
Abstract
Six compounds were isolated from Penicillium sp. SG-W3, a marine-derived fungus, and their inhibitory activities against target enzymes relating to neurological diseases were evaluated. Compound 1 (pannorin) was a potent and selective monoamine oxidase (MAO)-A inhibitor with a 50% inhibitory concentration (IC50) of 1.734 μM and a selectivity index (SI) of > 23.07 versus MAO-B, and it showed an efficient antioxidant activity. All compounds showed weak inhibitory activities against acetylcholinesterase, butyrylcholinesterase, and β-secretase. The inhibition constant (Ki) of 1 for MAO-A was 1.049 ± 0.030 μM with competitive inhibition. Molecular docking simulation predicted that compound 1 forms hydrogen bonds with MAO-A, and binds more tightly to MAO-A than to MAO-B (− 25.02 and − 24.06 kcal/mol, respectively). These results suggest that compound 1 is a selective, reversible, and competitive MAO-A inhibitor that can be a therapeutic candidate for treating neurological diseases.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD) is a major concerned-neurodegenerative disease, leading cause of dementia [1]. Recently, more than 50 million people have been affected by AD, and it is expected to double every 20 years owing to industrialization and aging of the population [1]. The most typical symptoms of AD are memory, intellectual, speech, movement, and space–time disorders [2, 3]. Despite the increasing number of patients with AD, no effective drugs are available to treat AD [3]. However, its pathogenesis remains unclear. Several studies have shown that monoamine oxidase (MAO) level increase, acetylcholine (ACh) level reduction, neuro-inflammation, β-amyloid (Aβ) accumulation, and tau phosphorylation play important roles in AD etiology [1, 4]. Oxidative deamination of various amines catalyzed by MAO (EC 1.4.3.4) has been reported to be a source of reactive oxygen species (ROS) and a cause of various diseases [4]. Moreover, Aβ accumulation in the brain also causes AD, due to abnormal amyloid precursor protein (APP) lysis by β-secretase (BACE1) and neurofibromins tangling by tau hyperphosphorylation [5].
Parkinson’s disease (PD), along with AD, are major neurological diseases. Many studies have suggested that the degeneration of dopaminergic neurons, specially, in the nigrostriatal pathway causes PD, due to oxidative stress [6, 7]. Dopamine metabolism involves in ROS formation in the brain as a major process. In general, dopamine levels are mostly regulated by MAO-A, locating in catecholaminergic neurons. Furthermore, increased levels of MAO-B, a major enzyme involved in dopamine catabolism inside glial cells, lead to neuronal degeneration [8, 9].
Therefore, dopamine degradation by MAO-B produces ROS, which causes oxidative stress and PD. Additionally, increased MAO-B activity correlates with cognitive impairment in patients with AD [10, 11].
MAOs are located in the mitochondrial outer membrane in two isoforms, i.e., MAO-A and MAO-B, catalyzing the oxidative deamination of neurotransmitters, such as monoamines [12, 13]. In addition, MAOs are also associated with Aβ accumulation in the brain of AD patients [14]. MAO inhibitors, moclobemide and clorgyline inhibit MAO-A, whereas lazabemide, pargyline, selegiline, and rasagiline inhibit MAO-B [15]. Among them, MAO-A inhibitors have been used to treat anxiety and depression, and MAO-B inhibitors have been used to treat PD; study to find new MAO inhibitors as neurological disorder treatments is ongoing [16, 17].
ACh, one of neurotransmitters in the brain, has important functions in both central and peripheral nervous systems. Cholinesterases (ChEs) include acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), which commonly decompose ACh into choline and acetyl coenzyme A [18, 22]. AChE catalyzes ACh, whereas BChE catalyzes both ACh and butyrylcholine (BCh), but has a higher preference for BCh than ACh. These ChEs are involved in serine hydrolysis in the body and regulate ACh levels in the glial cells, the hippocampus, and the temporal nerve cortex [17, 19, 20]. Several studies have reported that AChE and BChE inhibitors that increase ACh levels in the brain as treatment candidates for AD [21, 22]. FDA-approved ChE inhibitors such as donepezil, galantamine, and rivastigmine elevate ACh levels and alleviate AD symptoms [20, 23, 24].
In the healthy brain, neuronal APPs are typically digested by α- and γ-secretase enzymes with three domains such as cell, cell membrane, and extracellular space. The normal Aβ fragments are soluble polypeptides produced through this digestive reaction and can be recycled in cells. However, Aβ fragments produced by BACE1 and γ-secretase are not recycled and can cause AD through accumulation in brain neurons. Until recently, many studies have focused on BACE1 inhibitors, however, there has been no success in drug approval, while several monoclonal antibodies as Aβ aggregation inhibitors like aducanumab [25] and lecanemab [26] have been developed. Nevertheless, BACE1 inhibitor development remains worthwhile. In addition, multi-target inhibitors that improve cognitive function by simultaneously inhibiting MAOs, ChEs, and BACE1 to increase monoamine and acetylcholine levels and decrease Aβ plaque formation have been developed to treat AD [17, 27].
Marine and freshwater fungi have distinct metabolite profiles [28]. Marine fungi have garnered interest owing to the secondary metabolites produced, which often have distinct structural features and interesting biological and pharmacological characteristics [29]. Marine Penicillium species produce various secondary metabolites with anti-viral, anti-bacterial, anti-tumor, and anti-inflammatory activities [30, 31]. Though various MAO inhibitors of marine origin were described [32], only few MAO inhibitors have been isolated from Penicillium spp. till date.
This study describes the inhibitory activities of six compounds isolated from the marine Penicillium sp. SG-W3 against the enzymes MAO, AChE, BChE, and BACE1.
Materials and methods
General experimental procedures
NMR spectra were obtained using NMR spectrometers, a Varian Inova (Varian Medical Systems, Inc., Charlottesville, VA, USA; 500 and 125 MHz for 1H and 13C, respectively) and a Bruker (Bruker, Middlesex, MA, USA; 300 MHz for 1H), using the residual solvent signals as internal references, dimethyl sulfoxide-d6 (DMSO-d6) (δH 2.50 ppm and δC 39.5 ppm) and deuterated methanol (CD3OD) (δH 4.87 and 3.31 ppm). Low-resolution LC/MS analysis was performed using an Agilent Technologies 1260 quadrupole (Santa Clara, CA, USA) and Waters Micromass ZQ LC/MS system (Milford, MA, USA) using a reverse-phase column (Phenomenex Luna C18 (2) 100 Å, 50 × 4.6 mm2, 5 µm; Torrance, CA, USA) at 1.0 mL/min at NanoBioEnergy Materials Center (Ewha Womans University). Column chromatography (CC) was carried out using reversed-phase silica gel C18 column (70–230 mesh, Merck, Germany) with a step gradient solvent of water and methanol. The fractions were further purified using the reverse-phase HPLC C18 column (Phenomenex Luna C18 (2)).
Isolation and identification of a marine fungal strain SG-W3
The fungal strain SG-W3 was isolated as previously described [33] with a few modifications such as growing the fungus on PDA containing 1% (w/v) NaCl at 28 °C for 7 to 14 days. Seawater was collected from Dangjin, Chungcheongnam-do, Republic of Korea (36º53ʹ19.1ʺN, 126º49ʹ36.6ʺE) on March 25, 2020, transported to the laboratory, and filtered using a membrane filter (0.45-μm, Hyundai Micro Co., Korea). The filters were then incubated on potato dextrose agar (PDA; BD, USA) and yeast mold agar (YM; BD) supplemented with 3% (w/v) NaCl, 0.1% (w/v) ampicillin, and 0.1% (w/v) streptomycin at 20 ºC for 7–14 days and the SG-W3 fungal colony was isolated and transferred on fresh PDA media. SG-W3 spores were stored in 20% glycerol at – 80 ºC and deposited at the Microbial Marine Bio Bank (MMBB) of the National Marine Biodiversity Institute of Korea (MABIK).
Fungal DNA extraction, polymerase chain reaction (PCR), and PCR product purification were performed as previously described [33]. Briefly, DNA was isolated from SG-W3 mycelia, and the partial beta-tubulin gene sequence was amplified, as a molecular marker for fungal identification [34]. The beta-tubulin segment was sequenced by Macrogen Inc. (Seoul, Korea), and used as a query sequence to search for close sequence matches using BLASTN in GenBank. Phylogenetic tree was constructed using MEGA version 6 [35].
Culture and isolation of compounds
The strain SG-W3 was cultured in 12 Ultra Yield Flasks (2.5 L) containing 1 L PDB SW medium (24 g potato dextrose broth and 39.4 g sea salt in 1 L distilled water) at 27 ℃ with shaking at 120 rpm for 7 days, and the medium (12 L) was extracted with the equal volume of ethyl acetate (EtOAc). It was concentrated to yield 2.2 g crude extract using a rotary vacuum evaporator. Then, 10 L crude extract (1.9 g) was fractionated by flash CC on C18 resin eluted with 200 mL H2O/CH3OH (80/20, 60/40, 50/50, 40/60, 30/70, 20/80, and 0/100) to obtain seven fractions (F1–F7). Fraction 4 was further purified by reversed-phase HPLC equipped with a Phenomenex Luna C-18(2) column (250 × 100 mm2, 2.0 mL/min, 5 μm, 100 Å, UV = 254 nm) under an isocratic condition with 35% aqueous CH3CN to yield pannorin (1, 15.8 mg, tR = 19.0 min), 2,5-dimethyl-6,8-dihydroxy-chromone (2, 6.5 mg, tR = 21.2 min), penimethavone A (3, 11.8 mg, tR = 24.0 min), calyxanthone (4, 2.6 mg, tR = 28.6 min), and endocrocin (5, 15.8 mg, tR = 37.5 min). In addition, 2 L extract (0.3 g) was fractionated by open-CC on silica gel and eluted with a step gradient of CH2Cl2 /CH3OH (98/2, 90/10, 0/100) to obtain three fractions (F1–F3). Fraction 1 was purified using reverse-phase HPLC (Phenomenex Luna C-18 (2)) under isocratic conditions of 65% CH3CN in water to obtain hydroxyviocristin (6, 1.1 mg, tR = 19.0 min).
Pannorin (1): brownish powder, 1H NMR (500 MHz, DMSO-d6): δH 12.12 (s, 4-OH), 10.00 (s, 8-OH), 9.86 (s, 10-OH), 7.19 (s, H-6), 6.55 (d, J = 2.2 Hz, H-7), 6.52 (d, J = 2.2 Hz, H-9), 5.51 (s, H-3), 2.65 (s, CH3-5); 13C NMR (125 MHz, DMSO-d6): δc 169.7 (C-4), 161.2 (C-2), 158.9 (C-8), 156.6 (C-10), 154.6 (C-10b), 138.1 (C-6a), 132.4 (C-5), 124.1 (C-6), 107.7 (C-4a), 106.5 (C-10a), 102.7 (C-9), 100.9 (C-7), 88.9 (C-3), 23.5 (CH3-5); LR-ESI–MS m/z = 259.91 [M + H]+.
2,5-Dimethyl-6,8-dihydroxy-chromone (2): brownish powder, 1H NMR (500 MHz, DMSO-d6): δH 10.15 (s, 8-OH), 10.04 (s, 6-OH), 7.20 (s, H-10), 6.57 (d, J = 2.2 Hz, H-9), 6.55 (d, J = 2.2 Hz, H-7), 6.18 (s, H-3), 2.71 (s, CH3-5), 2.37 (s, CH3-2); 13C NMR (125 MHz, DMSO-d6): δc 178.4 (C-4), 163.3 (C-2), 159.2 (C-8), 156.8 (C-6),156.6 (C-14), 138.6 (C-5), 134.3 (C-11), 124.7 (C-10), 116.1 (C-13), 112.0 (C-3), 107.1 (C-12), 102.8 (C-7), 101.1 (C-9), 22.9 (CH3-5), 19.4 (CH3-2); LR-ESI–MS m/z = 257.94 [M + H]+.
Penimethavone A (3): yellowish powder, 1H NMR (500 MHz, DMSO-d6): δH 12.91 (s, OH-5), 10.81 (s, OH-7), 9.81 (s, OH- 2′), 9.71 (s, OH-4′), 6.35 (d, J = 1.8 Hz, H-8), 6.27 (d, J = 1.5 Hz, H-3′), 6.20 (d, J = 1.8 Hz, H-6), 6.21 (d, J = 1.5 Hz, H-5′), 6.21 (s, H-3), 2.13 (s, H-7′); 13C NMR (125 MHz, DMSO-d6): δc 181.8 (C-4), 164.2 (C-7), 164.1 (C-2), 161.6 (C-5), 159.9 (C-4′), 158.2 (C-8a), 157.0 (C-2′), 139.0 (C-6′), 111.5 (C-3), 111.2 (C-1′), 108.7 (C-5′), 103.8 (C-4a), 100.2 (C-3′), 98.7 (C-6), 93.8 (C-8), 19.8 (C-7′); LR-ESI–MS m/z = 201.10 [M + H]+.
Calyxanthone (4): yellow powder, 1H NMR (400 MHz, DMSO-d6): 7.34 (s, H-4), 7.06 (s, H-5), 6.35 (d, J = 2.1 Hz, H-2), 6.17 (d, J = 2.1 Hz, H-7), 2.44 (s, H-11); LR-ESI–MS m/z = 287.10 [M + H]+.
Endocrocin (5): orange powder, 1H NMR (400 MHz, DMSO-d6): δH 12.67 (s, 1-OH), 7.40 (s, H-4), 7.09 (s, H-5), 6.59 (s, H-7), 2.48 (s, CH3-3); LR-ESI–MS m/z = 315.04 [M + H]+.
Hydroxyviocristin (6): yellow powder, 1H NMR (400 MHz, CD3OD): δH 7.60 (s, H-9), 7.21 (s, H-3), 7.20 (d, J = 2.0 Hz, H-8), 6.58 (d, J = 2.0 Hz, H-6), 2.45 (s, CH3-3); LR-ESI–MS m/z = 271.10 [M + H]+.
Chemicals
All the enzymes used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA) [13]. Mono- and di-basic sodium phosphate (anhydrous) were obtained from Daejung Chemicals and Metals (Siheung, South Korea). A dialyzer DiaEasy™ (6–8 kDa) was purchased from BioVision (St. Grove, MA, USA).
Enzyme assays and kinetics
MAO-A human (recombinant, M7316, Sigma-Aldrich, 527 amino acids natively) and MAO-B human (recombinant, M7441, Sigma-Aldrich, 520 amino acids natively) activities were measured using the continuous method at 316 and 250 nm for 45 min, respectively, using a UV/Vis spectrophotometer (OPTIZEN POP, K-LAB, Daejeon, Republic of Korea). The absorbance was measured using 0.5 mL volume in a 1 mL quartz cuvette containing 50 mM sodium phosphate (pH 7.2) at room temperature [13, 36]. The substrates used were kynuramine (0.06 mM) and benzylamine (0.3 mM), respectively. AChE (electric eel, C2888, Sigma-Aldrich,) and BChE (equine serum, C7512, Sigma-Aldrich,) activities were analyzed using the continuous assay method at 412 nm for 15 min with 0.50 mM acetylthiocholine iodide and butyrylthiocholine iodide as substrate, respectively [37, 38]. BACE1 activity was measured by BACE1 assay kit (human, CS0010, Sigma-Aldrich) using a microplate spectrophotometer (Varioskan LUX, Thermo Fisher Scientific, Inc., Waltham, MA, USA).
For enzyme kinetics, MAO-A activity was assayed at 0.0075–0.12 μM kynuramine [13, 36]. The kinetic parameters, Km and Vmax, were determined using Lineweaver–Burk (LB) plots.
MAO, ChE, and BACE1 inhibition studies
In the primary screening, the inhibitory activity of 10 µM test compounds against MAOs, ChEs, and BACE1 were evaluated. The 50% inhibitory concentration (IC50) of the compounds were calculated up to 40 µM by using GraphPad Prism software 5 (San Diego, CA, USA) [13]. The selectivity index (SI) was calculated using IC50 MAO-B/IC50 MAO-A. The reference inhibitors were included: toloxatone and clorgyline for MAO-A, safinamide and pargyline for MAO-B, and donepezil for AChE or BChE [49, 50]. For the inhibition kinetics, compound 1 for MAO-A was used at approximately 0.5-, 1.0-, and 1.5-times the IC50 [13, 36], and its inhibition constant (Ki) was determined using the secondary plot of their slopes in the LB plots.
Antioxidant activity assay
The antioxidant activity was analyzed by measuring the absorbance at 517 nm, after 15 min preincubation of the test compound (50 μM) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) (0.1 mM) [39].
Reversibility studies
The reversibility patterns of compound 1 for MAO-A inhibition were analyzed by measuring the undialyzed (AU) and dialyzed (AD) residual activities after pre-incubation for 30 min, as previously described. The inhibition type was determined by comparing to the reference compounds [13, 36].
Molecular docking and dynamics simulation
MAO-A and MAO-B structures (PDB ID: 2Z5X and 2V5Z, respectively) were obtained from the Protein Data Bank [40]. The three-dimensional structures of compounds 1 (PubChem ID: 54692973), 2, and 4 (PubChem ID: 23902332) were generated from the SMILES string in PubChem [41] and in-house program using OpenBabel [42]. AutoDock Vina [43] was used to predict the binding positions of molecules. A docking box for each protein was defined as a cube with a length of 22.5 Å along each axis. The distance between voxel points was set to 0.375 Å. AutoDock4 [44] affinity maps were calculated. The receptors and ligands were prepared using AutoDockTools [44].
To explain the selectivity of compound 1 to MAO-A and MAO-B, predicted complex structures were further prepared for molecular dynamics (MD) simulations using CHARMM-GUI [45]. Cubic boxes with a periodic boundary condition were created by extending 10 Å from the docked complexes along each axis. TIP3P water molecules were used to solvate the boxes, and K+ and Cl− ions were then added to neutralize them. Proteins and ligands were parameterized using AMBER FF19SB [46] and GAFF [47], respectively. Both systems were prepared in AMBER input format using CHARMM-GUI [48].
AMBER20 [49] was used to simulate the system. The system was initialized with a minimization of 2500 steepest descent steps, followed by 2500 conjugate gradient steps. Subsequently, the NVT equilibration was performed for 125 ps using a Langevin thermostat. FAD and compound 1 were subjected to position restraints of 1.0 kcal/mol/Å2 during both steps. Three independent 100 ns MD simulations were conducted for both systems using the SHAKE algorithm [50]. The binding affinities of compound 1 and MAOs were determined using MMGBSA [51] for all MD production trajectories. MMGBSA is one of the widely binding affinity prediction methods using MD simulation. Since it was first proposed in 1998 by P. A. Kollman [PMID: 10052623], a couple of thousand papers have been published [PMID: 25835573] to study the binding affinity of biomolecular complexes. The method calculates the interaction between protein and ligand using molecular mechanics (MM), and solvation of each molecule using generalized-Born (GB) and solvent accessible area (SA) for polar and nonpolar atoms, respectively. Compared to the docking score, it shows higher correlation with the experimentally observed binding affinities [PMID: 23268595]. In this study, the igb option was set to 2, and the interaction between the compound and binding-site residues was analyzed by decomposing MMGBSA to the residue level.
Results and discussion
Identification of the fungal strain SG-W3
The beta-tubulin segment of a seawater-derived strain SG-W3 was analyzed for molecular identification, and the resulting 402-bp sequence was deposited in GenBank (accession number OR639834). Beta-tubulin sequence-based neighbor-joining phylogenetic analysis showed that SG-W3 was closely related to Penicillium cyclopium CBS 144.45 (MN969380), Penicillium polonicum CBS 222.28 (MN969392), and Penicillium melanoconidium CBS 115506 (MN969387) with 97.77%, 97.52%, and 97.52% sequence identity, respectively (Additional file 1: Fig. S1). In the phylogenetic tree, SG-W3 was clustered separately and thus not identified at the species level. Based on these data, SG-W3 was assigned as Penicillium.
Identification of the compounds
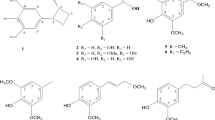
Compound 1 was isolated as a brownish powder, and LR-ESI–MS spectroscopic data showed an ionic peak at m/z 259.91 [M + H]+. The 1H NMR spectrum of compound 1 displayed meta-coupled aromatic protons δH at 6.55 (d, J = 2.2 Hz, H-7) and 6.52 (d, J = 2.2 Hz, H-9); an olefinic proton δH at 5.51 (s, H-3); an methyl proton at δH 2.65 (s, CH3-5); an upfielded proton at δH 7.19 (s, H-6); and three hydroxy protons at δH 9.86 (s, 10-OH), 10.00 (s, 8-OH), and 12.12 (s, 4-OH) (Additional file 1: Fig. S2). The 13C NMR spectra showed one ester carbonyl carbon at δC 161.2 (C-2); one methyl carbon at δC 23.5 (CH3-5); three aromatic methine carbons at δC 100.9 (C-7), 102.7 (C-9), and 124.1 (C-6); five quaternary carbons at δC 106.5 (C-10a), 107.7 (C-4a), 132.4 (C-5), 138.1 (C-6a), and 154.6 (C-10b); three aromatic carbons bearing a hydroxyl group at δC 156.6 (C-10), 158.9 (C-8) and 169.7 (C-4); and an olefinic methine carbon at δC 88.9 (C-3) (Additional file 1: Fig. S3). Based on a comparison of NMR data with the literature, compound 1 was identified as pannorin [52]. Compound 1 inhibited the GSK-3 isoform and HMG-CoA reductase [52, 53].
Compound 2 was isolated (a brownish powder), and LR-ESI–MS spectroscopic data showed an ionic peak at m/z 257.94 [M + H]+. The 1H NMR spectrum of compound 2 showed two methyl groups at δH 2.37 (s, CH3-2), 2.71 (s, CH3-5), two hydroxyl group at δH 10.04 (s, 6-OH), 10.15 (s, 8-OH), a pair of meta-coupled protons at δH 6.55 (d, J = 2.2 Hz, H-7), 6.57 (d, J = 2.2 Hz, H-9), two singlet aromatic protons at δH 7.20 (s, H-10) and 6.18 (s, H-3) (Additional file 1: Fig. S4). The 13C NMR spectrum of compound 2 indicated the presence of 15 carbon signals, including a carbonyl group at δc 178.4 (C-4); four aromatic carbons connected with oxygen atoms at δc 163.3 (C-2), 159.2 (C-8), 156.8 (C-6),156.6 (C-14); four aromatic tertiary carbons at δc 101.1 (C-9), 102.8 (C-7), 112.0 (C-3), 124.7 (C-10); and four quaternary carbons at δc 107.1 (C-12), 116.1 (C-13), 134.3 (C-11), 138.6 (C-5), revealing that compound 2 had a benzochromone skeleton (Additional file 1: Fig. S5). Based on a comparison of the NMR data with the data previously reported, we identified compound 2 as 2, 5-dimethyl-6, 8-dihydroxy-chromone [54].
Compound 3 was isolated as a yellow powder, and LR-ESI–MS revealed an ionic peak at m/z 201.10 [M + H]+. The 1H NMR spectrum of compound 3 exhibited four exchangeable protons at δH 12.91 (s, OH-5), 10.81 (s, OH-7), 9.81 (s, OH- 2′), 9.71 (s, OH-4′) and two pairs of meta-coupled aromatic signals at δH 6.35 (d, J = 1.8 Hz, H-8), 6.27 (d, J = 1.5 Hz, H-3′), 6.20 (d, J = 1.8 Hz, H-6), 6.21 (d, J = 1.5 Hz, H-5′). Additionally, one olefinic singlet at δH 6.21 (s, H-3) and one methyl singlet at δH 2.13 (s, H-7′) were observed (Additional file 1: Fig. S6). Compound 3 was identified as penimethavone A by comparing its NMR data with those reported previously [55]. Various biological activities of compound 3, including anti-cancer, anti-bacterial, and anti-SARS-CoV-2 activities, have been reported [55,56,57].
Compound 4 was isolated as a yellow powder, and LR-ESI–MS spectroscopic data showed an ionic peak at m/z 287.10 [M + H]+. The 1H NMR spectrum of compound 4 showed a pair of meta-coupled protons at δH 6.35 (d, J = 2.1 Hz, H-2) and 6.17 (d, J = 2.1 Hz, H-7), two singlet aromatic protons at δH 7.34 (s, H-4) and 7.06 (s, H-5), and one methyl proton at δH 2.44 (s, H-12) (Additional file 1: Fig. S7). Compound 4 was identified as a calyxanthone based on a comparison of its 1H NMR data with previously reported data [56]. In addition, compound 4 exhibits anti-proliferative activity [56].
Compound 5 was isolated (an orange powder), and the LR-ESI–MS spectroscopic data showed an ionic peak at m/z 315.04 [M + H]+. The 1H NMR spectrum of compound 5 showed one hydroxy proton δH at 12.67 (s, 1-OH); three singlet aromatic protons at δH 7.40 (s, H-4), 7.09 (s, H-5), and 6.59 (s, H-7); and one methyl singlet at δH 2.48 (s, H-12) (Additional file 1: Fig. S8). It was identified as endocrocin by comparing the 1H NMR data with literature [56]. Compound 5 exhibits anti-inflammatory activity and may act as a GDH inhibitor [58, 59].
Compound 6 was isolated as a yellow powder, and the LR-ESI–MS spectroscopic data revealed an ionic peak at m/z 271.10 [M + H]+. The 1H NMR spectrum of compound 6 displayed four aromatic protons at δH 7.60 (s, H-9), 7.21 (s, H-3), 7.20 (d, J = 2.0 Hz, H-8), 6.58 (d, J = 2.0 Hz, H-6), and one methyl proton at δH 2.45 (s, CH3-3) (Additional file 1: Fig. S9). Compound 6 was identified as hydroxyviocristin based on comparison of its 1H NMR data with previously reported data [37].
Thus, six compounds, pannorin (1), 2, 5-dimethyl-6, 8-dihydroxy-chromone (2), penimethavone A (3), calyxanthone (4), endocrocin (5), and hydroxyviocristin (6), were isolated from Penicillium sp. SG-W3 (Fig. 1). Pannorin (1) has been previously produced by Aspergillus sp. [52]. Compound 2,5-dimethyl-6,8-dihydroxy-chromone (2) was previously isolated from the roots of Rheum palmatum [54], and this is the first report of its isolation from Penicillium sp. [55]. Penimethavone A (3), calyxanthone (4), and endocrocin (5) were isolated from Penicillium spp. [56], and hydroxyviocristin (6) was isolated from Aspergillus cristatus [60].

Chemical structures of compounds 1–6
Based on the structural features of compounds 1–6, the plausible biosynthetic pathway of them was proposed to begin with the non-reducing polyketide synthase for the generating C16-octaketide. Then, this undergoes cyclization to form atrochryone carboxylic acid, which can be autoxidized to produce endocrocin (5). The emodin derived from this process undergoes enzymatic transformation, involving oxidative ring-opening between C-4 and C-5. Subsequent dehydration leads to the formation of intermediate calyxanthone (4) [61].
Inhibitory activity against the enzymes
Inhibitory activities of six compounds against MAOs (MAO-A and MAO-B), ChEs (AChE and BChE), and BACE1 were evaluated (Table 1). In primary screening, most 10 μM compounds showed higher inhibition against MAO-A at than that against other enzymes (Table 1). Among them, compounds 1 and 2 most potently inhibited MAO-A (IC50: 1.734 and 4.290 μM, respectively). These IC50 values of MAO-A were 3.40- and 1.38-times lower than naphthopyrone rubrofusarin (IC50 = 5.90 μM), respectively [62]. The SI of compound 1 was relatively high, > 23.07 for MAO-A vs. MAO-B (IC50 > 40 μM) (Table 1). Most compounds had high IC50 (> 40 μM) for MAO-B, except compounds 2 and 6 (IC50: 23.29 and 11.82 μM, respectively; Table 1). Regarding the structure–activity relationship (SAR), the naphthopyrone (also named benzocoumarin) scaffold (1) showed higher MAO-A inhibition than other scaffolds such as anthracene (2, 4, 5, and 6) and flavone (3) (Fig. 1, Table 1). In contrast, most compounds weakly inhibited ChEs and BACE1 (Table 1). These results suggested that compound 1 is a potent selective MAO-A inhibitor, comparable to the reference toloxatone (IC50: 1.25 μM).
Antioxidant activity
Compounds 1 and 2 showed antioxidant activity, with 37.8% and 33.1% inhibition in the DPPH assay, respectively (Fig. 2). The activity of compound 1 was similar to that of the reference Trolox (40.7%), indicating that compound 1 is an efficient antioxidant.

Antioxidant activity of compounds 1–6 using for 2,2-dipheny-1-picrylhydrazyl (DPPH) assay. The results are the means ± standard errors of triplicate experiments. Trolox was used as a reference inhibitor. The % inhibition was calculated as (absorbance of control − absorbance of reaction mixture)/absorbance of control × 100
Inhibition kinetics
MAO-A inhibition by compound 1 was analyzed using five substrate concentrations (kynuramine) and three inhibitor concentrations. In LB plots, the lines of compound 1 intersected at a point on y-axis and compound 1 was determined to be a competitive MAO-A inhibitor (Fig. 3A), and its secondary plot showed that its Ki was 1.049 ± 0.030 μM (Fig. 3B). These results suggested that compound 1 is a potent competitive MAO-A inhibitor.

A Lineweaver–Burk (LB) plots and B its secondary plot of the slope vs inhibitor concentrations for MAO-A inhibition by compound 1. The experiments were conducted using five kynuramine concentrations and three inhibitor concentrations
Reversibility studies
Inhibition reversibility was evaluated by the dialysis method after pre-incubating the enzyme (two times the IC50) and inhibitor for 30 min. Recovery types were analyzed by comparing the undialyzed (AU) and dialyzed (AD) relative residual activities. MAO-A inhibition by compound 1 recovered from 32.70% (AU) to 75.00% (AD) (Fig. 4). The relative residual activities of compound 1 was similar to those of toloxatone, a reversible inhibitor (AU 29.53%; AD 80.00%), and different from those of clorgyline, an irreversible inhibitor (AU 36.10%; AD 33.85%). These results indicate that compound 1 is a reversible MAO-A inhibitor.

Recovery of MAO-A inhibition by compound 1 using dialysis. Toloxatone, clorgyline, and compound 1 concentrations used were approximately two times their 50% inhibitory concentration (IC50; 2.50, 0.026, and 3.40 μM, respectively). After 30 min pre-incubation, the mixtures were dialyzed for 6 h with twice of buffer change
Molecular docking analysis of three compounds
Docking simulation was performed to investigate the activity trend of compounds 1, 2, and 4. The predicted binding affinities are shown in Additional file 1: Table S1. Compound 1 exhibits the lowest binding affinity to MAO-A, and compound 2 shows the lowest binding affinity to MAO-B, as observed in IC50 values. According to the Cheng-Prusoff equation [PMID: 4202581], lower IC50 corresponds to the lower binding affinity, although they are not directly related. The predicted Ki values of compound 1 to MAO-A and MAO-B from the docking scores are 501 nM and 989 nM, respectively. Su et al. [PMID: 30481020] benchmarked the correlation between the experimentally observed binding affinity and the docking score. AutoDock, which is employed in this study, shows the Pearson’s corelation coefficient as 0.604. Based on the benchmark, compound 1 might have a larger Ki to MAO-B than to MAO-A.
Molecular dynamics simulations to explain selectivity
The docking poses of compound 1 to MAO proteins, as the starting structure for MD simulations, are displayed in Fig. 5. The hydrophobic scaffolds of compound 1 in both binding poses substantially overlapped since both protein-binding pockets comprised conserved hydrophobic residues. G67 (58), Y69 (61), F177 (168), V182 (173), Y197 (188), I207 (198), I325 (326), T336 (327), L337 (328), M350 (341), F352 (343), Y407 (398), T408 (399), G443 (434), and Y444 (435) were structurally and sequentially conserved hydrophobic residues in both MAO proteins at 5 Å from the docked poses (MAO-B residue numbers provided in parentheses).

Predicted binding positions of compound 1 to A MAO-A and B MAO-B. The proteins, compound 1, and FAD are indicated in gold, blue, and pink, respectively
In both docking poses, the hydroxyl groups were oriented toward opposite sides owing to N181 (MAO-A) substitution with C172 (MAO-B). The hydroxyl groups of compound 1 formed hydrogen bonds with the side chains in region between Q215 and N181 of MAO-A, but with C172 of MAO-B. The distance between Cδ of Q215 and Cγ of N181 in MAO-A was 7.7 Å. Moreover, the bulky side chain of Y326 (MAO-B) brought it closer to FAD, whereas I335 (MAO-A), which occupied the same position, offered more space to place the molecule.
During the 100 ns MD simulation, the RMSDs of MAO-A and MAO-B in complex with compound 1 fluctuated around 3–4 and 2–3 Å after 30 ns, respectively (Fig. 6). These shifts may be owing to the C-terminal helix of the proteins. Specifically, the long helix in MAO-A, with approximately 14 additional residues or four helix turns, resulted in significant fluctuations during the simulation. The RMSD for the C-terminal helix of MAO-A (V498–L524) and MAO-B (V489–I501) were 1.1–6.2 and 0.8–3.4 Å, respectively. The RMSD of compound 1 was < 0.5 Å in all trajectories, with approximately 0.2 Å fluctuations. Guterres and Im [63] have reported that the RMSD threshold for active molecules was < 3 Å. Therefore, compound 1 may function as an active molecule for both MAO proteins.

RMSD plots of MD simulations for A MAO-A, MAO-B (B), and pannorin-docked C MAO-A and D MAO-B. Complex and ligand RMSDs calculated from the same trajectory are presented as the same color codes
The binding affinities calculated by MMGBSA showed that MAO-A (− 25.02 kcal/mol) was a stronger binding partner than MAO-B (− 24.06 kcal/mol). Converting the predicted affinities to Ki gives 1.03 × 10–18 M and 5.03 × 10–18 M for MAO-A and MAO-B, respectively. The predicted Ki for MAO-A and compound 1 complex is much smaller than the experimentally determined one. Due to the enthalpy nature of MMGBSA method, the binding affinity might be overestimated. However, the method is good at relative ranking the compounds [PMID: 25835573]. Since the IC50 values and predicted binding affinities are correlated as discussed, the predicted values are consistent with the experimentally observed IC50.
Residue-wise pair decomposition analysis using MMGBSA identified hotspots for compound 1. Table 2 lists the top 10 residues interacting with the compound in both complexes. Hydrophobic amino acids constituted most residues in both proteins. For MAO-A, Q215 (− 4.12 kcal/mol) and N181 (− 3.95 kcal/mol) were the top two residues driven by electrostatic potential (− 2.16 and − 4.29 kcal/mol, respectively). C172 of MAO-B occupied the same position as N181 of MAO-A and was ranked within the top 10 with a score of − 2.81 kcal/mol. However, the van der Waals interaction between C172 and compound 1 was dominant (− 1.39 kcal/mol). The interaction between MAO-B Y326 and compound 1 was ranked third with − 3.53 kcal/mol. Thus, this interaction placed the compound closer to FAD in MAO-B than that in MAO-A and the interaction between FAD and compound 1 was ranked fourth with − 3.27 kcal/mol. From the decomposition analysis, the selectivity of compound 1 for MAO-A over MAO-B was mainly caused by MAO-A N181/MAO-B C172 and MAO-A I335/MAO-B Y326.
In conclusion, marine fungi-produced secondary metabolites often exhibit distinct structural features and various biological and pharmacological characteristics. In this study, six compounds were isolated from marine Penicillium sp. SG-W3 and their inhibitory activities against MAOs, ChEs, and BACE1 related to neurological diseases were evaluated. Among them, compound 1 (pannorin) showed the highest inhibitory activity against MAO-A (IC50 = 1.734 μM) and SI > 23.07. The kinetic study showed compound 1 was a competitive reversible MAO-A inhibitor with 1.049 ± 0.030 μM Ki. In addition, compound 1 exhibited antioxidant activity in the DPPH assay. However, all the compounds weakly inhibited MAO-B, AChE, BChE, and BACE1, except compound 6 against MAO-B (IC50: 11.82 μM). Molecular docking simulation predicted that compound 1 formed hydrogen bonds with MAO-A at Q215 and N181, and have stronger binding energy to MAO-A (− 25.02 kcal/mol) than that to MAO-B (− 24.06 kcal/mol) using MMGBSA. These results suggested that compound 1 is a potent, reversible, competitive, and selective MAO-A inhibitor that could be used as a therapeutic candidate for treating neurological diseases.
Availability of data and materials
All data generated or analyzed during the present study are included in this published article.
References
Cao Z, Song Q, Yu G, Liu Z, Cong S, Tan Z, Deng Y (2021) Novel 3-Benzylidene/benzylphthalide mannich base derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem 35:116074
Behl T, Kaur D, Sehgal A, Singh S, Sharma N, Zengin G, Andronie-Cioara FL, Toma MM, Bungau S, Bumbu AG (2021) Role of monoamine oxidase activity in Alzheimer’s disease: an insight into the therapeutic potential of inhibitors. Molecules 26:3724
Dhapola R, Hota SS, Sarma P, Bhattacharyya A, Medhi B, Reddy DH (2021) Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 29:1669–1681
Santin Y, Resta J, Parini A, Mialet-Perez J (2021) Monoamine oxidases in age-associated diseases: new perspectives for old enzymes. Ageing Res Rev 66:101256
Ashrafian H, Zadeh EH, Khan RH (2021) Review on Alzheimer’s disease: inhibition of amyloid beta and tau tangle formation. Int J Biol Macromol 167:382–394
Olanow CW, Tatton WG (1999) Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 22:123–144
Chang KH, Chen CM (2020) The role of oxidative stress in Parkinson’s disease. Antioxidants (Basel) 9:597
Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, MacGregor R (1997) Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 18:431–435
Saura J, Andrés N, Andrade C, Ojuel J, Eriksson K, Mahy N (1997) Biphasic and region-specific MAO-B response to aging in normal human brain. Neurobiol Aging 18:497–507
Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47:S161-170
Carradori S, Secci D, Bolasco A, Chimenti P, D’Ascenzio M (2012) Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin Ther Pat 22:759–801
Ramsay RR, Albreht A (2018) Kinetics, mechanism, and inhibition of monoamine oxidase. J Neural Transm 125:1659–1683
Oh JM, Jang HJ, Kang MG, Mun SK, Park D, Hong SJ, Kim MH, Kim SY, Yee ST, Kim H (2022) Medicarpin and homopterocarpin isolated from Canavalia lineata as potent and competitive reversible inhibitors of human monoamine oxidase-B. Molecules 28:258
Schedin-Weiss S, Inoue M, Hromadkova L, Teranishi Y, Yamamoto NG, Wiehager B, Bogdanovic N, Winblad B, Sandebring-Matton A, Frykman S (2017) Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res Ther 9:57
Youdim MBH, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7:295–309
Finberg JP, Youdim MB (1983) Selective MAO A and B inhibitors: their mechanism of action and pharmacology. Neuropharmacology 22:441–446
Ramsay RR, Tipton KF (2017) Assessment of enzyme inhibition: a review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 22:E1192
Sakayanathan P, Loganathan C, Kandasamy S, Ramanna RV, Poomani K, Thayumanavan P (2019) In vitro and in silico analysis of novel astaxanthin-S-allyl cysteine as an inhibitor of butyrylcholinesterase and various globular forms of acetylcholinesterases. Int J Biol Macromol 140:1147–1157
Darvesh S, Hopkins DA, Geula C (2003) Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 4:131–138
Kumar A, Pintus F, Di Petrillo A, Medda R, Caria P, Matos MJ, Viña D, Pieroni E, Delogu F, Era B (2018) Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci Rep 8:4424
Dos Santos TC, Gomes TM, Pinto BAS, Camara AL, de Paes AMA (2018) Naturally occurring acetylcholinesterase inhibitors and their potential use for Alzheimer’s disease therapy. Front Pharmacol 9:1192
Lane RM, Potkin SG, Enz A (2006) targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J Neuropsychopharmacol 9:101–124
Li S, Li AJ, Travers J, Xu T, Sakamuru S, Klumpp-Thomas C, Huang R, Xia M (2021) Identification of compounds for butyrylcholinesterase inhibition. SLAS Discovery 26:1355–1364
Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL (1995) Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem 64:749–760
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537:50–56
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S (2023) Lecanemab in early Alzheimer’s disease. N Engl J Med 388:9–21
Chowdhury S, Kumar S (2021) Inhibition of BACE1, MAO-B, cholinesterase enzymes, and anti-amyloidogenic potential of selected natural phytoconstituents: Multi-target-directed ligand approach. J Food Biochem 45:e13571
Imhoff JF (2016) natural products from marine fungi–still an underrepresented resource. Mar Drugs 14:19
Rateb ME, Ebel R (2011) Secondary metabolites of fungi from marine habitats. Nat Prod Rep 28:290–344
Ma HG, Liu Q, Zhu GL, Liu HS, Zhu WM (2016) Marine natural products sourced from marine-derived Penicillium fungi. J Asian Nat Prod Res 18:92–115
Liu S, Su M, Song SJ, Jung JH (2017) Marine-derived Penicillium species as producers of cytotoxic metabolites. Mar Drugs 15:329
Hong A, Tu LC, Yang I, Lim KM, Nam SJ (2020) Marine natural products with monoamine oxidase (MAO) inhibitory activity. Pharm Biol 58:716–720
Chung D, Baek K, Bae SS, Jung J (2019) Identification and characterization of a marine-derived chitinolytic fungus, Acremonium sp. YS2-2. J Microbiol 57:372–380
Gavazzi F, Pigna G, Braglia L, Gianì S, Breviario D, Morello L (2017) Evolutionary characterization and transcript profiling of β-tubulin genes in flax (Linum usitatissimum L.) during plant development. BMC Plant Biol 17:237
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Oh JM, Jang HJ, Kim WJ, Kang MG, Baek SC, Lee JP, Park D, Oh SR, Kim H (2020) Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int J Biol Macromol 151:441–448
Jeong GS, Kaipakasseri S, Lee SR, Marraiki N, Batiha GES, Dev S, Palakkathondi A, Kavully FS, Gambacorta N, Nicolotti O (2020) Selected 1,3-benzodioxine-containing chalcones as multipotent oxidase and acetylcholinesterase inhibitors. Chem Med Chem 15:2257–2263
Oh JM, Kang Y, Hwang JH, Park JH, Shin WH, Mun SK, Lee JU, Yee ST, Kim H (2022) Synthesis of 4-substituted benzyl-2-triazole-linked-tryptamine-paeonol derivatives and evaluation of their selective inhibitions against butyrylcholinesterase and monoamine oxidase-B. Int J Biol Macromol 217:910–921
Brand-Williams W, Marie-Elisabeth C, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. Food Sci Technol Lebensm Wiss Technol 28:25–30
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B (2023) PubChem 2023 update. Nucleic Acids Res 51:D1373–D1380
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminform 3:33
Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Jo S, Kim T, Iyer VG, Im W (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29:1859–1865
Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q (2020) ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput 16:528–552
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12:405–413
Case DA et al (2020) Amber 2021. University of California, San Francisco
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA.Py: an efficient program for end-state free energy calculations. J Chem Theory Comput 8:3314–3321
Ogawa H, Hasumi K, Sakai K, Murakawa S, Endo A (1991) Pannorin, a new 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor produced by Chrysosporium pannorum. J Antibiot (Tokyo) 44:762–767
Wiese J, Imhoff JF, Gulder TAM, Labes A, Schmaljohann R (2016) Marine fungi as producers of benzocoumarins, a new class of inhibitors of glycogen-synthase-kinase 3β. Mar Drugs 14:200
Li L, Zhang C, Xiao YQ, Chen DD, Tian GF, Wang Y (2011) Two new compounds from the roots of Rheum palmatum. Chin J Nat Med 9:0410–0413
Hou XM, Wang CY, Gu YC, Shao CL (2016) Penimethavone A, a flavone from a gorgonian-derived fungus Penicillium chrysogenum. Nat Prod Res 30:2274–2277
Yong K, Kaleem S, Yi W, Wu B, Zhang Z (2021) new polyhydroxanthones from the marine-associated fungus Penicillium sp ZZ1750. Teterahedron Lett 81:153354
Sayed AM, Alhadrami HA, El-Gendy AO, Shamikh YI, Belbahri L, Hassan HM, Abdelmohsen UR, Rateb ME (2020) Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 8:970
Gautam R, Karkhile KV, Bhutani KK, Jachak SM (2010) Anti-inflammatory, cyclooxygenase (COX)-2, COX-1 inhibitory, and free radical scavenging effects of Rumex nepalensis. Planta Med 76:1564–1569
Song C, Wu M, Zhang Y, Li J, Yang J, Wei D, Li H, Guo L, Qin J (2022) Bioactive monomer and polymer polyketides from edible mushroom Cortinarius caerulescens as glutamate dehydrogenase inhibitors and antioxidants. J Agric Food Chem 70:804–814
Laatsch H, Anke H (1982) Stoffwechselprodukte von mikroorganismen, 214 viocristin, isoviocristin und hydroxyviocristin—struktur und synthese natürlich vorkommender 1,4-anthrachinone. Liebigs Ann Chem 12:2189–2215
Yang BJ, Chen GD, Li YJ, Hu D, Guo LD, Xiong P, Gao H (2016) A new xanthone glycoside from the endolichenic fungus Sporormiella irregularis. Molecules 21:764
Paudel P, Seong SH, Shrestha S, Jung HA, Choi JS (2019) In vitro and in silico human monoamine oxidase inhibitory potential of anthraquinones, naphthopyrones, and naphthalenic lactones from Cassia obtusifolia linn seeds. ACS Omega 4:16139–16152
Guterres H, Im W (2020) Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J Chem Inf Model 60:2189–2198
Acknowledgements
Not applicable.
Funding
This work was supported by National Research Foundation of Korea (NRF) grant funded by the Korean Government (NRF-2022R1A2B5B01002536) and by National Marine Biodiversity Institute of Korea (2023M00600).
Author information
Authors and Affiliations
Contributions
Conceptualization: HK; Compound isolation: QG, E-YL, DC, GC; Biological assay: JMO; Molecular docking and dynamic analysis: WHS; Writing–original draft preparation: JMO, QG, DC, WHS; Writing–review and editing: S-JN, HK; Supervision: S-JN, HK. All the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Phylogenetic analysis of SG-W3. Figure S2. 1H NMR spectrum of pannorin (1) in DMSO-d6. Figure S3. 13C NMR Spectrum of pannorin (1) in DMSO-d6. Figure S4. 1H NMR spectrum of 2, 5-dimethyl-6, 8-dihydroxy-chromone (2) in DMSO-d6. Figure S5. 13C NMR Spectrum of 2, 5-dimethyl-6, 8-dihydroxy-chromone (2) in DMSO-d6. Figure S6. 1H NMR spectrum of penimethavone A (3) in DMSO-d6. Figure S7. 1H NMR spectrum of calyxanthone (4) in DMSO-d6. Figure S8. 1H NMR spectrum of endocrocin (5) in DMSO-d6 S9. Figure S9. 1H NMR spectrum of hydroxyviocristin (6) in Methanol-d4. Table S1. Predicted binding affinities of the compounds.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, J.M., Gao, Q., Shin, WH. et al. Pannorin isolated from marine Penicillium sp. SG-W3: a selective monoamine oxidase A inhibitor. Appl Biol Chem 67, 26 (2024). https://doi.org/10.1186/s13765-024-00878-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00878-7