
Abstract
Parkinson’s disease (PD), the second most common age-associated neurodegenerative disorder, is characterized by the loss of dopaminergic (DA) neurons and the presence of α-synuclein-containing aggregates in the substantia nigra pars compacta (SNpc). Chronic neuroinflammation is one of the hallmarks of PD pathophysiology. Post-mortem analyses of human PD patients and experimental animal studies indicate that activation of glial cells and increases in pro-inflammatory factor levels are common features of the PD brain. Chronic release of pro-inflammatory cytokines by activated astrocytes and microglia leads to the exacerbation of DA neuron degeneration in the SNpc. Besides, peripheral immune system is also implicated in the pathogenesis of PD. Infiltration and accumulation of immune cells from the periphery are detected in and around the affected brain regions of PD patients. Moreover, inflammatory processes have been suggested as promising interventional targets for PD and even other neurodegenerative diseases. A better understanding of the role of inflammation in PD will provide new insights into the pathological processes and help to establish effective therapeutic strategies. In this review, we will summarize recent progresses in the neuroimmune aspects of PD and highlight the potential therapeutic interventions targeting neuroinflammation.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is an age-related neurodegenerative disorder characterized clinically by resting tremor, slowness of movement, rigidity and postural instability and pathologically by the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) [1–4]. Deposition of protein aggregates containing α-synuclein (termed Lewy bodies) is evident in multiple brain regions of advanced PD patients [5]. The etiology of PD has not yet been fully understood. Although a variety of possible pathogenetic mechanisms have been proposed over the years, including excessive release of oxygen free radicals during enzymatic dopamine breakdown, impairment of mitochondrial function, loss of trophic support, abnormal kinase activity, disruption of calcium homeostasis, dysfunction of protein degradation and neuroinflammation, the pathogenesis of PD is still largely uncertain [6–8]. However, emerging evidence indicates that sustained inflammatory responses, T cell infiltration and glial cell activation are common features of both human PD patients and animal models of PD and play vital roles in the degeneration of DA neurons [9, 10], which suggests the possibility of developing potential therapies for PD by targeting the inflammatory processes. In this review, we will focus on the role of inflammation in the progression of PD and the potential application of anti-inflammatory medications in the treatment of this devastating disorder.
Microglia-mediated inflammation in PD
Microglia is one of the major cell types which are involved in the inflammatory responses in the central nervous system (CNS) [11]. In 1988, McGeer et al. showed the presence of reactive microglia in the SNpc of human post-mortem brain tissue, which is the evidence revealed for the first time suggesting the involvement of neuroinflammation in PD pathogenesis [12]. Positron emission tomography (PET) studies also indicate that there is pronounced activation of microglia in various regions of PD brain [13, 14]. Moreover, activation of microglia in the SNpc and striatum is profound in various types of PD animal models [15–17]. Further biochemical analysis reveals higher levels of pro-inflammatory mediators including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interferon-gamma (IFN-γ) in the midbrain of PD patients. These data strongly suggest the involvement of immune components in PD pathogenesis.
Under physiological conditions, the quiescent state of microglia is maintained by a variety of immunomodulators, such as CX3CL1, CD200, CD22, CD47, CD95 and neural cell adhesion molecule (NCAM), which are produced mainly by neuronal cells [18–26]. Interestingly, the receptors for these molecules are almost exclusively expressed by microglia in the CNS, indicating the critical role of neuron-microglia interactions in the regulation of neuroinflamamtion [18–26]. For instance, CX3CL1-CX3CR1 signaling negatively regulates microglial activation and protects DA neurons from degeneration induced by neurotoxins [27, 28]. Deficiency of CX3CL1 or CX3CR1 in vivo results in increased neurotoxicity induced by systemic lipopolysaccharide (LPS) treatment and enhanced cell death of DA neurons in the SNpc of animal PD models [28, 29]. Likewise, dysfunction of CD200-CD200R signaling also increases the activation of microglia and exacerbates the degeneration of DA neurons in rat PD models [30, 31].
It has been proposed that activated microglia may be beneficial to the host, at least in the early phase of neurodegeneration process [12, 32–34]. For instance, it has been shown that suppression of Jmjd3, which is essential for M2 microglia polarization, in the substantia nigra (SN) in vivo dramatically causes microglial overactivation and exacerbated dopamine (DA) neuron death in a PD animal model [35], indicating a protective role of M2 microglia in this process. However, long-term over-activation of microglia in the PD brain significantly up-regulates the expression of a large group of pro-inflammatory cytokines including TNF-α, IL-1β, interleukin-6 (IL-6) and IFN-γ, which contribute to the acceleration of nigral DA neuron degeneration [36, 37]. As the disease progresses, molecules such as α-synuclein, ATP and metalloproteinase-3 (MMP-3) released from the degenerating DA neurons will further enhance microglia activation, amplify the neuroinflammatory responses in the brain, and result in the deterioration of the neurodegenerative processes [11, 38] forming a vicious cycle of neurodegeneration. Activated microglia accumulate around the α-synuclein-positive aggregates in many regions of PD brain [39]. These cells are likely activated by over-produced [38, 40], mutants or misfolded α-synuclein leading to increases in the production and release of the pro-inflammatory cytokines [38, 41, 42]. Thus, the neurotoxicity induced by excessive or misfolded α-synuclein may be partially caused by microglia-mediated inflammatory responses.
ATP, a purinergic neurotransmitter, is also able to robustly modulate various functions of microglia [43, 44]. The migration of microglia to the injured and inflammatory areas is controlled by ATP released from the damaged neurons and neighboring astrocytes [44]. In addition, ATP binds to the P2Y receptor which is mainly expressed by microglia in the brain and induces the production of high levels of IL-1β, TNF-α and nitric oxide (NO) [45]. Another protein produced by degenerating neurons is MMP-3 which also plays important roles in the regulation of the activation states of microglia, at least in vitro. Overexpression of MMP-3 in microglia-neuron co-cultures induces prominent activation of microglia and increased the oxidative stress reaction. In contrast, MMP-3−/− mice administrated with N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) display attenuated nigrostriatal DA neuronal degeneration, microglial activation, and superoxide generation [46]. These data support the notion that microglia is a major player in neuroinflammation in the context of PD pathogenesis and MMP-3 plays a pivotal role in dopaminergic neuronal degeneration.
Microglia activation phenotypes in PD
Mounting evidence indicates that microglia has two alternative activation phenotypes, termed the M1 (pro-inflammatory) phenotype and the alternative M2 (anti-inflammatory) phenotype. These different activation statuses of microglia are characterized by secretion of different arrays of cytokines [47]. It has been demonstrated that LPS/IFN-γ treatment induces M1 activation, while IL-4/IL-13 treatment triggers M2 activation in microglia. The classical M1 activation of microglia is featured by the production of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-12, and other cytotoxic molecules such as superoxide, NO and reactive oxygen species (ROS), contributing to the amplification of the pro-inflammatory responses during injuries and infections. Conversely, M2 microglia plays an immunosuppressive role by antagonizing the classic M1 microglia and promoting tissue repair. The M2 microglia produces a variety of cytokines with anti-inflammatory property, such as IL-4, IL-13, IL-10, and TGF-β. The different activation forms of microglia can be distinguished by their characteristic gene expression pattern. For example, Arg1, FIZZ1 (also known as RELM-α), Chi3l3 (also known as YM1) and CD206 were expressed in mouse M2 phase microglia [48]. Expression of Arg1, FIZZ1 and Chi3l3 may be regulated by cytokines, since their levels are significantly increased in primary cultured microglia or the striatal and frontal cortical regions of mouse brain following IL-4 stimulation [49].
What factors affect M1/M2 microglia phenotype in the context of PD? Mis-folded proteins and environmental toxins induce the activation of microglia toward M1 phenotype in PD animal models [38, 50, 51]. Chronic MPTP administration leads to progressive reduction of CD206 expression, which suggests the down-regulation of M2 phase activation of microglia in the progression of PD [47]. Conversely, IL-4 treatment up-regulates microglial expression of histone H3K27me3 demethylase (Jmjd3) which is involved in the regulation of epigenetic modification of chromosomes and contributes to various human diseases. Expression levels of M2 marker genes, such as Arg1 and CD206, are significantly down-regulated after Jmjd3 knockdown in N9 microglia cell line, indicating the essential role of Jmjd3 for M2 microglia polarization. Knockdown of Jmjd3 in vivo exacerbates the DA neuron loss in the SNpc of MPTP-induced mouse PD model by revoking M2 activation of microglia [35].
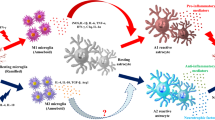
Conditioned medium (CM) from M1 phase N9 microglia results in increased death of DA neurons, whereas CM mixture from both M1 and M2 cells reverses the neurotoxicity elicited by the M1-CM [35]. Previous investigations indicated that a majority of the activated microglia express M2 associated genes at the early stages following injury in various models. M1 signature genes, however, gradually become predominant in later stages [32]. These interesting observations suggest that it is important to balance different microglia activation phenotypes in PD (Fig. 1). It appears to be a promising strategy to intervene the progression of PD by manipulating the transition of microglia activation statuses. Even though current data suggest differential role of M1 and M2 in the pathogenesis of PD in its animal models, similar results from patients are lacking. Future work should warrant research in this aspect.

Diagrammatic representation of inflammatory mechanisms involved in PD pathogenesis. Microglia become activated M1 phenotype in PD under pathological conditions such as protein aggregation, gene mutations, environmental factors and cytokines released from infiltrated T cells. The pro-inflammatory mediators from M1 microglia activate astrocytes leading to elevated production of proinflammatory factors, nitric oxide and superoxide radical, contributing to degeneration of DA neurons. The molecules released from degenerative DA neurons can further cause activation of glia and enhanced inflammatory response. At certain stage of PD, subpopulation of microglia may become activated M2 phenotype releasing anti-inflammatory factors, including TGF-β, and exert a neuroprotective effect in PD
Astrocyte-mediated neuroinflammation in PD
A great body of studies show that astrocytes also play vital roles in the neuroinflammatory processes in PD. Like microglia, astrocytes respond to the inflammatory stimulations such as LPS, IL-1β and TNF-α by producing pro-inflammatory cytokines both in vitro and in vivo [11, 52]. Reactive astrogliosis characterized by the increased expression levels of glial fibrillary acidic protein (GFAP) and hypertrophy of cell body and cell extensions have been reported in various PD animal models. Importantly, astrogliosis also exists in the affected brain regions of patients with PD, indicating the possible involvement of astrocytes in the immune processes in PD [53].
It has been observed that astrocytic responses are relatively slower than microglial activation after stimulations. Microglia may initiate the inflammatory responses after immune stimulations such as LPS treatment and α-synuclein aggregation. Astrocytes are then activated by a variety of molecules including pro-inflammatory mediators released from activated microglia and these immunosignals are further amplified by astrocytes [11]. Uncontrolled neuroinflammation caused by the synergic activation of microglia and astroyctes ultimately contributes to the enhanced death of DA neurons in the SNpc during neurodegeneration [11, 54]. The expression levels of TNF-α and IL-6 of primary cultured astrocytes are increased significantly after α-synuclein treatment in vitro [55]. Specific overexpression of mutant α-synuclein in astrocytes causes widespread astrogliosis, microglial activation and degeneration of DA neurons and motor neurons in mice [56].
One of the long-standing questions in PD research is how the neuroinflammation is developed and the roles of astrocytes in this process. Human post-mortem and animal experimental results suggest a progressive decline of the dopaminergic neuronal system with age [57]. Whether the reduction of Drd2 in aging brain has significant impact on brain function and ultimately contribute to the development of early/mid-stage PD remains poorly understood. This reduction primarily reflects the changes in neuronal cells. In sharp contrast, the contribution from glial Drd2 in overall levels is minor. Indeed, our own studies have revealed that astrocytic Drd2 is of very low abundance in overall levels of Drd2 in the striatal tissue [58]. Given that astroglial and microglial cells are the ‘sensors’ in the brain which constantly monitor brain activities, it is likely that glial Drd2 is more vulnerable to microenvironment changes, despite that glial Drd2 abundance is very low in the brain. Nevertheless, the biological consequence of glial Drd2 loss may be significant. Ablation of astrocytic Drd2 causes dramatic reduction of anti-inflammation protein alphaB-crystallin in the CNS [58, 59]. Thus, glial Drd2 may be an important player in the maintenance of immune homeostasis (Fig. 1). It is plausible that down-regulation of Drd2, presumably including those in glial cells, in aging brain compromise the immune homeostasis contributing to PD pathogenesis. Future studies may be required to develop new technology that can specifically label and visualize glial dopamine receptors in aging and PD brain.
The idea that Drd2 plays important roles in the modulation of neuroinflammation is supported by a recent study in which Drd2 activation by Drd2 agonists quinpirole and ropinirole reduces expression levels of IL-1β and monocyte chemoattractant protein-1 as well as microglia/macrophages activation in experimental intracerebral hemorrhage brain injury model [60]. These results further suggest the anti-inflammatory effects of Drd2 in certain CNS disorders.
PD-associated genes and neuroinflammation
Increasing evidence indicates that some PD-associated genes are involved in the regulation of immune responses of microglia and astrocytes in the CNS. One of such genes is α-synuclein (SCNA), the missense mutations of which results in a familial form of PD [61]. Abnormal accumulation of α-synuclein in neuronal cytoplasm and neurites is one of the pathological hallmarks of PD [62]. Wild-type or pathogenic forms of α-synuclein induces pronounced microglial activation in vitro [63]. Furthermore, oligomers of α-synuclein could elicit microglia responses through the activation of toll-like receptor 2 (TLR2)-mediated signaling [64]. In α-synuclein transgenic mice, there is prominent activation of microglia and upregulation of TLRs in the brainstem and SNpc [65, 66].
In the last several years, the common variations in leucine-rich repeat kinase 2 (LRRK2) gene have been identified as risk factors of both familial and sporadic PD [67–70]. Inflammatory stimuli such as LPS could drastically enhance the expression levels of LRRK2 in primary cultured microglia [71], while knockdown of LRRK2 reduces LPS-induced TNF-α and iNOS production and also decreases the activation of nuclear factor κ B (NF-κB) transcriptional activity in microglia [72]. Moreover, the expression levels of pro-inflammatory cytokines are higher in microglia isolated from LPS-challenged transgenic mice overexpressing R1441G mutation of LRRK2 than in microglia from LPS-treated WT mice [73]. These data suggest that LRRK2 regulates microglia activation thus might contribute to the progression of PD through neuroinflammatory pathways.
Of note, Lrrk2 has been shown to play crucial roles in periphery inflammation. LRRK2 is relatively abundant in peripheral blood mononuclear cells and macrophages in the immune system. Expression levels of LRRK2 are significantly increased in the process of THP-1 monocyte-to-macrophage differentiation following IFN-γ treatment [74, 75]. LRRK2 deficiency results in enhanced susceptibility to experimental colitis in mice [76], indicating the important role of LRRK2 in periphery inflammation and its potential association with PD.
Parkin is another PD-associated gene which encodes an E3-ubiquitin ligase. Mutations in parkin are the most common causes of recessively inherited PD [77]. Systemic LPS treatment induces more prominent degeneration of DA neurons in the SNpc of parkin knockout mice, comparing with littermate controls [78]. Aged parkin knockout mice display increased astrogliosis in the striatum and aberrant microglial activation in the midbrain [79]. Co-culture of microglia from parkin knockout mice and wild-type neurons increase sneuronal vulnerability to rotenone toxicity [80], suggesting that parkin-null microglia produce and release soluble factors that are detrimental to neuronal cells. Indeed, these microglia express higher levels of pro-inflammatory cytokines such as TNF-α, IL-6 and iNOS after LPS challenge [81], implicating that parkin plays an important role in the regulation of PD-associated inflammation.
Missense mutations in the PTEN-induced putative kinase1 (PINK1) gene cause the early-onset, recessively inherited form of PD [82]. Under pathological conditions, PINK1 directly phosphorylates parkin to enhance its activity and causes mitochondria damages [83]. PINK1 is also involved in the regulation of inflammatory cytokine production. Under basal condition and following systemic LPS treatment, mice null for PINK1 produce higher levels of pro-inflammatory cytokines including IL-1β, IL-12 and TNF-α [84, 85]. The underlying molecular mechanisms of PINK1’s effect remain elusive. PINK1 may regulate IL-1β-mediated NF-κB activity through interacting with IL-1 receptor-associated kinase 1 (IRAK1) and toll-interacting protein (Tollip) [86].
It has been shown that DJ-1, one of PD genes, is mainly expressed in astrocytes and microglia in human brain and the expression levels of DJ-1 are strongly upregulated in reactive astrocytes in PD patients [87]. Astrocytes from DJ-1 knockout mice produce higher levels of cyclooxygenase-2 (COX2) and IL-6 following LPS treatment [4]. Primary neurons plated on DJ-1-null astrocytes shows severe apoptosis following LPS treatment, which indicates that loss of DJ-1 might contribute to PD pathogenesis by astrocyte-mediated neuroinflammatory response [4]. Likewise, microglia with DJ-1 knockdown also exhibits hyper-responsiveness to LPS [88]. Taken together, it is highly likely that mutation, down-regulation or overexpression of PD associated genes perturb the expression of pro-inflammatory cytokines in glial cells after LPS treatment. The question that how PD genes regulate inflammatory responses in PD should be addressed in the future.
Peripheral immune cell-mediated inflammation in PD
Under physiological conditions, the peripheral immune cells such as T- and B-lymphocytes are hardly detectable in the CNS. Following infections or tissue injuries, blood monocytes and tissue-resident macrophages are quickly activated and secret an array of inflammatory cytokines like IL-1β, TNF-α and IL-6 as well as chemokines. These cytokines and chemokines could enter the brain and stimulate microglia to initiate the neuroinflammatory reactions [89]. The CNS has been considered as immunologically privileged because of the existence of blood brain barrier (BBB). It is known that the neurovascular unit is formed by tight junctions between endothelial cells and the surrounding components of the CNS, including pericytes, astrocytes and the basement membrane. BBB limits the entrance of pathogens and peripheral immune cells into the brain parenchyma. BBB breakdown leads to increased infiltration of peripheral immune cells into the CNS, which has been identified as one of the major contributing factors for neurodegenerative diseases including PD [90, 91]. There are dramatically morphological changes of endothelial cells in the SNpc of PD brain, suggesting the possible dysfunction of BBB in the pathological processes of PD [91–93]. Several studies show a strong correlation between the BBB disruption and loss of DA neurons in mice by intranigral injection of vascular endothelial growth factor (VEGF) which is a potent inducer of BBB damages [94, 95]. Furthermore, the expression levels of VEGF are drastically increased in both PD patients and the MPTP-induced mouse PD model [94].
The number of activated microglia and infiltrated B and T-lymphocytes is significantly increased in the SNpc of AAV-mediated α-synuclein overexpressing mice. Moreover, nitrated α-synuclein can get through the BBB, enter cervical lymph node causing activation of T-lymphocytes and increases in the expression of the class II major histocompatibility complex in MPTP-treated mice. Transplantation of T cells from mice immunized with nitrated α-synuclein into the brain significantly exacerbates the neuroinflammatory responses and degeneration of DA neurons. Conversely, mice deficient in T- and B-lymphocytes are more resistant to the neurotoxicity caused by MPTP intoxication [16, 96]. Furthermore, MPTP-induced neurotoxicity is markedly attenuated in the SNpc of CD4 −/− mice. Peripheral inflammation caused by ulcerative colitis increase LPS-induced DA neuron degeneration, microglial activation, production of pro-inflammatory cytokines and permeability of the BBB [97]. These data indicate that there is a close relationship between the peripheral immune system and the progression of PD. A better understanding of the relationship between the CNS and immune system and the molecular mechanisms underlying their crosstalk may help to further clarify the pathological processes of PD.
It has been reported that systemic infection could contribute to the etiology and the progress of PD [98, 99]. Indeed, the main causes of death in PD patients are respiratory infections [98]. Gastrointestinal infections could lead to a worsening of PD [100, 101]. Moreover, infection of helicobacter pylori (HP), a bacterium found on the luminal surface of the gastric epithelium, is associated with clinical diagnosis and worse severity of motor function in PD patients [102, 103]. These data indicate a link between systemic infection and PD. It would be very interesting to see whether control of systemic infections helps to reduce the exacerbation of PD symptoms and delay the disease progression.
Anti-inflammatory therapies in PD
Given the important roles of neuroinflammation in the initiation and progression of PD, it is desirable to develop intervening therapies for this devastating disorder by targeting the inflammatory pathways mediated by activated glial cells. For example, soluble TNF-α is known to contribute to the progressive degeneration of DA neurons in rodents induced by stereotactic injection of 6-hydroxydopamine (6-OHDA) or LPS. Overexpression of dominant-negative TNF-α specifically inhibits TNF signaling in the SNpc and attenuates activation of microglia, thereby reducing loss of DA neurons and improving locomotor ability in 6-OHDA-induced rat PD model [104, 105].
Minocycline is a semi-synthetic, second-generation tetracycline analog, which, as a lipophilic molecule, can easily get through the BBB and has been reported to have anti-inflammatory and neuroprotective properties in multiple inflammation-related neurological diseases [106, 107]. Minocycline treatment effectively protects DA neurons from degeneration and decreases glial cell activation in the SNpc of LPS and 6-OHDA challenged mice [108, 109]. Administration of minocycline also blocks MPTP-induced neurotoxicity of DA neurons in vivo by directly or indirectly inhibiting the phosphorylation of p38 MAPK, which is a key regulator of the expression of inflammatory genes [110]. It is often disappointing to note that, although there are many encouraging studies, the therapeutic potential of minocycline in PD is still controversial. In addition to beneficial effect of mincycline, it is also found to exacerbate the loss of DA neurons and behavior deficits in MPTP-induced rodent and non-human primate models [111]. The reason for these discrepancies is not clear. One of the probabilities is that it may due to the different drug dosages and durations used in different studies.
Besides minocycline, some other compounds with anti-inflammatory properties also show significant protective functions for DA neuron in PD models. Glucocorticoids are well known for their broad ranges of anti-inflammatory effects and have been widely used in clinical studies for brain inflammation. Glucocorticoid receptor expressed on microglia is found to regulate the expression of the transcription factors such as NF-κB and activator protein-1 (AP-1) which are key regulators of inflammatory gene expression [112, 113]. Administration of the synthetic steroid dexamethasone shows a protective effect for DA neurons and decreased the activation of glial cells in the SNpc of MPTP- or LPS-treated mice [112, 114]. Moreover, opioid antagonist naloxone has been shown to inhibit LPS-induced activation of microglia and expression of pro-inflammatory cytokines in vitro, attenuating neuronal loss. The compound dramatically reverses the acute dystonia and parkinsonism following general anaesthesia [115, 116].
Several non-steroidal anti-inflammatory drugs (NSAIDS), such as aspirin, salicylic acid (SA), ibuprofen and celecoxib, have also been shown to have protective effects on DA neurons in PD [117–120]. Aspirin is a potent inhibitor of COX2, which plays important roles in the processes of neuroinflammation and neuronal degeneration [121]. Aspirin promotes the resolution of inflammation [122] and prevents dopamine depletion in the striatum in MPTP-induced mouse and 6-OHDA-induced rat PD models [117]. SA is a metabolite product of aspirin, which is widely used for the treatment of myocardial infarction, cardiovascular diseases [118]. SA administration in MPTP mice alleviates the neurotoxin-induced behavioral impairments as well as dopamine depletion [118, 123]. Similarly, ibuprofen and celecoxib also show protective roles for DA neurons in MPTP- and 6-OHDA-induced PD models [119, 120]. Long-term use of NSAIDs, especially the use of ibuprofen, decreased PD risk by 21 %, suggesting high therapeutic potentials of NSAIDs for PD [120, 124, 125].
Concluding remarks
Animal experiments and clinical studies have generated an array of evidence supporting the involvement of inflammation in the progression of PD (Fig. 1). However, the precise role of inflammation in PD is still not fully understood. Moreover, the inflammatory responses in PD refer to both glial activation and peripheral immune cell infiltration, but the relationship between these two different inflammatory pathways is still unclear. These issues significantly hamper the development of PD-modifying therapeutics by targeting inflammatory pathways. Moreover, the different forms of microglia activation further increase the difficulty and complexity of manipulation of microglial responses in PD. A better understanding of how these two activated phenotypes of microglia contribute to PD progression may provide a basis for future drug discoveries. So far, many candidate drugs targeting inflammation in PD have been developed and tested. Although some of them have been reported to attenuate the behavior deficits and loss of DA neuron in PD animal models, the clinical studies of these candidates only show moderate effects. Future studies should focus on identifying more specific drug targets by extending the understanding of fundamental processes of inflammation at different stages of the disease progression.
References
Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14.
von Campenhausen S, Bornschein B, Wick R, Botzel K, Sampaio C, Poewe W, et al. Prevalence and incidence of Parkinson’s disease in Europe. Eur Neuropsychopharmacol. 2005;15:473–90.
Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26 Suppl 1:S1–58.
Waak J, Weber SS, Waldenmaier A, Gorner K, Alunni-Fabbroni M, Schell H, et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009;23:2478–89.
Mori F, Piao YS, Hayashi S, Fujiwara H, Hasegawa M, Yoshimoto M, et al. Alpha-synuclein accumulates in Purkinje cells in Lewy body disease but not in multiple system atrophy. J Neuropathol Exp Neurol. 2003;62:812–9.
Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29:1583–90.
Dick FD. Parkinson’s disease and pesticide exposures. Br Med Bull. 2006;79–80:219–31.
Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98.
Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:S210–S2.
Lv Y, Zhang Z, Hou L, Zhang L, Zhang J, Wang Y, et al. Phytic acid attenuates inflammatory responses and the levels of NF-kappaB and p-ERK in MPTP-induced Parkinson’s disease model of mice. Neurosci Lett. 2015;597:132–6.
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–91.
Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57–9.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–12.
Leal MC, Casabona JC, Puntel M, Pitossi FJ. Interleukin-1beta and tumor necrosis factor-alpha: reliable targets for protective therapies in Parkinson’s Disease? Front Cell Neurosci. 2013;7:53.
Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376.
Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25.
Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3:130181.
Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, et al. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–42.
Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 2000;290:1768–71.
Mott RT, Ait-Ghezala G, Town T, Mori T, Vendrame M, Zeng J, et al. Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia. 2004;46:369–79.
Numakawa T, Ishimoto T, Suzuki S, Numakawa Y, Adachi N, Matsumoto T, et al. Neuronal roles of the integrin-associated protein (IAP/CD47) in developing cortical neurons. J Biol Chem. 2004;279:43245–53.
Smith RE, Patel V, Seatter SD, Deehan MR, Brown MH, Brooke GP, et al. A novel MyD-1 (SIRP-1alpha) signaling pathway that inhibits LPS-induced TNFalpha production by monocytes. Blood. 2003;102:2532–40.
Chang RC, Hudson P, Wilson B, Liu B, Abel H, Hemperly J, et al. Immune modulatory effects of neural cell adhesion molecules on lipopolysaccharide-induced nitric oxide production by cultured glia. Brain Res Mol Brain Res. 2000;81:197–201.
Vernet-der Garabedian B, Derer P, Bailly Y, Mariani J. Innate immunity in the Grid2Lc/+ mouse model of cerebellar neurodegeneration: glial CD95/CD95L plays a non-apoptotic role in persistent neuron loss-associated inflammatory reactions in the cerebellum. J Neuroinflammation. 2013;10:65.
Chang RC, Hudson P, Wilson B, Haddon L, Hong JS. Influence of neurons on lipopolysaccharide-stimulated production of nitric oxide and tumor necrosis factor-alpha by cultured glia. Brain Res. 2000;853:236–44.
Morganti JM, Nash KR, Grimmig BA, Ranjit S, Small B, Bickford PC, et al. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J Neurosci. 2012;32:14592–601.
Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:9.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–24.
Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:154.
Wang XJ, Zhang S, Yan ZQ, Zhao YX, Zhou HY, Wang Y, et al. Impaired CD200-CD200R-mediated microglia silencing enhances midbrain dopaminergic neurodegeneration: roles of aging, superoxide, NADPH oxidase, and p38 MAPK. Free Radic Biol Med. 2011;50:1094–106.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70.
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–74.
Cagnin A, Kassiou M, Meikle SR, Banati RB. In vivo evidence for microglial activation in neurodegenerative dementia. Acta Neurol Scand Suppl. 2006;185:107–14.
Tang Y, Li T, Li J, Yang J, Liu H, Zhang XJ, et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014;21:369–80.
Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis. 2006;24:183–93.
McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, et al. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci. 2006;26:9365–75.
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–42.
Yamada T. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:5.
Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–58.
Beraud D, Maguire-Zeiss KA. Misfolded alpha-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat Disord. 2012;18 Suppl 1:S17–20.
Rojanathammanee L, Murphy EJ, Combs CK. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J Neuroinflammation. 2011;8:44.
Koizumi S, Ohsawa K, Inoue K, Kohsaka S. Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia. 2013;61:47–54.
George J, Goncalves FQ, Cristovao G, Rodrigues L, Meyer Fernandes JR, Goncalves T, et al. Different danger signals differently impact on microglial proliferation through alterations of ATP release and extracellular metabolism. Glia. 2015.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8.
Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ, et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007;21:179–87.
Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis. 2014;71:280–91.
Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2015.
Pepe G, Calderazzi G, De Maglie M, Villa A, Vegeto E. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. J Neuroinflammation. 2014;11:1031.
Kim HG, Ju MS, Ha SK, Lee H, Kim SY, Oh MS. Acacetin protects dopaminergic cells against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neuroinflammation in vitro and in vivo. Biol Pharm Bull. 2012;35:1287–94.
Ha SK, Moon E, Lee P, Ryu JH, Oh MS, Kim SY. Acacetin attenuates neuroinflammation via regulation the response to LPS stimuli in vitro and in vivo. Neurochem Res. 2012;37:1560–7.
Tanaka T, Kai S, Matsuyama T, Adachi T, Fukuda K, Hirota K. General anesthetics inhibit LPS-induced IL-1beta expression in glial cells. PLoS One. 2013;8:e82930.
Yamada T, Kawamata T, Walker DG, McGeer PL. Vimentin immunoreactivity in normal and pathological human brain tissue. Acta Neuropathol. 1992;84:157–62.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34.
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–60.
Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain. 2010;3:12.
Antonini A, Leenders KL. Dopamine D2 receptors in normal human brain: effect of age measured by positron emission tomography (PET) and [11C]-raclopride. Ann N Y Acad Sci. 1993;695:81–5.
Shao W, Zhang SZ, Tang M, Zhang XH, Zhou Z, Yin YQ, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature. 2013;494:90–4.
Liu Y, Zhou Q, Tang M, Fu N, Shao W, Zhang S, et al. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol Aging. 2015;36:1686–91.
Zhang Y, Chen Y, Wu J, Manaenko A, Yang P, Tang J, et al. Activation of Dopamine D2 Receptor Suppresses Neuroinflammation Through alphaB-Crystalline by Inhibition of NF-kappaB Nuclear Translocation in Experimental ICH Mice Model. Stroke. 2015;46:2637–46.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29:1690–701.
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One. 2013;8:e55375.
Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics. 2012;9:297–314.
Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, et al. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. 2012;237:318–34.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7.
Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32:1602–11.
Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, et al. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One. 2012;7:e34693.
Gillardon F, Schmid R, Draheim H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–8.
Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–85.
Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118:795–808.
Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–70.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8.
Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–34.
Rodriguez-Navarro JA, Casarejos MJ, Menendez J, Solano RM, Rodal I, Gomez A, et al. Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J Neurochem. 2007;103:98–114.
Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem. 2006;97:934–46.
Tran TA, Nguyen AD, Chang J, Goldberg MS, Lee JK, Tansey MG. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS One. 2011;6:e23660.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60.
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21.
Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, et al. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One. 2011;6:e16038.
Kim J, Byun JW, Choi I, Kim B, Jeong HK, Jou I, et al. PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp Neurobiol. 2013;22:38–44.
Lee HJ, Chung KC. PINK1 positively regulates IL-1beta-mediated signaling through Tollip and IRAK1 modulation. J Neuroinflammation. 2012;9:271.
Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain. 2004;127:420–30.
Trudler D, Weinreb O, Mandel SA, Youdim MB, Frenkel D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem. 2014;129:434–47.
Blatteis CM. Role of the OVLT in the febrile response to circulating pyrogens. Prog Brain Res. 1992;91:409–12.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97.
Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, et al. Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–9.
Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet. 1999;353:981–2.
Guan J, Pavlovic D, Dalkie N, Waldvogel HJ, O’Carroll SJ, Green CR, et al. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013;23:154–64.
Yasuda T, Fukuda-Tani M, Nihira T, Wada K, Hattori N, Mizuno Y, et al. Correlation between levels of pigment epithelium-derived factor and vascular endothelial growth factor in the striatum of patients with Parkinson’s disease. Exp Neurol. 2007;206:308–17.
Rite I, Machado A, Cano J, Venero JL. Blood–brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem. 2007;101:1567–82.
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–92.
Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson’s disease. J Neurochem. 2010;114:1687–700.
Beyer MK, Herlofson K, Arsland D, Larsen JP. Causes of death in a community-based study of Parkinson’s disease. Acta Neurol Scand. 2001;103:7–11.
Bu XL, Wang X, Xiang Y, Shen LL, Wang QH, Liu YH, et al. The association between infectious burden and Parkinson’s disease: a case–control study. Parkinsonism Relat Disord. 2015;21:877–81.
Weller C, Oxlade N, Dobbs SM, Dobbs RJ, Charlett A, Bjarnason IT. Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol Med Microbiol. 2005;44:129–35.
Przuntek H, Muller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm. 2004;111:201–16.
Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B. Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol. 2012;19:864–9.
Tan AH, Mahadeva S, Marras C, Thalha AM, Kiew CK, Yeat CM, et al. Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord. 2015;21:221–5.
Harms AS, Barnum CJ, Ruhn KA, Varghese S, Trevino I, Blesch A, et al. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson’s disease. Mol Ther. 2011;19:46–52.
McCoy MK, Ruhn KA, Martinez TN, McAlpine FE, Blesch A, Tansey MG. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol Ther. 2008;16:1572–9.
Cheng S, Hou J, Zhang C, Xu C, Wang L, Zou X, et al. Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Rep. 2015;5:10535.
Noble W, Garwood CJ, Hanger DP. Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion. 2009;3:78–83.
Tomas-Camardiel M, Rite I, Herrera AJ, de Pablos RM, Cano J, Machado A, et al. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood–brain barrier, and damage in the nigral dopaminergic system. Neurobiol Dis. 2004;16:190–201.
He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909:187–93.
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–71.
Diguet E, Fernagut PO, Wei X, Du Y, Rouland R, Gross C, et al. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004;19:3266–76.
Castano A, Herrera AJ, Cano J, Machado A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J Neurochem. 2002;81:150–7.
Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin Jr AS. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–53.
Kurkowska-Jastrzebska I, Litwin T, Joniec I, Ciesielska A, Przybylkowski A, Czlonkowski A, et al. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int Immunopharmacol. 2004;4:1307–18.
Iselin-Chaves IA, Grotzsch H, Besson M, Burkhard PR, Savoldelli GL. Naloxone-responsive acute dystonia and parkinsonism following general anaesthesia. Anaesthesia. 2009;64:1359–62.
Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–17.
Di Matteo V, Pierucci M, Di Giovanni G, Di Santo A, Poggi A, Benigno A, et al. Aspirin protects striatal dopaminergic neurons from neurotoxin-induced degeneration: an in vivo microdialysis study. Brain Res. 2006;1095:167–77.
Mohanakumar KP, Muralikrishnan D, Thomas B. Neuroprotection by sodium salicylate against 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-induced neurotoxicity. Brain Res. 2000;864:281–90.
Sanchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2004;1:6.
Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology. 2011;76:863–9.
Starke RM, Chalouhi N, Ding D, Hasan DM. Potential Role of Aspirin in the Prevention of Aneurysmal Subarachnoid Hemorrhage. Cerebrovasc Dis. 2015;39:332–42.
Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9.
Aubin N, Curet O, Deffois A, Carter C. Aspirin and salicylate protect against MPTP-induced dopamine depletion in mice. J Neurochem. 1998;71:1635–42.
Manthripragada AD, Schernhammer ES, Qiu J, Friis S, Wermuth L, Olsen JH, et al. Non-steroidal anti-inflammatory drug use and the risk of Parkinson’s disease. Neuroepidemiology. 2011;36:155–61.
Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74:995–1002.
Acknowledgments
This work was supported by grants from the National Key Basic Research Program of China (2011CB504102), Natural Science Foundation of China (31430036, 31123002, 31321091).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
QW and YL wrote the manuscript. JZ conducted the manuscript editing. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Q., Liu, Y. & Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener 4, 19 (2015). https://doi.org/10.1186/s40035-015-0042-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-015-0042-0