
Abstract
Melanoma is one of the most aggressive and lethal skin cancers. Lucidadiol is a triterpenoid isolated from Ganoderma lucidum and is known to have various biological functions, including antibacterial effects. However, the anti-cancer effects and mechanism of action of lucidadiol in malignant melanoma are unknown. In this study, lucidadiol significantly reduced B16 melanoma cell viability in a dose- and time-dependent manner. In addition, lucidadiol induced apoptosis and suppressed cell mobility in B16 melanoma cells. Moreover, our findings revealed that lucidadiol remarkably downregulated phospho-Akt/ERK/JNK, but not p38. Taken together, our results suggest that lucidadiol could exerts its anti-cancer effects by inducing apoptosis via modulation of the Akt/MAPK pathway. Therefore, lucidadiol may be a potential cancer therapeutic agent for malignant melanoma.
Similar content being viewed by others

Introduction
Melanoma is one of the most lethal types of skin cancer, developing from melanocytes located in the basal layer of epithelial surfaces [1]. Although it accounts for only 4% of all skin cancers, melanoma-associated mortality is estimated to be approximately 80% [2]. The main risk factors for developing melanoma are the number of melanocytic nevi, genetic susceptibility, mutagenesis, and exposure to ultraviolet radiation, which induces genotoxic effects [3]. Current therapies include surgery, radiotherapy, and chemotherapy; however, these are only effective for early-stage melanoma, which is less invasive [4]. Notably, melanoma has a poor prognosis at later stages of tumor progression, often progressing to an unresectable stage or advanced metastatic disease. Moreover, the current therapeutic agents used to treat melanoma have several side effects, often inducing resistance to conventional chemotherapy and radiation [5]. Thus, there is an urgent need to develop an early diagnosis system and improve therapeutic agents for melanoma.
Natural products are a critical source for the development of novel anti-cancer drugs, as they are considered to be less toxic and have fewer side effects than synthetic drugs [6]. As such, interest in chemotherapy drugs originating from natural sources has been progressively increasing for various cancer types, including melanoma [7,8,9].
Lucidadiol (Fig. 1A) is a lanostane-type triterpenoid isolated from Garnoderma lucidum, commonly known as Lingzhi. This species has been previously used as a medicinal mushroom to treat various diseases, as well as for its life-prolonging effects [10]. Its anti-cancer properties are mainly attributed to triterpenoids, which are one of the main constituents [11]. Until now, lucidadiol has been reported to have antiviral activity against influenza virus A and herpes simplex virus (HSV) 1 [12]. However, the anticancer mechanism of lucidadiol in melanoma cells has yet to be investigated.

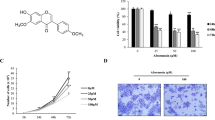
The inhibitory effects of lucidadiol on the viability of B16 melanoma cells. A The structure of lucidadiol. B, C Cells were treated with lucidadiol at various concentrations for 24 h or treated with 15 µM for 0, 12, 24, and 48 h. Cell viability was measured using the MTT assay. D, E Cell viability was determined via trypan blue and crystal violet staining, after treatment with/without lucidadiol for 24 and 48 h. scale bar = 100 µm. F The colonies were stained with crystal violet and analyzed using a microplate reader. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group
Recent studies have found that melanoma has the highest mutation frequency among cancers analyzed to determine genetic factors of malignancies [13]. In particular, the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways are among the most important signaling pathways which might play a role in the pathogenesis of melanoma [14,15,16]. The PI3K/Akt pathway is one of the most critical signaling networks in cancer development and is frequently activated in melanomas, with an N-RAS mutation being exhibited by 20% of cases [17]. In addition, the MAPK signaling pathways include ERK, c-Jun NH2-terminal kinase (JNK), and p38 MAPK [18]. MAPK pathways modulate various cellular functions such as proliferation, survival, and invasion in melanoma [19,20,21,22]. Notably, ERK was shown to be hyperactivated in approximately 90% of melanoma cases, which is induced by several factors, such as growth factors and genetic alteration of upstream factors, including Ras and Raf [23]. Accordingly, dysregulation of these signaling pathways is commonly identified in almost all cancers, including melanoma, due to amplification and the cascade of genetic mutations, which lead to enhanced growth, survival, and metabolism of cancer cells [24, 25]. Thus, the Akt/MAPK signaling pathway may be an important therapeutic target in malignant melanoma.
In this study, we explored the anti-cancer effects of lucidadiol in melanoma by investigating the molecular mechanisms underlying its anti-cancer effects. We demonstrated that lucidadiol significantly decreased cell viability by inducing apoptosis and suppressed cancer cell colony formation and migration by inducing apoptosis. Furthermore, we found that the lucidadiol-mediated cancer suppressive effects in melanoma cells could be controlled by the Akt/MAPK signaling pathway. Thus, the results of this study indicate that lucidadiol could be a potential novel and universal drug for melanoma treatment.
Materials and methods
Chemicals and reagent
Lucidadiol was purchased from ChemFaces, dissolved in dimethyl sulfoxide (DMSO), and diluted in serum-free medium (SFM) to achieve the required concentration at the time of use. Dulbecco's Modified Eagle’s medium (DMEM, high glucose) was purchased from Hyclone and fetal bovine serum (FBS) was purchased from Corning. Penicillin- streptomycin-glutamine was purchased from Gibco. MTT [3-(4,5-dimethylthiazol-2-yl) − 2,5-diphenyltetrazolium bromide] and DMSO were purchased from Sigma-Aldrich Co. Primary antibodies was purchased from Cell Signaling Technology (Danvers, MA, USA), Abcam (Cambrige, MA, USA) and Invitrogen (Middlesex Country, MA, USA). Goat anti-rabbit IgG (5220-0036) and goat anti-mouse IgG (5220-0341) antibodies were purchased from Seracare.
Cell culture
Mouse melanoma B16 cells were cultured in DMEM high glucose medium supplemented with 10% FBS and 1% streptomycin/penicillin/glucose at 5% CO2 at 37 °C. The medium was renewed every 3 days.
Cell viability and proliferation assay
B16 melanoma cells were cultured in 96-well plates at 5 × 104 cells/mL for 24 h. Cells were treated with various concentrations of lucidadiol for 12, 24, and 48 h. Afterwards, 40 µL of MTT solution (1 mg/mL) was added to the cells and incubated at 37 °C for 2 h. Then, the MTT solution was removed and 100 µL of DMSO was added to dissolve the formazan crystals. Absorbance was measured using a microplate reader at 570 nm.
Trypan blue and crystal violet staining were performed to assess cell viability, according to a published method with modifications [26]. For Trypan blue staining, B16 melanoma cells were cultured in 6-well plates with/without lucidadiol for 24 and 48 h. The cells were trypsinized and counted using a hemocytometer after staining with trypan blue. For crystal violet staining, B16 melanoma cells were cultured in 24-well plates as described above. The cells were fixed with 4% paraformaldehyde for 10 min and stained with 0.05% crystal violet in 20% methanol for 5 min. The cells were then washed with distilled water and dried for 1 d at room temperature (RT). The cell images were observed under a microscope at × 200 magnification.
Colony-forming assay
B16 melanoma cells were cultured in 6-well plates with/without lucidadiol for 48 h, after which the medium was removed and growth medium was added. The cells were further cultured to form a colony for 9 days with the growth medium being renewed every 3 days. After 9 days, the colonies were fixed with 4% paraformaldehyde and stained with 0.05% crystal violet, as described above. The colonies were dissolved in 10% acetic acid for 20 min. The absorbance was measured using a microplate reader at 570 nm.
Cell cycle analysis
As mentioned above, the cells were cultured in 6-well plates at 1 × 105 cells/mL with/without lucidadiol. Then, the cell pellets were collected after trypsinization and fixed with cold 70% ethanol at − 20 °C overnight. RNase (100 µg/mL) was added to the cell pellet in 0.1% Triton X-100 for 2 h at 37 °C after washing with cold phosphate-buffered saline (PBS) twice and then the PI solution (20 µg/mL) was added for 5 min at RT. The DNA content was measured via flow cytometry (BD FACSVerse™, BD Bioscience) and was analyzed with Flowjo software.
Apoptosis detection
As mentioned above, the cells were cultured in 6-well plates at 1 × 105 cells/mL with/without lucidadiol. The cell pellets were collected after trypsinization and washed twice with cold PBS. The cell pellets were then incubated with a binding buffer and stained with Annexin V-FITC/PI for 15 min. The percentage of apoptotic cells was measured by flow cytometry (BD FACSVerse™, BD Bioscience) and was analyzed with Flowjo software.
Wound healing assay
B16 melanoma cells were seeded in 60 mm dish at 5 × 106 cells/dish and cultured until confluence. After 24 h, the cell monolayer was scratched with a sterile 1 mL pipette tip, followed by changing the medium to one with/without lucidadiol. Cell migration was evaluated by observing them at intervals of 0, 12, and 24 h under a microscope. Images were recorded and the area of the wound gap was measured using the ImageJ software.
Western blot analysis
For protein analysis, total protein was extracted by lysing cells in lysis buffer [10 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA pH 8, 1% Triton X-100, 1% sodium deoxycholate, 30 mM NaF, 1.5 mM NaVO4, 1 mM PMSF, and 1 mg/mL each of aprotinin, leupeptin, and pepstatin A] with sonication for 10 s. The cell lysates were then centrifuged at 13,000 rpm for 10 min at 4 °C and quantified using the Bradford assay. Then, the proteins (25 µg) were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 5% skimmed milk in Tris-buffered saline supplemented with 0.1% Tween-20 (TBS-T) at RT for 30 min, the membrane was incubated with appropriate primary antibodies overnight at RT with gentle shaking and then washed three times with TBS-T for 10 min. Subsequently, the membrane was incubated with secondary horseradish peroxidase (HRP)-conjugated anti-rabbit/mouse IgG antibodies for 1 h at RT. β-Actin was used as the loading control. The membranes were then incubated with an ECL reagent and bands were visualized using a Chemidoc and analyzed via ImageJ. The primary antibodies used in this study were as follows: anti-β-actin (#8457), anti-p53 (#MA5-12453), anti-Bax (#2772), anti-PARP (#9532), anti-p-ERK1/2 (#4370), anti-pJNK (#9255), anti-p38 (#9216), anti-pAKT (#4060), anti-caspase3 (#9662), and anti-MMP-9 (#Ab38898).
Statistical analysis
The results are presented as mean ± standard deviation (SD). Differences between groups were analyzed using Dunnett’s test. Statistical significance was set at p < 0.05.
Result
Lucidadiol inhibits cell viability and colony-formation in B16 melanoma cells
To evaluate the effects of lucidadiol on the viability of B16 melanoma cells, the MTT assay was performed at various lucidadiol concentrations (0–50 µM) and incubation times (0, 12, 24, and 48 h). The results showed that lucidadiol decreased the viability of B16 melanoma cells in a concentration-dependent manner (IC50 values of 48.42 µM at 24 h) (Fig. 1B) and time-dependent manner (with 15 and 25 µM) (Fig. 1C). Similarly, we tested the cell survival rate after lucidadiol treatment using trypan blue and crystal violet staining. The number of crystal violet-stained cells and trypan blue-unstained cells was similar to the number of viable cells (MTT assay) (Fig. 1D and E). Moreover, we assessed the anti-proliferative capacity of lucidadiol using a colony formation assay. Our findings indicate that lucidadiol reduces the number of B16 melanoma cell colonies (Fig. 1F). Thus, these data indicate that lucidadiol treatment may decrease the viability and growth of melanoma cells.
Lucidadiol treatment induces the accumulation of Sub-G1 phase in B16 melanoma cells
Previous studies have suggested that the inhibition of cancer cell viability is often mediated by regulating the cell cycle phase distribution [27,28,29]. Thus, to investigate the possible mechanism underlying the lucidadiol-induced decrease in cancer cell survival, the cell cycle distribution after lucidadiol treatment was measured via flow cytometry with propidium iodide (PI) staining. The cells were treated with 0, 15, and 25 µM lucidadiol for 24 and 48 h. Our results showed that lucidadiol-treated cells accumulated in the Sub-G1 phase (apoptotic cells) at 48 h post-treatment when compared to the control group (Fig. 2A). The percentage of cells in the Sub G1 phase, which indicates apoptosis, was found to be significantly increased, from 1.16% in control cells to 3.26% and 3.43% in cells treated with 15 and 25 µM lucidadiol, respectively (Fig. 2B). These results suggest that lucidadiol suppressed cell viability by inducing the accumulation of Sub-G1 phase, leading to an increased number of apoptotic melanoma cells.

Lucidadiol treatment induced the accumulation of Sub-G1 phase in B16 melanoma cells. A The cell cycle distribution of B16 melanoma cells after treatment with lucidadiol for 24 and 48 h was measured via flow cytometry. B Statistical analysis of each cell cycle population expressed via graphs. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group
Lucidadiol induces apoptosis in B16 melanoma cells
Despite different patterns of interaction with intracellular targets, induction of apoptosis is one of the characteristic mechanisms of action of most cytotoxic anticancer agents that ultimately induce cancer cell death [30]. Therefore, we used Annexin V/PI double staining to measure the type of lucidadiol-induced cell death. Annexin V, an indicator of apoptotic cells, can bind with phosphatidylserine, which is exposed from the inner leaflet to the outer leaflet of the plasma membrane during apoptosis [31]. The results showed that the population of Annexin V/PI positive apoptotic cells increased in lucidadiol-treated cells in a time-dependent manner (Fig. 3A). The apoptotic cell ratio in B16 melanoma cells treated with lucidadiol was 7.46% (0 µM), 18.18% (15 µM), and 18.9% (25 µM) at 24 h post-treatment, and 6.36% (0 µM), 24.5% (15 µM), and 25.23% (25 µM) at 48 h post-treatment (Fig. 3B). To elucidate the potential molecular mechanism underlying lucidadiol-induced apoptosis of B16 melanoma cells, the expression level of apoptosis-related proteins was analyzed by western blot. In no treatment condition as control, apoptotic-related proteins (Bax, p53, PARP, and procaspase-3) indicated slightly apoptotic status due to serum starvation for 48 h (Fig. 4). Bax (pro-apoptotic) expression levels were shown to be increased slightly in lucidadiol (15 µM)-treated cells, when compared to control cells (Fig. 4A). Furthermore, lucidadiol promoted the cleavage of procaspase‑3 and PARP, as well as the expression of p53 (tumor suppressor protein) (Fig. 4B). These results suggest that lucidadiol decreases cell viability by inducing apoptosis in B16 melanoma cells.

Apoptosis of B16 melanoma cells induced by lucidadiol. A The distribution of apoptotic B16 melanoma cells was measured via flow cytometry at 24 and 48 h post-lucidadiol treatment. B Statistical analysis of apoptotic cell populations expressed via a graph. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group

Expression levels of apoptosis-associated proteins in lucidadiol-treated B16 melanoma cells. A Representative blots of Bax expression levels in B16 melanoma cells treated with lucidadiol (0 and 15 µM) for 0 and 48 h. The graph represents the Bax expression level with/without lucidadiol treatment at 48 h. B Representative blots of p53, PARP, and caspase-3 expression levels in B16 melanoma cells treated with lucidadiol (0 and 15 µM) for 0 and 48 h. The graph represents p53, PARP, and caspase-3 expression levels in cells with/without lucidadiol treatment at 48 h. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group
Lucidadiol blocks the migration of B16 melanoma cells
Cell migration is an important phenomenon that is one of the key determinants of malignant cancers, including melanoma, breast, and lung cancer [32]. Thus, we investigated the effect of lucidadiol on B16 melanoma cell mobility, which is a hallmark of tumor progression and metastasis, by using a concentration lower than the cytotoxic IC50 (48.42 µM for 24 h). The results showed that the migration of cells treated with lucidadiol (15 and 25 µM) was decreased to 46.3% and 59.7%, respectively, when compared to control cells (Fig. 5A). Matrix metalloproteinases (MMPs) are proteins that modulate cancer cell adhesion, migration, and invasion [33]. MMP-9 is known to contribute to cancer cell invasion and metastasis by degrading the extracellular matrix [34]. Notably, we found that MMP-9 expression levels were significantly decreased in B16 melanoma cells treated with lucidadiol (15 and 25 µM) via western blot analysis (Fig. 5B). Therefore, these results suggest that lucidadiol regulates cell migration in B16 melanoma cells.

Lucidadiol inhibits the migration of B16 melanoma cells. A The effects of lucidadiol on B16 melanoma cell migration were quantified via a wound-healing assay for 12 and 24 h and statistical analysis of cell migration ability for each group is presented in the graph. B Representative blots of MMP-9 protein levels after treatment with lucidadiol (0 and 15 µM) for 0 and 48 h. The graph presents MMP-9 protein levels in B16 melanoma cells with/without lucidadiol treatment 48 h. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group
Lucidadiol modulates Akt/MAPK pathway in B16 melanoma cells
Finally, we attempted to uncover the molecular pathways underlying the anti-cancer effects of lucidadiol. Considering that the Akt/MAPK signaling pathway plays a critical role in melanoma progression by affecting cell proliferation, migration, and apoptosis [35], the phosphorylation level of proteins associated with the Akt/MAPK signaling pathway in lucidadiol-treated B16 melanoma cells was assessed by western blotting. The results indicated that Akt, ERK, and JNK phosphorylation levels were markedly decreased following lucidadiol treatment (Fig. 6A) but p38 phosphorylation levels were significantly increased (Fig. 6B). These results suggest that lucidadiol could induces apoptosis and suppression of migration in B16 melanoma cells by regulating the Akt/MAPK signaling pathways.

Lucidadiol modulates the Akt/MAPK signaling pathway in B16 melanoma cells. A Representative blot of p-Akt and p-MAPK in B16 melanoma cells treated with lucidadiol at 0 and 15 µM for 0 and 48 h. B Statistical analyses of phosphorylated Akt, ERK, JNK, and p38 expression levels with/without lucidadiol treatment are represented in the graphs. The data are presented as the mean ± SD. *p < 0.05 when compared with the control group
Discussion
Lucidadiol was isolated from Ganoderma lucidum, which has been used in oriental medicine for approximately 2000 years [12, 36]. Lucidadiol was shown to have diverse biological functions, including antibacterial effects [12, 37]. However, its anti-cancer activity against melanoma and its mechanism of action has yet to be uncovered. In this study, we demonstrated for the first time that lucidadiol exhibits anti-cancer activity against B16 melanoma cells by modulating the Akt/MAPK pathway.
To investigate the molecular mechanism underlying the lucidadiol-induced decrease in melanoma cell viability, we first examined whether lucidadiol affected cell cycle progression. We discovered that lucidadiol treatment significantly induced an increased population in the Sub-G1 phase (only at 48 h). The populations of B16 melanoma cells in each cell cycle phase, including G1, S, and G2/M, were only shown slightly different after lucidadiol treatment, but the difference was not significant. The accumulation of cells in the Sub-G1 phase is considered to be a biomarker of DNA damage and is also associated with apoptosis [38]. Therefore, these results suggest that the lucidadiol-induced decrease in cell viability is related to apoptosis.
Apoptosis, a form of physiological cell death, occurs as a homeostatic and defense mechanism during developmental and immune reactions or when cells are damaged by various stimuli, such as disease [39]. The characteristics of apoptotic cells include chromatin condensation, cell shrinkage, membrane lipid rearrangement, caspase activation, DNA fragmentation, and cell fragmentation [40]. p53 is a tumor suppressor protein and may be a potential molecular target for regulating cell growth, by promoting apoptosis and DNA repair [41]. Activation of p53 is sufficient to trigger apoptosis by inducing the pro-apoptotic Bax protein family and inhibiting the anti-apoptotic Bcl-2 protein family, leading to the activation of caspase-3 (effector caspase) [42, 43]. Consequently, cleaved PARP levels are increased by cleaved caspase-3 (activated form), which plays a role in apoptotic processes, including DNA repair and cell cycle regulation [44]. As shown in Fig. 3, lucidadiol induces a potential increase in the apoptotic cell population through an apoptosis-related molecular mechanism. Overall, our findings indicate that the lucidadiol-induced decrease in melanoma cell viability is mediated by apoptosis and cell cycle arrest.
Given that distant metastasis is a leading cause of death in melanoma patients, it is important to elucidate the mechanisms underlying cancer cell metastasis [45]. Cancer metastasis is a complex process involving multiple steps such as migration, invasion, and adhesion [46]. MMPs are involved in cancer cell invasion and metastasis by cleaving extracellular matrix proteins [34]. In particular, MMP-9 has been found to be an indicator of melanoma invasiveness [47]. Our findings showed that lucidadiol treatment reduced the migration of melanoma cells by suppressing MMP9 expression levels. This suggests that lucidadiol may also have anti-metastasis effects in melanoma.
The Akt and ERK pathways have been shown to play crucial roles in the tumorigenesis of many cancers, including melanoma, by promoting the translation of target genes associated with cell proliferation, migration, and invasion [48]. Therefore, several studies have emphasized that inhibition of the PI3K/Akt and ERK signaling pathways may be a promising strategy for melanoma treatment [49,50,51]. In this study, lucidadiol treatment decreased the phosphorylation of Akt and ERK1/2 in B16 melanoma cells. As a result, we speculated that the alteration of biological activity induced by lucidadiol treatment in B16 melanoma could be, in part, attributed to its ability to inhibit the Akt/ERK pathway. In addition, the JNK signaling pathways, which are a subfamily of MAPK, have been shown to have paradoxical roles in carcinogenesis, with both tumor growth and tumor suppressor properties [52]. Alexaki et al. reported that JNK supports the viability of melanoma cells by modulating cell cycle arrest and apoptosis [53], while Qin et al. reported that a decrease in JNK activation is involved in suppressing melanoma cell growth [54]. Accordingly, our results indicate that lucidadiol induces apoptosis and cell cycle arrest through the inhibition of JNK activation. Another subfamily, p38 MAPK, is also activated by diverse pro-inflammatory and stressful stimuli. Although the role of p38 MAPK in tumors is complicated, many studies have suggested that p38 MAPK functions as a tumor suppressor associated with apoptosis in some cell systems [21, 55]. In addition, She et al. reported that p38 MAPK activates p53 and p53-induced apoptosis [56]. Moreover, activation of the ERK and p38 pathways is inversely regulated and high p38 activity levels inhibit ERK activity and prevent tumorigenesis [18]. Overall, our findings suggest that lucidadiol induces apoptosis of melanoma cells via cell cycle arrest and blocking of migration by suppressing Akt, ERK, and JNK activation while activating p38 MAPK.
To the best of our knowledge, this is the first demonstration of the anti-cancer mechanism of lucidadiol in melanoma cells. Taken together, our findings not only reveal the important role of the Akt/MAPK pathway in lucidadiol-induced bioactivity in melanoma cells but also suggest that lucidadiol may be a promising novel therapeutic agent for melanoma treatment.
Availability of data and materials
The datasets that support the finding of this study are available from the corresponding author on reasonable request.
References
Cichorek M, Wachulska M, Stasiewicz A, Tyminska A (2013) Skin melanocytes: biology and development. Postepy Dermatol Alergol 30:30–41
Pal HC, Hunt KM, Diamond A, Elmets CA, Afaq F (2016) Phytochemicals for the management of melanoma. Mini Rev Med Chem 16:953–979
Rastrelli M, Tropea S, Rossi CR, Alaibac M (2014) Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 28:1005–1011
Erdei E, Torres SM (2010) A new understanding in the epidemiology of melanoma. Expert Rev Anticancer Ther 10:1811–1823
Lee B, Mukhi N, Liu D (2012) Current management and novel agents for malignant melanoma. J Hematol Oncol 5:3
Pan MH, Ho CT (2008) Chemopreventive effects of natural dietary compounds on cancer development. Chem Soc Rev 37:2558–2574
Danciu C, Soica C, Antal D, Alexa E, Pavel IZ, Ghiulai R, Ardelean F, Babuta RM, Popescu A, Dehelean CA (2018) Natural compounds in the chemoprevention of malignant melanoma. Anticancer Agents Med Chem 18:631–644
Rejhova A, Opattova A, Cumova A, Sliva D, Vodicka P (2018) Natural compounds and combination therapy in colorectal cancer treatment. Eur J Med Chem 144:582–594
Muller AG, Sarker SD, Saleem IY, Hutcheon GA (2019) Delivery of natural phenolic compounds for the potential treatment of lung cancer. Daru 27:433–449
Sohretoglu D, Huang S (2018) Ganoderma lucidum polysaccharides as an anti-cancer agent. Anticancer Agents Med Chem 18:667–674
Wu GS, Guo JJ, Bao JL, Li XW, Chen XP, Lu JJ, Wang YT (2013) Anti-cancer properties of triterpenoids isolated from Ganoderma lucidum—a review. Expert Opin Investig Drugs 22:981–992
Lindequist U, Julich WD, Witt S (2015) Ganoderma pfeifferi—a European relative of Ganoderma lucidum. Phytochemistry 114:102–108
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421
Daphu I, Horn S, Stieber D, Varughese JK, Spriet E, Dale HA, Skaftnesmo KO, Bjerkvig R, Thorsen F (2014) In vitro treatment of melanoma brain metastasis by simultaneously targeting the MAPK and PI3K signaling pathways. Int J Mol Sci 15:8773–8794
Lugovic-Mihic L, Cesic D, Vukovic P, Novak Bilic G, Situm M, Spoljar S (2019) Melanoma development: current knowledge on melanoma pathogenesis. Acta Dermatovenerol Croat 27:163–168
Aasen SN, Parajuli H, Hoang T, Feng Z, Stokke K, Wang J, Roy K, Bjerkvig R, Knappskog S, Thorsen F (2019) Effective treatment of metastatic melanoma by combining MAPK and PI3K signaling pathway inhibitors. Int J Mol Sci 20:4235
Davies MA (2012) The role of the PI3K-AKT pathway in melanoma. Cancer J 18:142–147
Junttila MR, Li SP, Westermarck J (2008) Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J 22:954–965
Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27:6245–6251
Zhang JY, Selim MA (2012) The role of the c-Jun N-terminal kinase signaling pathway in skin cancer. Am J Cancer Res 2:691–698
Yong HY, Koh MS, Moon A (2009) The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs 18:1893–1905
Estrada Y, Dong J, Ossowski L (2009) Positive crosstalk between ERK and p38 in melanoma stimulates migration and in vivo proliferation. Pigment Cell Melanoma Res 22:66–76
Yajima I, Kumasaka MY, Thang ND, Goto Y, Takeda K, Yamanoshita O, Iida M, Ohgami N, Tamura H, Kawamoto Y et al (2012) RAS/RAF/MEK/ERK and PI3K/PTEN/AKT signaling in malignant melanoma progression and therapy. Dermatol Res Pract. https://doi.org/10.1155/2012/354191
Mahapatra DK, Asati V, Bharti SK (2017) MEK inhibitors in oncology: a patent review (2015-present). Expert Opin Ther Pat 27:887–906
Savoia P, Fava P, Casoni F, Cremona O (2019) Targeting the ERK signaling pathway in melanoma. Int J Mol Sci. https://doi.org/10.3390/ijms20061483
He Y, Zhou JW, Xu L, Gong MJ, He TC, Bi Y (2013) Comparison of proliferation and differentiation potential between mouse primary hepatocytes and embryonic hepatic progenitor cells in vitro. Int J Mol Med 32:476–484
Li TH, Yan HX (2018) Antitumor and apoptosisinducing effects of pomolic acid against SKMEL2 human malignant melanoma cells are mediated via inhibition of cell migration and subG1 cell cycle arrest. Mol Med Rep 17:1035–1040
Yu R, Zhang Y, Xu Z, Wang J, Chen B, Jin H (2018) Potential antitumor effects of panaxatriol against DU-15 human prostate cancer cells is mediated via mitochondrial mediated apoptosis, inhibition of cell migration and sub-G1 cell cycle arrest. J BUON 23:200–204
Bao J, Xia L, Zhao Y, Xia R (2020) Scutellarin exerts anticancer effects on human leukemia cells via induction of Sub-G1 cell cycle arrest, apoptosis and also inhibits migration and invasion by targeting Raf/MEK/ERK signalling pathway. J BUON 25:1050–1055
Hickman JA (1992) Apoptosis induced by anticancer drugs. Cancer Metastasis Rev 11:121–139
Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ (2016) Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb Protoc. https://doi.org/10.1101/pdb.prot087288
Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y (2020) Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther 5:28
Klein T, Bischoff R (2011) Physiology and pathophysiology of matrix metalloproteases. Amino Acids 41:271–290
Huang H (2018) Matrix metalloproteinase-9 (MMP-9) as a cancer biomarker and MMP-9 biosensors: recent advances. Sensors. https://doi.org/10.3390/s18103249
Pappalardo F, Russo G, Candido S, Pennisi M, Cavalieri S, Motta S, McCubrey JA, Nicoletti F, Libra M (2016) Computational modeling of PI3K/AKT and MAPK signaling pathways in melanoma cancer. PloS ONE 11:e0152104
Unlu A, Nayir E, Kirca O, Ozdogan M (2016) Ganoderma Lucidum (Reishi Mushroom) and cancer. J BUON 21:792–798
Peng X, Liu J, Xia J, Wang C, Li X, Deng Y, Bao N, Zhang Z, Qiu M (2015) Lanostane triterpenoids from Ganoderma hainanense J. D. Zhao. Phytochemistry 114:137–145
Wang HM, Chiu CC, Wu PF, Chen CY (2011) Subamolide E from Cinnamomum subavenium induces sub-G1 cell-cycle arrest and caspase-dependent apoptosis and reduces the migration ability of human melanoma cells. J Agric Food Chem 59:8187–8192
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Zhang JH, Xu M (2000) DNA fragmentation in apoptosis. Cell Res 10:205–211
Kanapathipillai M (2018) Treating p53 mutant aggregation-associated cancer. Cancers 10:154
Amaral JD, Xavier JM, Steer CJ, Rodrigues CM (2010) Targeting the p53 pathway of apoptosis. Curr Pharm Des 16:2493–2503
Chowdhury I, Thompson WE, Welch C, Thomas K, Matthews R (2013) Prohibitin (PHB) inhibits apoptosis in rat granulosa cells (GCs) through the extracellular signal-regulated kinase 1/2 (ERK1/2) and the Bcl family of proteins. Apoptosis 18:1513–1525
Agarwal A, Mahfouz RZ, Sharma RK, Sarkar O, Mangrola D, Mathur PP (2009) Potential biological role of poly (ADP-ribose) polymerase (PARP) in male gametes. Reprod Biol Endocrinol 7:143
Cazes A, Ronai ZA (2016) Metabolism in melanoma metastasis. Pigment Cell Melanoma Res 29:118–119
Yang H, Cao J, Grossniklaus HE (2015) Uveal melanoma metastasis models. Ocul Oncol Pathol 1:151–160
Salemi R, Falzone L, Madonna G, Polesel J, Cina D, Mallardo D, Ascierto PA, Libra M, Candido S (2018) MMP-9 as a candidate marker of response to BRAF inhibitors in melanoma patients With BRAF(V600E) mutation detected in circulating-free DNA. Front Pharmacol 9:856
Revathidevi S, Munirajan AK (2019) Akt in cancer: mediator and more. Semin Cancer Biol 59:80–91
Krajarng A, Chulasiri M, Watanapokasin R (2017) Etlingera elatior extract promotes cell death in B16 melanoma cells via down-regulation of ERK and Akt signaling pathways. BMC Complement Altern Med 17:415
Peng X, Wang Z, Liu Y, Peng X, Liu Y, Zhu S, Zhang Z, Qiu Y, Jin M, Wang R et al (2018) Oxyfadichalcone C inhibits melanoma A375 cell proliferation and metastasis via suppressing PI3K/Akt and MAPK/ERK pathways. Life Sci 206:35–44
Meng L, Ji R, Dong X, Xu X, Xin Y, Jiang X (2019) Antitumor activity of ginsenoside Rg3 in melanoma through downregulation of the ERK and Akt pathways. Int J Oncol 54:2069–2079
Hammouda MB, Ford AE, Liu Y, Zhang JY (2020) The JNK signaling pathway in inflammatory skin disorders and cancer. Cells. https://doi.org/10.3390/cells9040857
Alexaki VI, Javelaud D, Mauviel A (2008) JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment Cell Melanoma Res 21:429–438
Qin Y, Milton DR, Oba J, Ding Z, Lizee G, Ekmekcioglu S, Grimm EA (2015) Inflammatory IL-1beta-driven JNK activation in stage III melanoma. Pigment Cell Melanoma Res 28:236–239
Bradham C, McClay DR (2006) p38 MAPK in development and cancer. Cell Cycle 5:824–828
She QB, Chen N, Dong Z (2000) ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem 275:20444–20449
Acknowledgements
This research was supported by a grant from Next-Generation BioGreen21 Program (PJ01327302), Rural Development Administration of Korea, the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (Grant Number: 2020R1F1A1070844), and the Korea Polar Research Institute (KOPRI, PE21900).
Author information
Authors and Affiliations
Contributions
S-AS, JSL, BJJ, GR, MH, HK, JA and MHK performed experiments. S-AS and CSL wrote the manuscript with guidance from CSL. UJY, JHL, and HHP provided intellectual contributions in this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, SA., Lee, J.S., Joo, B.J. et al. Anti-cancer effects of lucidadiol against malignant melanoma cells. Appl Biol Chem 64, 75 (2021). https://doi.org/10.1186/s13765-021-00647-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-021-00647-w