
Abstract
Among 276 herbal extracts, a methanol extract of Castanopsis cuspidata var. sieboldii stems was selected as an experimental source for novel acetylcholinesterase (AChE) inhibitors. Five compounds were isolated from the extract by activity-guided screening, and their inhibitory activities against butyrylcholinesterase (BChE), monoamine oxidases (MAOs), and β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) were also evaluated. Of these compounds, 4′-O-(α-l-rhamnopyranosyl)-3,3′,4-tri-O-methylellagic acid (3) and 3,3′,4-tri-O-methylellagic acid (4) effectively inhibited AChE with IC50 values of 10.1 and 10.7 µM, respectively. Ellagic acid (5) inhibited AChE (IC50 = 41.7 µM) less than 3 and 4. In addition, 3 effectively inhibited MAO-B (IC50 = 7.27 µM) followed by 5 (IC50 = 9.21 µM). All five compounds weakly inhibited BChE and BACE-1. Compounds 3, 4, and 5 reversibly and competitively inhibited AChE, and were slightly or non-toxic to MDCK cells. The binding energies of 3 and 4 (− 8.5 and − 9.2 kcal/mol, respectively) for AChE were greater than that of 5 (− 8.3 kcal/mol), and 3 and 4 formed a hydrogen bond with Tyr124 in AChE. These results suggest 3 is a dual-targeting inhibitor of AChE and MAO-B, and that these compounds should be viewed as potential therapeutics for the treatment of Alzheimer’s disease.
Similar content being viewed by others
Introduction
Cognitive dysfunctions, such as learning, memory, processing information speed, visual perception, mental flexibility, and persistent attention-deficit dysfunctions, are the primary symptoms of Alzheimer's disease (AD). Acetylcholine (ACh) regulates cognitive functions, especially learning and memory, via neurotransmission and is synthesized from choline and acetyl co-enzyme A in presynaptic neurons and then released into the synaptic gaps. Acetylcholinesterase (AChE) terminates the ACh-mediated neurotransmission and is mostly found in neurons1,2. The therapeutic efficacies of AChE inhibitors in AD have been shown to be due to augment synaptic ACh levels in the cerebral cortex and improve cholinergic transmissions3. In AD, levels of ACh are low and cause central nervous system (CNS) disorders, which are characterized by gradual declines in cognition, memory, and cognitive functions. Currently, AChE inhibitors approved by the FDA for the treatment of AD include donepezil, galantamine, and rivastigmine. Like AChE, butyrylcholinesterase (BChE) also importantly contributes to cholinergic neurotransmission. BChE is present in glial cells, hippocampus, and the temporal nerve cortex, and is involved in cognitive function. BChE has less substrate specificity than AChE, but both enzymes effectively hydrolyze ACh2.
Importantly, the formation and aggregation of β-amyloid peptide (Aβ) are associated with the hydrolysis of amyloid precursor protein (APP) by β-site amyloid precursor protein cleaving enzyme 1 (BACE-1; β-secretase-1), and since the anionic site of AChE is involved in Aβ aggregation, studies on dual AChE and BACE-1 inhibitors are being actively pursued4.
On the other hand, monoamine oxidases (MAOs) are involved in the pathways leading to catecholamine and 5-hydroxytryptamine inactivation, and thus, MAOs are recognized targets in diseases associated with these pathways. Furthermore, selective MAO-A inhibitors have anti-depressant activity, and selective MAO-B inhibitors are recognized developmental targets for the treatment of AD and Parkinson's disease (PD)5. Irreversible MAO-B inhibitors such as rasagiline and selegiline are used for the treatment of PD6,7,8, alongside levodopa, dopamine agonists, and catechol-O-methyltransferase inhibitors.
Recently, a multi-targeting treatment strategy was devised to target MAO-B and AChE, and it has been reported that MAO and AChE inhibitors can improve the cognitive functions and relieve symptoms in AD by increasing monoamine and choline ester levels9.
During our on-going efforts to identify potent natural inhibitors of MAO-A, MAO-B, and AChE in a herbal extract library, we found that rhamnocitrin isolated from the leaves of Prunus padus var. seoulensis, potently and selectively inhibits human MAO-A10, and that calycosin isolated from Maackia amurensis potently and selectively inhibits human MAO-B11. In this study, we screened Castanopsis cuspidata var. sieboldii as a potent AChE inhibiting herbal source, and isolated AChE inhibitors by activity-guided fractionation.
C. cuspidata is an evergreen broad-leaved tree that is widely distributed in western Japan12, and has been reported to contain galloyl shikimic acid13, ellagitannins14, and dehydrodiallic acid, cretanin, chesnatin, chestanin15, galloyl ester triterpenoid, and hexahydorxydiphenic acid conjugated triterpenoid16. Furthermore, extract of C. cuspidata has antioxidant17,18, anticancer, and anti-inflammatory18, and anti-fungal19 properties.
In this study, C. cuspidata was selected as an AChE inhibitor resource from a library of herbal extracts, five compounds were isolated and identified in an extract of the stems of the plant. Their inhibitory activities against AChE, BChE, MAO-A, MAO-B, and BACE-1 were evaluated, and kinetic and reversibility studies, cytotoxicity tests, in silico pharmacokinetics, and docking simulations were performed to identify novel candidate compounds for the treatment of AD and PD.
Materials and methods
General experimental procedures
1H and 13C NMR spectroscopic data were recorded using JEOL ECZ500R and JEOL ECA600 (JEOL, Tokyo, Japan) instruments, respectively. HR-ESI-MS data were acquired using an ACQUITY UPLC I-Class/Vion IM-QTOF system (Waters, Milford, MA, USA) coupled with ACQUITY BEH C18 column (Waters, 2.1 × 100 mm, 1.7 µm). Preparative HPLC was performed using a YMC K-Prep LAB 300 (YMC, Kyoto, Japan) equipped with a DAD-50-700S column (50.0 × 700 mm) packed with YMC ODS AQ HG resin (YMC, 10 µm, 500 g) and a Gilson HPLC system (GX271, a 321 pump, and a 172 diode array detector, Gilson, Middleton, WI, USA) coupled with an Acclaim Polar Advantage II C18 column, 250 × 21.2 mm, 5 µm (Thermo Fisher Scientific, Waltham, MA, USA), respectively.
Plant material and the isolation of compounds 1–5
A methanol extract of the stems of C. cuspidata var. sieboldii was provided by the Korean Plant Extract Bank at the Korea Research Institute of Bioscience and Biotechnology (KRIBB, Daejeon, Korea) under agreement on Jan 4, 2020. The plant was identified by Dr. Gwanpil Song, and collected from Aewol-eup, Jeju-do, Korea in 2016. A voucher specimen (KRIB 0084147) was deposited at the KRIBB herbarium. All the experimental protocols adhered to the relevant ethical guidelines/regulations on the usage of plants. The extract (50 g) was separated by preparative reverse phase column chromatography eluting using a H2O/MeOH gradient (0–15 min, 5% MeOH; 15–110 min, 5 → 20% MeOH; 110–140 min, 20 → 50% MeOH; 140–150 min, 50 → 100% MeOH; and 150–180 min, 100% MeOH) to obtain ten fractions (1–10) using the YMC K-Prep LAB300 unit (YMC, DAD-50-700S, flow rate: 100 mL/min). Of these fractions, two (CCS8 and CCS9) identified by bioactivity screening for AChE inhibition were purified by preparative HPLC. CCS8 (2.9 g) was subjected to reverse phase column chromatography using a YMC K-Prep LAB300 and a solvent gradient (0 → 100% MeOH over150 min at 100 mL/min) to yield ten sub-fractions (CCS8-1–CCS8-10). Sub-fraction CCS8-1 (179.6 mg) was purified by preparative HPLC (Acclaim Polar Advantage II, 15 → 20% ACN over 50 min at 15 mL/min) to obtain compound 1 (62.1 mg). Compound 2 (4.5 mg) was obtained from sub-fraction CCS8-5 (297.1 mg) by preparative HPLC (Acclaim Polar Advantage II, 20 → 40% ACN over 50 min at 15 mL/min) and compound 3 (9.3 mg) was isolated from sub-fraction CCS8-6 (30.9 mg) by preparative HPLC (Acclaim Polar Advantage II, 25 → 100% ACN over 65 min at 15 mL/min). CCS-9 (3.6 g) was separated by preparative HPLC, YMC K-Prep LAB300 (YMC, DAD-50-700S, 40% ACN 0–10 min; 40 → 80% ACN 10–90 min; 80 → 100% ACN 90–95 min; 100% ACN 95–120 min at 100 mL/min) to produce ten sub-fractions (CCS9-1–CCS9-10). Compound 5 (6.8 mg) was obtained from sub-fraction CC-9–1-2 (64.7 mg) by preparative HPLC (Acclaim Polar Advantage II, 40 → 60% ACN over 75 min at 15 mL/min) and compound 4 (4.2 mg) was purified from sub-fraction CC-9–1-4 (50.7 mg) by preparative HPLC (Acclaim Polar Advantage II, 55 → 65% ACN over 45 min at 15 mL/min).
Chemicals and enzymes
Recombinant human MAO-A and MAO-B, kynuramine, benzylamine, toloxatone, lazabemide, AChE (Type VI-S from Electrophorus electricus), BChE (equine serum), acetylthiocholine iodide (ATCI), butyrylthiocholine iodide (BTCI), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), tacrine, donepezil, and the BACE-1 activity detection kit were purchased from Sigma-Aldrich (St. Louis, MO, USA). Clorgyline and pargyline were obtained from BioAssay Systems (Hayward, CA, USA).
Roswell Park Memorial Institute-1640 medium (RPMI-1640), Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), and penicillin/streptomycin solution were purchased from Hyclone Laboratories (San Ramon, CA, USA). The cell counting kit-8 (CCK-8) and dimethyl sulfoxide (DMSO) were obtained from Dojindo Laboratories (Kumamoto, Japan) and Sigma-Aldrich, respectively.
Enzyme assays
AChE and BChE inhibitory activities were measured after preincubating enzymes with inhibitors for 15 min before adding substrates (ATCI and BTCI, respectively) and DTNB. AChE activities were assayed as previously described20, with slight modification21,22. Reactions were performed using ~ 0.2 U/mL of enzyme in the presence of 0.5 mM substrate and 0.5 mM of DTNB in 0.5 mL reaction mixtures, which were continuously monitored for 15 min at 412 nm. MAO-A and MAO-B activities were determined using a continuous spectrophotometric method, as described previously23,24. The Km values of MAO-A for kynuramine and of MAO-B for benzylamine were 0.040 and 0.17 mM, respectively. The concentrations of kynuramine (0.06 mM) and benzylamine (0.3 mM) used were 1.5 times and 1.8 times Km values, respectively. Reaction rates are expressed as changes in absorbance per min. BACE-1 assays were performed using a β-secretase (BACE-1) activity detection kit at excitation and emission wavelengths of 320 and 405 nm, respectively, using a fluorescence spectrometer (FS-2, Scinco, Seoul, Korea), after reaction for 2 h at 37 °C with 7-methoxycoumarin-4-acetyl-[Asn670,Leu671]-amyloid β/A4 protein fragment 667–676-(2,4-dinitrophenyl)Lys-Arg-Arg amide trifluoroacetate as substrate25.
Analysis of inhibitor reversibility
The reversibilities of AChE and MAO-B inhibitions by 3, 4, and 5 were investigated by dialysis at concentrations of ~ 2 × IC50 values; tacrine and donepezil were used as reference AChE and BChE inhibitors22,26, and lazabemide and pargyline were used as reference reversible and irreversible MAO-B inhibitors, respectively21. After preincubating three compounds or reference inhibitors with enzymes for 15 min, residual activities of undialyzed (AU) and dialyzed (AD) samples were expressed as percentages of those of non-inhibitor treated controls. Reversibilities were assessed using AU and AD values and compared with those of reference compounds.
Inhibitory activities and enzyme kinetics
Inhibitions of AChE, BChE, MAO-A, MAO-B, and BACE-1 by compounds 1 to 5 were initially investigated at a concentration of 10 µM27,28. IC50 values of the five compounds and the reference compounds (tacrine and donepezil for AChE and BChE, toloxatone and clorgyline for MAO-A, lazabemide and pargyline for MAO-B, and quercetin for BACE-1) were determined. Kinetic parameters, inhibition types, and Ki values of the most potent AChE inhibitors (3 and 4) and MAO-B inhibitors (3 and 5) were analyzed. Kinetics of AChE inhibitions by 3, 4, and 5 and of MAO-B inhibitions by 3 and 5 were investigated at five different substrate concentrations (0.05, 0.10, 0.20, 0.50, and 1.0 mM for AChE and 0.0375, 0.075, 0.15, 0.3, and 0.6 mM for MAO-B) in the absence or presence of each inhibitor at concentrations of ~ 0.5 × , 1.0 × , and 2.0 × their IC50 values. Inhibitory patterns and Ki values were determined using Lineweaver–Burk (LB) plots and secondary plots of LB slopes.
Cytotoxicity test
Madin-Darby canine kidney (MDCK) cells and human acute promyelocytic leukemia (HL-60) cells were obtained from the Korean Cell Line Bank (Seoul, Korea). MDCK cells were cultured in DMEM, and HL-60 cells in RPMI-1640 medium containing 10% FBS, 1% penicillin/streptomycin, and 0.1% 2-mercaptoethanol. Cultures were maintained at 37 °C under 5% CO2, and media were changed every two days.
Cell viabilities were determined using the CCK-8 assay29. Briefly, MDCK cells or HL-60 cells were resuspended at 1 × 105 or 3 × 105 cells/mL and suspensions (100 µL) were added to wells of a 96-well plate and incubated in 5% CO2 atmosphere at 37 °C for 24 h. After incubation, 100 µL of each medium was treated with compounds at 1, 3, 10, 30, or 50 µM and incubated in 5% CO2 at 37 °C for 24 h. CCK-8 (10 µL/well) was then added to 100 µL aliquots from each cell and incubated 2–4 h. Absorbances were measured at 450 nm using a microplate reader (Versamax, Molecular Devices, San Jose, CA, USA).
Docking simulation of ellagic acid derivatives with AChE and MAO-B
To simulate dockings of 3, 4, and 5 with AChE and MAO-B, we used AutoDock Vina30, which has an automated docking facility. To define enzyme binding pockets, we used active sites defined by a complex of AChE with 4-carbamoyl-1-(3-{2-[(E)-(hydroxyimino)methyl]-1H-imidazol-1-yl}propyl)pyridin-1-ium (LND) (PDB ID: 6O5V) and a complex of MAO-B with (R)-rosiglitazone (RGZ) (PDB ID: 4A7A). To prepare 3, 4, and 5 for docking simulations, we performed the following steps: (1) created 2D structures of the three compounds, (2) converted these 2D structures into 3D structures, and (3) performed energy minimization using the ChemOffice program (http://www.cambridgesoft.com). Docking simulations of AChE or MAO-B with 3, 4, and 5 were performed using AutoDock Vina 1.1.231. Based on the docking results, we checked for possible hydrogen bonding interactions with bonding relaxation constraints of 0.4 Å and 10.0° using Chimera 1.15 program32.
Pharmacokinetics by SwissADME
Pharmacokinetic analyses for drug-like properties were performed in silico on 3, 4, and 5 using the SwissADME web tool at http://www.swissadme.ch/33.
Results
Isolation and identification of compounds in C. cuspidata extract by bioassay-guided fractionation using AChE inhibitory activity
Of the 276 herbal extracts tested, the methanol extract of C. cuspidata stems was selected based on its AChE inhibitory activity, novelty of the plant, and availability of raw material. During preparative HPLC of the extract, eluents were divided into ten fractions. AChE inhibitory analysis showed that fractions 8 (CCS-8) and 9 (CCS-9) had the lowest residual activities of 30.2 and 21.9%, respectively, at 50 µg/mL (Fig. 1).

AChE inhibitory activities of the methanol extract of C. cuspidata and the fractions obtained by the preparative HPLC. Residual activities were measured at extract and fraction concentrations of 50 µg/mL.
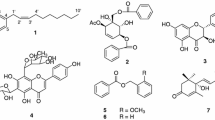
Compounds 1–5 were isolated from CCS-8 and CCS-9 by bioactivity-guided fractionation. The chemical structures of 1–5 were determined by comparing spectroscopic data, that is, 1H, 13C, 2D NMR (COSY, HMQC, and HMBC), and HRESIMS data with literature values. 1H, 13C, and MS data are provided in Supplementary Materials (Figs. S1–S10). Compounds 1–5 were identified as chestanin (1)34, 4′-O-(β-d-glucopyranosyl)-3,3′,4-tri-O-methylellagic acid (2)35, 4′-O-(α-l-rhamnopyranosyl)-3,3′,4-tri-O-methylellagic acid (3)36, 3,3′,4-tri-O-methylellagic acid (4)37, and ellagic acid (5)37. The purities of 1–5 were 95.2%, 99.1%, 99.1%, 92.0%, and 95.1% as determined by UPLC-PDA analysis (Fig. S11). A scheme for the purification process is provided in Fig. 2A, and the structures of the five isolated compounds are provided in Fig. 2B.

Scheme for the isolation of compounds 1–5 from C. cuspidata (A) and their chemical structures (B); chestanin (1), 4′-O-(β-d-glucopyranosyl)-3,3′,4-tri-O-methylellagic acid (2), 4′-O-(α-l-rhamnopyranosyl)-3,3′,4-tri-O-methylellagic acid (3), 3,3′,4-tri-O-methylellagic acid (4), and ellagic acid (5).
Chestanin (1) was previously isolated from the leaves of C. cuspidata and its structure elucidated, however, compound 5, that was ellagic acid, and compounds 2–4, which were identified as ellagic acid derivatives, were first isolated from C. cuspidata in this study. Ellagic acid, and ellagic acid derivatives may be considered secondary metabolites of the biosynthesis of gallic acid38.
Inhibitory activities of the five isolated compounds
The five compounds isolated (named 1 to 5) were assayed for inhibitory activities against five enzymes, namely, AChE, BChE, MAO-A, MAO-B, and BACE-1. 3 and 4 inhibited AChE by about 50% at 10 µM, whereas the other three compounds inhibited it by less than 30% (Table 1). 3 and 5 inhibited MAO-B by more than 50%. All five compounds weakly inhibited BChE, MAO-A, and BACE-1. Regarding IC50 values, 3 and 4 had similar IC50 values of 10.1 and 10.7 µM, respectively, for AChE (Table 1), and had moderate selectivity index (SI) values of 3.96 and 3.73, respectively, for AChE over BChE. In addition, 3 and 5 had IC50 values of 7.27 and 9.21 µM, respectively, for MAO-B. Collectively, these results suggest that 3 is a dual-acting inhibitor for AChE and MAO-B and that 4 and 5 are effective AChE and MAO-B inhibitors, respectively.
Reversibility analyses of AChE or MAO-B inhibitions by 3, 4, or 5
Reversibilities of AChE inhibitions by 3, 4, or 5 were investigated by dialysis. Inhibitions of AChE by 3, 4, and 5 were substantially recovered from 35.6% (AU) to 86.3% (AD), from 35.3% to 88.5%, and from 35.3% to 92.3%, respectively, and these values were similar to those observed for the reversible inhibitor tacrine (34.5 to 96.5%) (Fig. 3A). Inhibitions of MAO-B by 3 and 5 were markedly recovered by dialysis from 38.7 to 82.4%, from 34.5 to 82.3%, respectively, and these values were similar to those observed of the reversible inhibitor lazabemide (36.8 to 90.3%), but not to those of the irreversible inhibitor pargyline (34.3 to 35.2%) (Fig. 3B). These results indicate that 3, 4, and 5 are reversible inhibitors of AChE, and that 3 and 5 are reversible inhibitors of MAO-B.

Dialysis recoveries of AChE inhibitions by 3, 4, and 5 (A), and of MAO-B inhibitions by 3 and 5 (B). The concentrations of 3, 4, 5, and tacrine used were 25, 20, 80, and 0.60 µM, respectively, and the concentrations of 3, 5, lazabemide, and pargyline used were 14, 20, 0.28, and 0.06 µM, respectively. Tacrine was used as a reference reversible AChE inhibitor, and lazabemide and pargyline were used as reference reversible and irreversible MAO-B inhibitors, respectively. After preincubating the compounds with enzymes for 15 min, the mixtures were dialyzed for 6 h with two buffer changes. Results are the averages of duplicate or triplicate experiments.
Kinetics of AChE or MAO-B inhibitions by 3, 4, or 5
Modes of AChE inhibitions by 3, 4, or 5 were investigated using LB plots. According to these plots, AChE inhibitions by 3, 4, or 5 were competitive but partly mixed (Fig. 4A,C,E, respectively). Secondary plots of the slopes of LB plots against inhibitor concentrations showed that the Ki values of 3, 4, and 5 were 5.37 ± 0.62, 3.74 ± 0.26, and 9.43 ± 2.51 µM (Figs. 4B,D,F, respectively). These results indicate that compounds 3, 4, and 5 acted as competitive but partly mixed inhibitors of AChE.

Lineweaver–Burk (LB) plots of AChE inhibitions by 3, 4, and 5 (A, C, and E, respectively), and their respective secondary plots (B, D, and F, respectively) of the slopes of LB plots versus inhibitor concentrations. Substrate concentrations ranged from 0.05 to 1.0 mM. Experiments were carried out at three inhibitor concentrations, i.e., at ~ 0.5 × , 1.0 × , and 2.0 × IC50 values. Initial reaction rates are expressed as increases in absorbance per min.
Regarding the inhibition of MAO-B, 3 and 5 were competitive inhibitors (Fig. 5A,C), and secondary plots showed that their Ki values of 3 and 5 were 2.25 ± 0.01 and 7.51 ± 1.86 µM, respectively (Fig. 5B,D). These results suggest that 3 and 5 are competitive inhibitors of MAO-B.

LB plots of MAO-B inhibitions by 3 (A) and 5 (C), and their secondary plots (B and D, respectively) of LB slopes vs. inhibitor concentrations. Substrate concentrations ranged from 0.0375 to 0.6 mM. Experiments were carried out at three inhibitor concentrations, i.e., at ~ 0.5 × , 1.0 × , and 2.0 × IC50 values.
Cytotoxicities of 3, 4, and 5
The effects of 3, 4, and 5 on the viabilities of MDCK and HL-60 cells were investigated using the CCK-8 assay. 3 and 5 showed negligible effects on the viabilities of MDCK (normal cell line) or HL-60 (cancer cell line) cells at 50 µM (Fig. 6). However, at this concentration, 4 reduced MDCK and HL-60 viabilities to 67.9% and 84.6%, respectively. These results suggest that 3 and 5 are non-toxic to the normal and cancer cells and that 4 is slightly toxic to both cell types.

Effects of 3, 4, and 5 on the viabilities of MDCK (A) and HL-60 (B) cells. Both cell lines were treated with each compound (at 1, 3, 10, 30, or 50 µM) for 24 h. Culture supernatants were then removed and CCK-8 was added. C, control without compounds; D, control treated with 0.1% DMSO. Data are expressed as the means ± SDs of triplicate experiments.
Molecular docking simulations of 3, 4, or 5 with AChE or MAO-B
Docking simulations showed that 3, 4, and 5 were located properly at the binding sites of LND complexed with AChE and of RGZ complexed with MAO-B. AutoDock Vina predicted that the binding energies of 3, 4, and 5 to AChE were − 8.5, − 9.2, and − 8.3 kcal/mol, respectively, and those to MAO-B were − 7.3, − 4.7, − 8.9 kcal/mol, respectively (Table 2). Docking simulation results showed that 3 or 4 formed a hydrogen bond with Tyr124 of AChE at distances of 3.155 and 2.918 Å, respectively, but that 5 did not form a hydrogen bond with AChE (Fig. 7). On the other hand, 3 or 5 formed a hydrogen-bond with Cys172 of MAO-B at distances of 3.154 and 3.267 Å, respectively. 4 was not predicted to form a hydrogen bond with MAO-B (Fig. 8).

Docking simulations of 3 (A), 4 (B), and 5 (C) with AChE (6O5V). 3 and 4 formed a single hydrogen bond with Try124 of AChE at distances of 3.155 and 2.918 Å, respectively. 3, 4′-O-(α-l-rhamnopyranosyl)-3,3′,4-tri-O-methylellagicacid; 4, 3,3′,4-tri-O-methyl-ellagicacid; 5, ellagic acid. Docking simulations were performed using AutoDock Vina 1.1.2. In addition, the structures were visualized by Chimera 1.15 program.

Docking simulations of 3 (A), 4 (B), and 5 (C) with MAO-B (4A7A). 3 and 5 both formed a single hydrogen bond interaction with Cys172 of MAO-B at distances of 3.154 and 3.267 Å, respectively. Docking simulations were performed using AutoDock Vina 1.1.2. In addition, the structures were visualized by Chimera 1.15 program.
In silico pharmacokinetics of 3, 4, and 5 as determined by SwissADME
In silico pharmacokinetic studies predicted the gastrointestinal (GI) absorptions of 4 and 5 are high, but that of 3 is low (Table 3). The compounds were not found to be P-glycoprotein (P-gp) substrates and not capable of crossing the blood–brain barrier (BBB). 3 was predicted to have the highest skin permeability (-8.74 cm/s).
Discussion
In our previous studies, we isolated the MAO-A inhibitors, hispidol from Glycin max Merrill39 and decursin from Angelica gigas Nakai40, and the MAO-B inhibitors, maackiain from Sophora flavescens21 and calycosin from Maackia amurensis11. In the present study, of the 276 herbal extracts examined, the methanol extract of the stems of C. cuspidata was selected as a potential source of AChE inhibitors and five compounds were subsequently isolated using a bioassay-guided method and identified.
Of the five compounds isolated, 3 and 4 effectively inhibited AChE, and 3 and 5 effectively inhibited MAO-B. Our results suggest that 3 is a dual-acting inhibitor of AChE and MAO-B, 4 is an AChE inhibitor, and 5 is a MAO-B inhibitor. Little information is available on natural dual AChE/MAO-B inhibitors, though, recently, macelignan was identified as a dual AChE/MAO-B inhibitor with IC50 values of 4.16 and 7.42 µM, respectively22.
Ellagic acid, 5, is a natural polyphenol with anti-proliferative, anti-oxidant, anti-diabetic, anticancer, and apoptosis-inducing activities41,42. Recently, ellagic acid was reported to have neuroprotective and cognition-enhancing effects in sporadic AD based on behavioral investigations, however, its IC50 for AChE was 132.92 μM, which was 3.2 times higher than our result (IC50 = 41.7 μM)43. Furthermore, the previously reported IC50 of ellagic acid for MAO-B was 0.412 μM using rat brain mitochondrial fractions, which was much lower than our result (IC50 = 9.21 μM)44. The inhibitory activities of the ellagic acid derivatives 3 and 4 on AChE and MAO-B have not been previously described.
Natural AChE inhibitors have been classified into three groups according to their IC50 values, i.e., high potency, IC50 < 15 μM; moderate potency, 15 < IC50 < 50 μM; and low potency, 50 < IC50 < 1000 μM45. Based on this classification, 3 (IC50 = 10.1 µM) and 4 (IC50 = 10.7 µM) are highly potent AChE inhibitors. The potencies of 3 and 4 were ~ 5 times lower or ~ 2 times higher than that of galantamine [IC50 values = 2.16 µM46 or 21.1 µM (6.07 µg/ml)47], which is a natural compound and used for the treatment of AD. Polyphenols that target AChE have attracted research interest as potential therapeutics for AD48. The AChE inhibitory potencies of 3 and 4 were higher than those previously reported for other natural polyphenol, e.g., C-glucosylflavone, isovitexin-7-O-methyl ether (swertisin) (IC50 = 32.09 µg/mL, i.e., 71.9 µM) from Anthocleista vogelii49, the flavonoids tiliroside (IC50 = 23.5 µM) and quercetin (IC50 = 19.8 µM) from Agrimonia pilosa50, curcumin (IC50 = 23.5 µM) from Curcuma longa51, and epigallocatechin gallate from green tea (IC50 = 14.8 µM)52, but lower than that of a xanthonoid α-mangostin (IC50 = 2.4853 or 6.3 µM54), a hydroxycinnamoylated catechin (IC50 = 2.49 µM) from Camellia sinensis var. assamica55, and two resveratrol oligomers vitisin A (IC50 = 1.04 µM) and heyneanol A (IC50 = 1.66 µM) from Vitis amurensis56. The potencies of 3 and 4 were similar to those of two khellactone derivatives (IC50 = 9.28 and 10.0 μM) from Peucedanum japonicum Thunberg26.
3, 4, and 5 inhibited AChE in a competitive but partially mixed manner, whereas 3 and 5 inhibited MAO-B in a competitive manner. The majority of AChE inhibitors described to date are mixed or partially mixed inhibitors22,26,57, whereas the MAO-B inhibitors described are usually competitive type58,59,60.
Regarding structure–activity relationships (SARs), the 3,3′,4-tri-O-methyl group of 4 (IC50 = 10.7 μM) and the α-L-rhamnopyranosyl-3,3′,4-tri-O-methyl groups of 3 (IC50 = 10.1 μM) increased AChE inhibitory activity as compared with the 5 parent (IC50 = 41.7 μM). However, the β-D-glucopyranosyl group of 2 (IC50 = 46.4 μM), which replaced the α-L-rhamnopyranosyl group of 3 decreased AChE inhibitory activity to the level of 5. As regards MAO-B inhibitory activities, the α-L-rhamnopyranosyl of 3 (IC50 = 7.27 μM) and 5 (IC50 = 9.21 μM) increased the activity as compared with the 3,3′,4-tri-O-methyl group of 4 and the β-d-glucopyranosyl group of 2 (IC50 values > 40 μM).
Docking analysis showed that the binding energies of 3 (− 8.5 kcal/mol) and 4 (− 9.2 kcal/mol) for AChE were higher than that of 5 (− 8.3 kcal/mol), largely due to hydrogen bond formation, which was not predicted for 5, and these results concur with their IC50 values as determined by inhibition assays, (i.e., 10.1, 10.7, and 41.7 μM of 3, 4, and 5, respectively). Regarding docking to MAO-B, 3 and 5 both formed a hydrogen bond, whereas 4 did not, and the binding energies of 3 (− 7.3 kcal/mol) and 5 (− 8.9 kcal/mol) for MAO-B were higher than that of 4 (− 4.7 kcal/mol), in line with their IC50 values (i.e., 7.27, > 40, and 9.21 μM of 3, 4, and 5, respectively). We attribute these differences to a combination of hydrogen bond formation, electrostatic bonding, van der Waals forces, dissolvent effects61, and the structural flexibilities of the compounds as determined by AutoDock program62.
Our in silico pharmacokinetic studies also predicted that 3, 4 and 5 are absorbed well in the GI tract, are not substrates for P-gp, which causes drug efflux to gut lumen, and inhibit some cytochrome P450, but do not cross the BBB. In terms of cytotoxicity, 3, 4, and 5 were non-toxic or slightly toxic to the normal and cancer cells at 50 μM. Ellagic acid and its derivatives are able to react with a polycyclic aromatic hydrocarbon metabolite, the ultimate carcinogen, and prevent its covalent binding to DNA63. However, some of them exhibited DNA-damaging activity in DNA repair-deficient yeast64. Therefore, when compounds 3, 4, and 5 are used, careful doses should be needed, though the compounds were non- or slightly toxic.
In summary, we isolated the highly potent, reversible, selective AChE inhibitors 3 and 4 from Castanopsis cuspidata var. sieboldii, and show that these compounds have potential use for the treatment of neurological diseases like AD.
Conclusion
Five compounds were isolated from the methanol extract of the stems of Castanopsis cuspidata var. sieboldii by activity-guided screening for AChE inhibitors. Of these, 3 and 4 effectively and selectively inhibited AChE (IC50 = 10.1 and 10.7 µM, respectively). In addition, 3 effectively inhibited monoamine MAO-B (IC50 = 7.27 µM) followed by 5 (IC50 = 9.21 µM). Binding energies of 3 and 4 (− 8.5 and − 9.2 kcal/mol, respectively) for AChE were higher than that of 5 (− 8.3 kcal/mol) due to hydrogen bonding. In addition, compounds 3, 4, and 5 were slightly or non-toxic to MDCK cells. All five compounds weakly inhibited BChE and BACE-1. Our results show that 3 is a dual-targeting AChE and MAO-B inhibitor, 4 is an AChE inhibitor, and 5 is a MAO-B inhibitor, and suggest the potential use of these compounds for the treatment of AD.
References
Zhang, X. et al. Multi-targetable chalcone analogs to treat deadly Alzheimer’s disease: Current view and upcoming advice. Bioorg. Chem. 80, 86–93 (2018).
Sakayanathan, P. et al. In vitro and in silico analysis of novel astaxanthin-s-allyl cysteine as an inhibitor of butyrylcholinesterase and various globular forms of acetylcholinesterases. Int. J. Biol. Macromol. 140, 1147–1157 (2019).
Anand, P. & Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 36, 375–399 (2013).
Costanzo, P. et al. Design, synthesis, and evaluation of donepezil-like compounds as AChE and BACE-1 Inhibitors. ACS Med. Chem. Lett. 7, 470–475 (2016).
Schedin-Weiss, S. et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res. Ther. 9, 57 (2017).
Youdim, M. B., Gross, A. & Finberg, J. P. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol. 132, 500–506 (2001).
Knoll, J. The pharmacological basis of the beneficial effects of (-)deprenyl (selegiline) in Parkinson’s and Alzheimer’s diseases. J. Neural. Transm. Suppl. 40, 69–91 (1993).
Tandarić, T., Prah, A., Stare, J., Mavri, J. & Vianello, R. Hydride abstraction as the rate-limiting step of the irreversible inhibition of monoamine oxidase B by rasagiline and selegiline: A computational empirical valence bond study. Int. J. Mol. Sci. 21, 6151 (2020).
Ibrahim, M. M. & Gabr, M. T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 14, 437–440 (2019).
Baek, S. C. et al. Rhamnocitrin isolated from Prunus padus var. seoulensis: A potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem. 83, 317–325 (2019).
Oh, J. M. et al. Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int. J. Biol. Macromol. 151, 441–448 (2020).
Wakamatsu, H. et al. Reductive metabolism of ellagitannins in the young leaves of Castanopsis sieboldii. Molecules 20, 4279 (2019).
Nonaka, G., Ageta, M. & Nishioka, I. Tannins and related compounds XXV. A new class of gallotannins possessing a (-)-shikimic acid core from Castanopsis cuspidata var. sieboldii NAKAI (1). Chem. Pharm. Bull. 33, 96–101 (1985).
Ageta, M., Nonaka, G. & Nishioka, I. Tannins and related compounds. LXVII: Isolation and characterization of castanopsinins A–H, novel ellagitannins containing a triterpenoid glycoside core, from Castanopsis cuspidata var. sieboldii NAKAI (3). Chem. Pharm. Bull. 36, 1646–1663 (1988).
Ageta, M., Ishimaru, K., Nonaka, G. I. & Nishioka, I. Tannins and related compounds. LXIV: Six new phenol glucoside gallates from Castanopsis cuspidata var. sieboldii NAKAI (2). Chem. Pharm. Bull. 36, 870–876 (1987).
Kim, H. W. et al. Glucose uptake-stimulating galloyl ester triterpenoids from Castanopsis sieboldii. J. Nat. Prod. 83, 3093–3101 (2020).
Kim, N., Choi, M. H. & Shin, H. J. Antioxidant activity of Castanopsis cuspidate var. sieboldii extracts. Korean Soc. Biotechnol. Bioeng. 10, 308 (2020).
Kim, J. Y. et al. Comparison of antioxidant and anti-inflammatory activity on chestnut, chestnut shell and leaves of Castanea crenata extracts. Korean Soc. Med. Crop Sci. 22, 8–16 (2014).
Moon, S. H., Song, C. K., Kim, T. K., Oh, D. E. & Kim, H. C. Antifungal activity on the water extracts of five fagaceae plants. Korean J. Org. Agric. 25, 295–310 (2017).
Ellman, G. L., Courtney, K. D., Andres, V. Jr. & Feather-Stone, R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 (1961).
Lee, H. W. et al. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 26, 4714–4719 (2016).
Lee, J. P. et al. Potent inhibition of acetylcholinesterase by sargachromanol I from Sargassum siliquastrum and by selected natural compounds. Bioorg. Chem. 89, 103043 (2019).
Baek, S. C., Choi, B., Nam, S. J. & Kim, H. Inhibition of monoamine oxidase A and B by demethoxycurcumin and bisdemethoxycurcumin. J. Appl. Biol. Chem. 61, 187–190 (2018).
Baek, S. C. et al. Selective inhibition of monoamine oxidase A by chelerythrine, an isoquinoline alkaloid. Bioorg. Med. Chem. Lett. 28, 2403–2407 (2018).
Ali, S. et al. Fluoro-benzimidazole derivatives to cure Alzheimer’s disease: In-silico studies, synthesis, structure-activity relationship and in vivo evaluation for β secretase enzyme inhibiton. Bioorg. Chem. 88, 102936 (2019).
Heo, J. H. et al. Acetylcholinesterase and butyrylcholinesterase inhibitory activities of khellactone coumarin derivatives isolated from Peucedanum japonicum Thurnberg. Sci. Rep. 10, 21695 (2020).
Jeong, G. S. et al. Inhibition of butyrylcholinesterase and human monoamine oxidase-B by the coumarin glycyrol and liquiritigenin isolated from glycyrrhiza uralensis. Molecules 5, 3896 (2020).
Jeong, G. S. et al. Selective inhibition of human monoamine oxidase B by 5-hydroxy-2-methyl-chroman-4-one isolated from an endogenous lichen fungus Daldinia fissa. J. Fungi 7, 84 (2021).
Gan, Z. S. et al. Iron reduces M1 macrophage polarization in RAW264.7 macrophages associated with inhibition of STAT1. Mediat. Inflamm. 2017, 8570818 (2017).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Mills, J. E. & Dean, P. M. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol. Des. 10, 607–622 (1996).
Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Daina, A., Michielin, V. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Ageta, M., Ishimaru, K., Nonaka, G. I. & Nishioka, I. Tannins and related compounds. LXIV. Six phenol glucoside gallates from Castanopsis cuspidata var sieboldii Nakai. Chem. Pharm. Bull. 36, 870–876 (1988).
Khac, D. D., Tran-Van, S., Campos, A. M., Lallemand, J. Y. & Fetizon, M. Ellagic compounds from Diplopanax stachyanthus. Phytochemistry 29, 251–256 (1990).
Le, H. T. et al. Constituents from the stem barks of Canarium bengalense with cytoprotective activity against hydrogen peroxide-induced hepatotoxicity. Arch. Pharm. Res. 35, 87–92 (2012).
Bai, N. et al. Active compounds from Lagerstroemia speciosa, insulin-like glucose uptake-Stimulatory/Inhibitory and adipocyte differentiation-inhibitory activities in 3T3-L1 cells. J. Agric. Food. Chem. 56, 11668–11674 (2008).
Evtyugin, D. D., Magina, S. & Evtuguin, D. V. Recent advances in the production and applications of ellagic acid and its derivatives. A review. Molecules 25, 2745 (2020).
Baek, S. C. et al. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett. 28, 584–588 (2018).
Lee, H. W. et al. Potent inhibition of monoamine oxidase A by decursin from Angelica gigas Nakai and by wogonin from Scutellaria baicalensis Georgi. Int. J. Biol. Macromol. 98, 598–605 (2017).
Derosa, G., Maffioli, P. & Sahebkar, A. Ellagic acid and its role in chronic diseases. Adv. Exp. Med. Biol. 928, 473–479 (2016).
Shakeri, A., Zirak, M. R. & Sahebkar, A. Ellagic acid: A logical lead for drug development?. Curr. Pharm. Des. 24, 106–122 (2018).
Jha, A. B., Panchal, S. S. & Shah, A. Ellagic acid: Insights into its neuroprotective and cognitive enhancement effects in sporadic Alzheimer’s disease. Pharmacol. Biochem. Behav. 175, 33–46 (2018).
Khatri, D. K. & Juvekar, A. R. Kinetics of inhibition of monoamine oxidase using curcumin and ellagic acid. Pharmacogn. Mag. 2, S116–S120 (2016).
Dos Santos, T. C., Gomes, T. M., Pinto, B. A. S., Camara, A. L. & De Andrade, P. A. M. Naturally occurring acetylcholinesterase inhibitors and their potential use for Alzheimer’s disease therapy. Front. Pharmacol. 9, 1192 (2018).
Shaikh, S. et al. Design, synthesis and evaluation of dihydropyranoindole derivatives as potential cholinesterase inhibitors against Alzheimer’s disease. Bioorg. Chem. 110, 104770 (2021).
Alqahtani, Y. S. Bioactive stigmastadienone from Isodon rugosus as potential anticholinesterase, α-glucosidase and COX/LOX inhibitor: In-vitro and molecular docking studies. Steroids 172, 108857 (2021).
Jabir, N. R., Khan, F. R. & Tabrez, S. Cholinesterase targeting by polyphenols: A therapeutic approach for the treatment of Alzheimer’s disease. CNS Neurosci. Ther. 9, 753–762 (2018).
Ajayi, O. S., Aderogba, M. A., Obuotor, E. M. & Majinda, R. R. T. Acetylcholinesterase inhibitor from Anthocleista vogelii leaf extracts. J. Ethnopharmacol. 231, 503–506 (2019).
Jung, M. & Park, M. Acetylcholinesterase inhibition by flavonoids from Agrimonia pilosa. Molecules 12, 2130–2139 (2007).
Kalaycıoğlu, Z., Gazioğlu, I. & Erim, F. B. Comparison of antioxidant, anticholinesterase, and antidiabetic activities of three curcuminoids isolated from Curcuma longa L. Nat. Prod. Res. 31, 2914–2917 (2017).
Okello, E. J. & Mather, J. Comparative kinetics of acetyl- and butyryl-cholinesterase inhibition by green tea catechins| relevance to the symptomatic treatment of Alzheimer’s disease. Nutrients 12, 1090 (2020).
Khaw, K. Y., Kumar, P., Yusof, S. R., Ramanathan, S. & Murugaiyah, V. Probing simple structural modification of alpha-mangostin on its cholinesterase inhibition and cytotoxicity. Arch. Pharm. 353, e2000156 (2020).
Ryu, H. W. et al. Rapid identification of cholinesterase inhibitors from the seedcases of mangosteen using an enzyme affinity assay. J. Agric. Food Chem. 62, 1338–1343 (2014).
Wang, W. et al. Novel acetylcholinesterase inhibitors from Zijuan tea and biosynthetic pathway of caffeoylated catechin in tea plant. Food Chem. 237, 1172–1178 (2017).
Jang, M. H., Piao, X. L., Kim, J. M., Kwon, S. W. & Park, J. H. Inhibition of cholinesterase and amyloid-beta aggregation by resveratrol oligomers from Vitis amurensis. Phytother. Res. 4, 544–549 (2008).
Kamila, C., Malgorzata, G. & Pawel, K. Discovery of new cyclopentaquinoline analogues as multifunctional agents for the treatment of Alzheimer’s disease. Int. J. Mol. Sci. 20, 498 (2019).
Najla, O. Z., Suresh, K. E. & Karam, F. A. S. The benzopyrone biochanin-A as a reversible, competitive, and selective monoamine oxidase B inhibitor. BMC Complement Altern. Med. 17, 34 (2017).
Sasidharan, R., Manju, S. L., Uçar, G., Baysal, I. & Mathew, B. Identification of indole-based chalcones: Discovery of a potent, selective, and reversible class of MAO-B inhibitors. Arch. Pharm. 349, 627–637 (2016).
Chaurasiya, N. D., Ibrahim, M. A., Muhammad, I., Walker, L. A. & Tekwani, B. L Monoamine oxidase inhibitory constituents of propolis: Kinetics and mechanism of inhibition of recombinant human MAO-A and MAO-B. Molecules 19, 18936–18952 (2014).
Oh, J. M. et al. Potent and selective inhibition of human monoamine oxidase-B by 4-dimethylaminochalcone and selected chalcone derivatives. Int. J. Biol. Macromol. 137, 426–432 (2019).
Forli, S. et al. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11, 905–919 (2016).
Huetz, P., Mavaddat, N. & Mavri, J. Reaction between ellagic acid and an ultimate carcinogen. J. Chem. Inf. Model. 45, 1564–1570 (2005).
Deng, J. Z., Marshall, R., Jones, S. H., Johnson, R. K. & Hecht, S. M. DNA-damaging agents from Crypteronia paniculata. J. Nat. Prod. 65, 1930–1932 (2002).
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (NRF-2019R1A2C1088967), by the KRIBB Research Initiative Program funded by the Ministry of Science and ICT (MSIT) of the Republic of Korea, and by the Korea Institute of Toxicology, Republic of Korea (1711121217).
Author information
Authors and Affiliations
Contributions
J.M.O. and J.E.P. carried out biological experiments and wrote primary manuscript, H.-J.J. wrote the primary manuscript, S.S., D.Y.K., and J.H.K. isolated the compounds and determined their structures, J.-I.N. and S.-T.Y. perforemed cytotoxicity test, M.-G.K. and D.P. analysed docking simulation and wrote the primary manuscript, and H.K. supervised the study and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, J.M., Jang, HJ., Kang, MG. et al. Acetylcholinesterase and monoamine oxidase-B inhibitory activities by ellagic acid derivatives isolated from Castanopsis cuspidata var. sieboldii. Sci Rep 11, 13953 (2021). https://doi.org/10.1038/s41598-021-93458-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93458-4
- Springer Nature Limited
This article is cited by
-
Fortification in Jaggery for Anti-Oxidant and Neuroprotective Benefits: Standardization, Sensory Preference, and Chemometric Analysis
Sugar Tech (2024)
-
Chemo- and bio-informatics insight into anti-cholinesterase potentials of berries and leaves of Myrtus communis L., Myrtaceae: an in vitro/in silico study
BMC Complementary Medicine and Therapies (2023)