras PM, Grigoriadis DE, et al. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30:13005–15.
Naninck EF, Hoeijmakers L, Kakava-Georgiadou N, Meesters A, Lazic SE, Lucassen PJ, et al. Chronic early life stress alters developmental and adult neurogenesis and impairs cognitive function in mice. Hippocampus. 2015;25:309–28.
Li C, Liu Y, Yin S, Lu C, Liu D, Jiang H, et al. Long-term effects of early adolescent stress: dysregulation of hypothalamic-pituitary-adrenal axis and central corticotropin releasing factor receptor 1 expression in adult male rats. Behav Brain Res. 2015;288:39–49.
Liu H, Atrooz F, Salvi A, Salim S. Behavioral and cognitive impact of early life stress: insights from an animal model. Prog Neuropsychopharmacol Biol Psychiatry. 2017;78:88–95.
Lobo FM, Lunkenheimer E. Understanding the parent-child coregulation patterns shaping child self-regulation. Dev Psychol. 2020;56:1121–34.
Schore AN. Effects of a secure attachment relationship on right brain development, affect regulation, and infant mental health. Infant Mental Health J Off Publ World Assoc Infant Mental Health. 2001;22:7–66.
Paley B, Hajal NJ. Conceptualizing emotion regulation and coregulation as family-level phenomena. Clin Child Fam Psychol Rev. 2022;25:19–43.
Englund MM, Kuo SI, Puig J, Collins WA. Early roots of adult competence: the significance of close relationships from infancy to early adulthood. Int J Behav Dev. 2011;35:490–6.
Gregory A, Rimm-Kaufman S. Positive mother–child interactions in kindergarten: Predictors of school success in high school. Sch Psychol Rev. 2008;37(4):499–515.
Mueller SC, Maheu FS, Dozier M, Peloso E, Mandell D, Leibenluft E, et al. Early-life stress is associated with impairment in cognitive control in adolescence: an fMRI study. Neuropsychologia. 2010;48:3037–44.
Pesonen AK, Eriksson JG, Heinonen K, Kajantie E, Tuovinen S, Alastalo H, et al. Cognitive ability and decline after early life stress exposure. Neurobiol Aging. 2013;34:1674–9.
Hawkins MAW, Layman HM, Ganson KT, Tabler J, Ciciolla L, Tsotsoros CE, et al. Adverse childhood events and cognitive function among young adults: prospective results from the national longitudinal study of adolescent to adult health. Child Abuse Negl. 2021;115:105008.
Wade M, Zeanah CH, Fox NA, Nelson CA. Global deficits in executive functioning are transdiagnostic mediators between severe childhood neglect and psychopathology in adolescence. Psychol Med. 2020;50:1687–94.
Mackes NK, Golm D, Sarkar S, Kumsta R, Rutter M, Fairchild G, et al. Early childhood deprivation is associated with alterations in adult brain structure despite subsequent environmental enrichment. Proc Natl Acad Sci U S A. 2020;117:641–9.
Ma J, Yang Y, Wan Y, Shen C, Qiu P. The influence of childhood adversities on mid to late cognitive function: from the perspective of life course. PLoS ONE. 2021;16:e0256297.
Zhang K, Zhang W. Adverse childhood experiences and mild cognitive impairment in later life: exploring rural/urban and gender differences using CHARLS. J Appl Gerontol. 2022;41:1454–64.
Saleh A, Potter GG, McQuoid DR, Boyd B, Turner R, MacFall JR, et al. Effects of early life stress on depression, cognitive performance and brain morphology. Psychol Med. 2017;47:171–81.
Kalia V, Knauft K, Hayatbini N. Adverse childhood experiences (ACEs) associated with reduced cognitive flexibility in both college and community samples. PLoS ONE. 2021;16:e0260822.
Hoeijmakers L, Ruigrok SR, Amelianchik A, Ivan D, van Dam AM, Lucassen PJ, et al. Early-life stress lastingly alters the neuroinflammatory response to amyloid pathology in an Alzheimer’s disease mouse model. Brain Behav Immun. 2017;63:160–75.
Abbink MR, Kotah JM, Hoeijmakers L, Mak A, Yvon-Durocher G, van der Gaag B, et al. Characterization of astrocytes throughout life in wildtype and APP/PS1 mice after early-life stress exposure. J Neuroinflammation. 2020;17:91.
Lesuis SL, Weggen S, Baches S, Lucassen PJ, Krugers HJ. Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS1 mice. Transl Psychiatry. 2018;8:53.
Lesuis SL, Kaplick PM, Lucassen PJ, Krugers HJ. Treatment with the glutamate modulator riluzole prevents early life stress-induced cognitive deficits and impairments in synaptic plasticity in APPswe/PS1dE9 mice. Neuropharmacology. 2019;150:175–83.
Hui J, Feng G, Zheng C, Jin H, Jia N. Maternal separation exacerbates Alzheimer’s disease-like behavioral and pathological changes in adult APPswe/PS1dE9 mice. Behav Brain Res. 2017;318:18–23.
Tanaka T, Hirai S, Hosokawa M, Saito T, Sakuma H, Saido T, et al. Early-life stress induces the development of Alzheimer’s disease pathology via angiopathy. Exp Neurol. 2021;337:113552.
Martisova E, Aisa B, Guerenu G, Ramirez MJ. Effects of early maternal separation on biobehavioral and neuropathological markers of Alzheimer’s disease in adult male rats. Curr Alzheimer Res. 2013;10:420–32.
Norton MC, Smith KR, Ostbye T, Tschanz JT, Schwartz S, Corcoran C, et al. Early parental death and remarriage of widowed parents as risk factors for Alzheimer disease: the Cache County study. Am J Geriatr Psychiatry. 2011;19:814–24.
Donley GAR, Lonnroos E, Tuomainen TP, Kauhanen J. Association of childhood stress with late-life dementia and Alzheimer’s disease: the KIHD study. Eur J Public Health. 2018;28:1069–73.
Tani Y, Fujiwara T, Kondo K. Association between adverse childhood experiences and dementia in older japanese adults. JAMA Netw Open. 2020;3:e1920740.
Tani Y, Fujiwara T, Kondo K. Adverse childhood experiences and dementia: interactions with social capital in the japan gerontological evaluation study cohort. Am J Prev Med. 2021;61:225–34.
Schickedanz HB, Jennings LA, Schickedanz A. The association between adverse childhood experiences and positive dementia screen in American older adults. J Gen Intern Med. 2022;37:2398–404.
Roberts AL, Zafonte R, Chibnik LB, Baggish A, Taylor H, Baker J, et al. Association of adverse childhood experiences with poor neuropsychiatric health and dementia among former professional US football players. JAMA Netw Open. 2022;5:e223299.
Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63.
Gjerstad JK, Lightman SL, Spiga F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress. 2018;21:403–16.
Lind K, Edman A, Nordlund A, Olsson T, Wallin A. Increased saliva cortisol awakening response in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24:389–95.
Milligan Armstrong A, Porter T, Quek H, White A, Haynes J, Jackaman C, et al. Chronic stress and Alzheimer’s disease: the interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol Rev Camb Philos Soc. 2021;96:2209–28.
Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–56.
Baglietto-Vargas D, Medeiros R, Martinez-Coria H, LaFerla FM, Green KN. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol Psychiatry. 2013;74:357–66.
Belanoff JK, Jurik J, Schatzberg LD, DeBattista C, Schatzberg AF. Slowing the progression of cognitive decline in Alzheimer’s disease using mifepristone. J Mol Neurosci. 2002;19:201–6.
Banerjee SB, Arterbery AS, Fergus DJ, Adkins-Regan E. Deprivation of maternal care has long-lasting consequences for the hypothalamic-pituitary-adrenal axis of zebra finches. Proc Biol Sci. 2012;279:759–66.
Jafari Z, Mehla J, Kolb BE, Mohajerani MH. Prenatal noise stress impairs HPA axis and cognitive performance in mice. Sci Rep. 2017;7:10560.
Molenaar NM, Tiemeier H, van Rossum EFC, Hillegers MHJ, Bockting CLH, Hoogendijk WJG, et al. Prenatal maternal psychopathology and stress and offspring HPA axis function at 6 years. Psychoneuroendocrinology. 2019;99:120–7.
Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone secretion. J Neurosci. 1997;17:2626–36.
McIlwrick S, Rechenberg A, Matthes M, Burgstaller J, Schwarzbauer T, Chen A, et al. Genetic predisposition for high stress reactivity amplifies effects of early-life adversity. Psychoneuroendocrinology. 2016;70:85–97.
Aisa B, Tordera R, Lasheras B, Del Rio J, Ramirez MJ. Cognitive impairment associated to HPA axis hyperactivity after maternal separation in rats. Psychoneuroendocrinology. 2007;32:256–66.
Kumsta R, Schlotz W, Golm D, Moser D, Kennedy M, Knights N, et al. HPA axis dysregulation in adult adoptees twenty years after severe institutional deprivation in childhood. Psychoneuroendocrinology. 2017;86:196–202.
Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99:1877–2013.
Socala K, Doboszewska U, Szopa A, Serefko A, Wlodarczyk M, Zielinska A, et al. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol Res. 2021;172:105840.
Osadchiy V, Martin CR, Mayer EA. The gut-brain axis and the microbiome: mechanisms and clinical implications. Clin Gastroenterol Hepatol. 2019;17:322–32.
Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry. 2013;18:666–73.
Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, et al. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. 2017;81.
Zhuang ZQ, Shen LL, Li WW, Fu X, Zeng F, Gui L, et al. Gut microbiota is altered in patients with Alzheimer’s disease. J Alzheimers Dis. 2018;63:1337–46.
Guo M, Peng J, Huang X, Xiao L, Huang F, Zuo Z. Gut microbiome features of chinese patients newly diagnosed with Alzheimer’s disease or mild cognitive impairment. J Alzheimers Dis. 2021;80:299–310.
Li B, He Y, Ma J, Huang P, Du J, Cao L, et al. Mild cognitive impairment has similar alterations as Alzheimer’s disease in gut microbiota. Alzheimers Dement. 2019;15:1357–66.
Wu L, Han Y, Zheng Z, Peng G, Liu P, Yue S, et al. Altered gut microbial metabolites in amnestic mild cognitive impairment and Alzheimer’s disease: signals in host–microbe interplay. Nutrients. 2021. https://doi.org/10.3390/nu13010228.
Chen C, Liao J, Xia Y, Liu X, Jones R, Haran J, et al. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut. 2022;71:2233–52.
Kim MS, Kim Y, Choi H, Kim W, Park S, Lee D, et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut. 2020;69:283–94.
Tran TTT, Corsini S, Kellingray L, Hegarty C, Le Gall G, Narbad A, et al. APOE genotype influences the gut microbiome structure and function in humans and mice: relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019;33:8221–31.
Kim N, Jeon SH, Ju IG, Gee MS, Do J, Oh MS, et al. Transplantation of gut microbiota derived from Alzheimer’s disease mouse model impairs memory function and neurogenesis in C57BL/6 mice. Brain Behav Immun. 2021;98:357–65.
Yang X, Yu D, Xue L, Li H, Du J. Probiotics modulate the microbiota-gut-brain axis and improve memory deficits in aged SAMP8 mice. Acta Pharm Sin B. 2020;10:475–87.
Kim CS, Cha L, Sim M, Jung S, Chun WY, Baik HW, et al. Probiotic supplementation improves cognitive function and mood with changes in gut microbiota in community-dwelling older adults: a randomized, double-blind, placebo-controlled, multicenter trial. J Gerontol A Biol Sci Med Sci. 2021;76:32–40.
Liu Q, Xi Y, Wang Q, Liu J, Li P, Meng X, et al. Mannan oligosaccharide attenuates cognitive and behavioral disorders in the 5xFAD Alzheimer’s disease mouse model via regulating the gut microbiota-brain axis. Brain Behav Immun. 2021;95:330–43.
Deng Y, Zhou M, Wang J, Yao J, Yu J, Liu W, et al. Involvement of the microbiota-gut-brain axis in chronic restraint stress: disturbances of the kynurenine metabolic pathway in both the gut and brain. Gut Microbes. 2021;13:1–16.
O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65:263–7.
Enqi W, Jingzhu S, Lingpeng P, Yaqin L. Comparison of the gut microbiota disturbance in rat models of irritable bowel syndrome induced by maternal separation and multiple early-life adversity. Front Cell Infect Microbiol. 2020;10:581974.
Rincel M, Aubert P, Chevalier J, Grohard PA, Basso L, Monchaux de Oliveira C, et al. Multi-hit early life adversity affects gut microbiota, brain and behavior in a sex-dependent manner. Brain Behav Immun. 2019;80:179–92.
Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7:13537.
Haran JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, et al. Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. mBio. 2019;10.
Zhou Y, Wang Y, Quan M, Zhao H, Jia J. Gut microbiota changes and their correlation with cognitive and neuropsychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis. 2021;81:583–95.
Park HJ, Kim SA, Kang WS, Kim JW. Early-life stress modulates gut microbiota and peripheral and central inflammation in a sex-dependent manner. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22041899.
Ma X, Zhang Y, Xu T, Qian M, Yang Z, Zhan X, et al. Early-life intervention using exogenous fecal microbiota alleviates gut injury and reduce inflammation caused by weaning stress in piglets. Front Microbiol. 2021;12:671683.
Reid BM, Horne R, Donzella B, Szamosi JC, Coe CL, Foster JA, et al. Microbiota-immune alterations in adolescents following early life adversity: a proof of concept study. Dev Psychobiol. 2021;63:851–63.
Coley EJL, Mayer EA, Osadchiy V, Chen Z, Subramanyam V, Zhang Y, et al. Early life adversity predicts brain-gut alterations associated with increased stress and mood. Neurobiol Stress. 2021;15:100348.
Simons M, Nave KA. Oligodendrocytes: myelination and axonal support. Cold Spring Harb Perspect Biol. 2015;8:a020479.
Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927.
Pan S, Mayoral SR, Choi HS, Chan JR, Kheirbek MA. Preservation of a remote fear memory requires new myelin formation. Nat Neurosci. 2020;23:487–99.
Wang F, Ren SY, Chen JF, Liu K, Li RX, Li ZF, et al. Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat Neurosci. 2020;23:481–6.
Chen JF, Liu K, Hu B, Li RR, Xin W, Chen H, et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron. 2021;109:2292-307.e5.
Steadman PE, Xia F, Ahmed M, Mocle AJ, Penning ARA, Geraghty AC, et al. Disruption of oligodendrogenesis impairs memory consolidation in adult mice. Neuron. 2020;105:150-64.e6.
Chen X, Wang F, Gan J, Zhang Z, Liang X, Li T, et al. Myelin deficits caused by Olig2 deficiency lead to cognitive dysfunction and increase vulnerability to social withdrawal in adult mice. Neurosci Bull. 2020;36:419–26.
Boda E. Myelin and oligodendrocyte lineage cell dysfunctions: New players in the etiology and treatment of depression and stress-related disorders. Eur J Neurosci. 2021;53:281–97.
Bonnefil V, Dietz K, Amatruda M, Wentling M, Aubry AV, Dupree JL, et al. Region-specific myelin differences define behavioral consequences of chronic social defeat stress in mice. Elife. 2019;8.
Kokkosis AG, Madeira MM, Mullahy MR, Tsirka SE. Chronic stress disrupts the homeostasis and progeny progression of oligodendroglial lineage cells, associating immune oligodendrocytes with prefrontal cortex hypomyelination. Mol Psychiatry. 2022;27:2833–48.
Huang CX, Xiao Q, Zhang L, Gao Y, Ma J, Liang X, et al. Stress-induced myelin damage in the hippocampal formation in a rat model of depression. J Psychiatr Res. 2022;155:401–9.
Lutz PE, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, Zhou Y, et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Am J Psychiatry. 2017;174:1185–94.
Ziegler G, Moutoussis M, Hauser TU, Fearon P, Bullmore ET, Goodyer IM, et al. Childhood socio-economic disadvantage predicts reduced myelin growth across adolescence and young adulthood. Hum Brain Mapp. 2020;41:3392–402.
Bordner KA, George ED, Carlyle BC, Duque A, Kitchen RR, Lam TT, et al. Functional genomic and proteomic analysis reveals disruption of myelin-related genes and translation in a mouse model of early life neglect. Front Psychiatry. 2011;2:18.
Yang Y, Cheng Z, Tang H, Jiao H, Sun X, Cui Q, et al. Neonatal maternal separation impairs prefrontal cortical myelination and cognitive functions in rats through activation of wnt signaling. Cereb Cortex. 2017;27:2871–84.
Wang Y, Su Y, Yu G, Wang X, Chen X, Yu B, et al. Reduced oligodendrocyte precursor cell impairs astrocytic development in early life stress. Adv Sci (Weinh). 2021;8:e2101181.
Teissier A, Le Magueresse C, Olusakin J, Andrade da Costa BLS, De Stasi AM, Bacci A, et al. Early-life stress impairs postnatal oligodendrogenesis and adult emotional behaviour through activity-dependent mechanisms. Mol Psychiatry. 2020;25:1159–74.
Walker KA. Inflammation and neurodegeneration: chronicity matters. Aging. 2018;11:3–4.
Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation. 2018;15:276.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421.
Bradburn S, Murgatroyd C, Ray N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. Ageing Res Rev. 2019;50:1–8.
Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Immunol. 2018;9:2122.
Brosseron F, Traschutz A, Widmann CN, Kummer MP, Tacik P, Santarelli F, et al. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimers Res Ther. 2018;10:25.
Lopez-Rodriguez AB, Hennessy E, Murray CL, Nazmi A, Delaney HJ, Healy D, et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1beta drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 2021;17:1735–55.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–73.
Dong Y, Li S, Lu Y, Li X, Liao Y, Peng Z, et al. Stress-induced NLRP3 inflammasome activation negatively regulates fear memory in mice. J Neuroinflammation. 2020;17:205.
Jolodar SK, Bigdeli M, Moghaddam AH. Hypericin ameliorates maternal separation-induced cognitive deficits and hippocampal inflammation in rats. Mini Rev Med Chem. 2021;21:1144–9.
Mi X, Zeng GR, Liu JQ, Luo ZS, Zhang L, Dai XM, et al. Ganoderma lucidum triterpenoids improve maternal separation-induced anxiety- and depression-like behaviors in mice by mitigating inflammation in the periphery and brain. Nutrients. 2022;14:2268.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Hu X. Microglia/macrophage polarization: Fantasy or evidence of functional diversity? J Cereb Blood Flow Metab. 2020;40:134–S6.
Wang YL, Han QQ, Gong WQ, Pan DH, Wang LZ, Hu W, et al. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J Neuroinflammation. 2018;15:21.
Sugama S, Takenouchi T, Hashimoto M, Ohata H, Takenaka Y, Kakinuma Y. Stress-induced microglial activation occurs through beta-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. J Neuroinflammation. 2019;16:266.
Roque A, Ochoa-Zarzosa A, Torner L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav Immun. 2016;55:39–48.
Gomez-Gonzalez B, Escobar A. Altered functional development of the blood-brain barrier after early life stress in the rat. Brain Res Bull. 2009;79:376–87.
Welcome MO, Mastorakis NE. Stress-induced blood brain barrier disruption: molecular mechanisms and signaling pathways. Pharmacol Res. 2020;157:104769.
Bolton JL, Short AK, Othy S, Kooiker CL, Shao M, Gunn BG, et al. Early stress-induced impaired microglial pruning of excitatory synapses on immature CRH-expressing neurons provokes aberrant adult stress responses. Cell Rep. 2022;38:110600.
Anacker C, Hen R. Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat Rev Neurosci. 2017;18:335–46.
Moreno-Jimenez EP, Terreros-Roncal J, Flor-Garcia M, Rabano A, Llorens-Martin M. Evidences for adult hippocampal neurogenesis in humans. J Neurosci. 2021;41:2541–53.
Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85.
Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25:554–60.
Hollands C, Tobin MK, Hsu M, Musaraca K, Yu TS, Mishra R, et al. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol Neurodegener. 2017;12:64.
Xu L, Guo Y, Wang G, Sun G, Sun W, Li J, et al. Inhibition of adult hippocampal neurogenesis plays a role in sevoflurane-induced cognitive impairment in aged mice through brain-derived neurotrophic factor/tyrosine receptor kinase B and neurotrophin-3/tropomyosin receptor kinase C pathways. Front Aging Neurosci. 2022;14:782932.
Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. 2018;361:eaan8821.
Mirescu C, Peters JD, Gould E. Early life experience alters response of adult neurogenesis to stress. Nat Neurosci. 2004;7:841–6.
Ruiz R, Roque A, Pineda E, Licona-Limon P, Jose Valdez-Alarcon J, Lajud N. Early life stress accelerates age-induced effects on neurogenesis, depression, and metabolic risk. Psychoneuroendocrinology. 2018;96:203–11.
Suri D, Veenit V, Sarkar A, Thiagarajan D, Kumar A, Nestler EJ, et al. Early stress evokes age-dependent biphasic changes in hippocampal neurogenesis, BDNF expression, and cognition. Biol Psychiatry. 2013;73:658–66.
Youssef M, Atsak P, Cardenas J, Kosmidis S, Leonardo ED, Dranovsky A. Early life stress delays hippocampal development and diminishes the adult stem cell pool in mice. Sci Rep. 2019;9:4120.
Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. 2010;24:19–30.
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–7.
Chesnokova V, Pechnick RN, Wawrowsky K. Chronic peripheral inflammation, hippocampal neurogenesis, and behavior. Brain Behav Immun. 2016;58:1–8.
Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–77.
Hoban AE, Stilling RM, Ryan FJ, Shanahan F, Dinan TG, Claesson MJ, et al. Regulation of prefrontal cortex myelination by the microbiota. Transl Psychiatry. 2016;6:e774.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469-80.e12.
Sege RD, Hatmaker-Flanigan E, De Vos E, Levin-Goodman R, Spivak H. Anticipatory guidance and violence prevention: results from family and pediatrician focus groups. Pediatrics. 2006;117:455–63.
Shaefer HL, Collyer S, Duncan G, Edin K, Garfinkel I, Harris D, et al. A universal child allowance: a plan to reduce poverty and income instability among children in the United States. RSF. 2018;4:22–42.
Kempermann G. Environmental enrichment, new neurons and the neurobiology of individuality. Nat Rev Neurosci. 2019;20:235–45.
Ohline SM, Abraham WC. Environmental enrichment effects on synaptic and cellular physiology of hippocampal neurons. Neuropharmacology. 2019;145:3–12.
do Prado CH, Narahari T, Holland FH, Lee HN, Murthy SK, Brenhouse HC. Effects of early adolescent environmental enrichment on cognitive dysfunction, prefrontal cortex development, and inflammatory cytokines after early life stress. Dev Psychobiol. 2016;58:482–91.
Rule L, Yang J, Watkin H, Hall J, Brydges NM. Environmental enrichment rescues survival and function of adult-born neurons following early life stress. Mol Psychiatry. 2021;26:1898–908.
Menezes J, Souto das Neves BH, Goncalves R, Benetti F, Mello-Carpes PB. Maternal deprivation impairs memory and cognitive flexibility, effect that is avoided by environmental enrichment. Behav Brain Res. 2020;381:112468.
Joushi S, Esmaeilpour K, Masoumi-Ardakani Y, Esmaeili-Mahani S, Sheibani V. Effects of short environmental enrichment on early-life adversity induced cognitive alternations in adolescent rats. J Neurosci Res. 2021;99:3373–91.
Shahidi F, Ambigaipalan P. Omega-3 polyunsaturated fatty acids and their health benefits. Annu Rev Food Sci Technol. 2018;9:345–81.
Swanson D, Block R, Mousa SA. Omega-3 fatty acids EPA and DHA: health benefits throughout life. Adv Nutr. 2012;3:1–7.
Avallone R, Vitale G, Bertolotti M. Omega-3 fatty acids and neurodegenerative diseases: new evidence in clinical trials. Int J Mol Sci. 2019;20:4256.
Yam KY, Schipper L, Reemst K, Ruigrok SR, Abbink MR, Hoeijmakers L, et al. Increasing availability of omega-3 fatty acid in the early-life diet prevents the early-life stress-induced cognitive impairments without affecting metabolic alterations. FASEB J. 2019;33:5729–40.
Pusceddu MM, Kelly P, Ariffin N, Cryan JF, Clarke G, Dinan TG. n-3 PUFAs have beneficial effects on anxiety and cognition in female rats: effects of early life stress. Psychoneuroendocrinology. 2015;58:79–90.
Pusceddu MM, El Aidy S, Crispie F, O’Sullivan O, Cotter P, Stanton C, et al. N-3 polyunsaturated fatty acids (PUFAs) reverse the impact of early-life stress on the gut microbiota. PLoS ONE. 2015;10:e0139721.
Liao JF, Hsu CC, Chou GT, Hsu JS, Liong MT, Tsai YC. Lactobacillus paracasei PS23 reduced early-life stress abnormalities in maternal separation mouse model. Benef Microbes. 2019;10:425–36.
Cowan CSM, Stylianakis AA, Richardson R. Early-life stress, microbiota, and brain development: probiotics reverse the effects of maternal separation on neural circuits underpinning fear expression and extinction in infant rats. Dev Cogn Neurosci. 2019;37:100627.
Yang L, Wu C, Li Y, Dong Y, Wu CY, Lee RH, et al. Long-term exercise pre-training attenuates Alzheimer’s disease-related pathology in a transgenic rat model of Alzheimer’s disease. Geroscience. 2022;44:1457–77.
Huang X, Zhao X, Li B, Cai Y, Zhang S, Wan Q, et al. Comparative efficacy of various exercise interventions on cognitive function in patients with mild cognitive impairment or dementia: a systematic review and network meta-analysis. J Sport Health Sci. 2022;11:212–23.
Sadeghi M, Peeri M, Hosseini MJ. Adolescent voluntary exercise attenuated hippocampal innate immunity responses and depressive-like behaviors following maternal separation stress in male rats. Physiol Behav. 2016;163:177–83.
Zolfaghari FS, Pirri F, Gauvin E, Peeri M, Amiri S. Exercise and fluoxetine treatment during adolescence protect against early life stress-induced behavioral abnormalities in adult rats. Pharmacol Biochem Behav. 2021;205:173190.
Park SS, Kim TW, Park HS, Seo TB, Kim YP. Effects of treadmill exercise on activity, short-term memory, vascular dysfunction in maternal separation rats. J Exerc Rehabil. 2020;16:118–23.
Acknowledgements
The authors thank Dr. Lorelei Tucker for English editing.
Funding
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Number RF1AG058603. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
ZH reviewed the literature. ZH and QZ drafted the manuscript. JDJ and QZ conceived and designed the review, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Z., Jordan, J.D. & Zhang, Q. Early life adversity as a risk factor for cognitive impairment and Alzheimer’s disease. Transl Neurodegener 12, 25 (2023). https://doi.org/10.1186/s40035-023-00355-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00355-z