
Abstract
Stroke, one of the most debilitating cerebrovascular and nuerological diseases, is a serious life-threatening condition and a leading cause of long-term adult disability and brain damage, either directly or by secondary complications. Most effective treatments for stroke are time dependent such as the only FDA-approved therapy, reperfusion with tissue-type plasminogen activator; thus, improving tissue oxygenation with normobaric hyperoxia (NBO) has been considered a logical and potential important therapy. NBO is considered a good approach because of its potential clinical advantages, and many studies suggest that NBO is neuroprotective, reducing ischemic brain injury and infarct volume in addition to improving pathologic and neurobehavorial outcomes. However, increased reactive oxygen species (ROS) generation may occur when tissue oxygen level is too high or too low. Therefore, a major concern with NBO therapy in acute ischemic stroke is the potential increase of ROS, which could exacerbate brain injury. The purpose of this review is to critically review the current literature reports on the effect of NBO treatment on ROS and oxidative stress with respect to acute ischemic stroke. Considering the available data from relevant animal models, NBO does not increase ROS or oxidative stress if applied for a short duration; therefore, the potential that NBO is a viable neuroprotective strategy for acute ischemic stroke is compelling. The benefits of NBO may significantly outweigh the risks of potential increase in ROS generation for the treatment of acute ischemic stroke.
Similar content being viewed by others

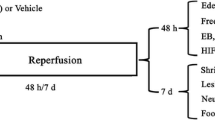
Introduction
Stroke is one of the most common causes of death and long-term disability of adults, and as the brain is highly sensitive to hypoxia insufficient brain oxygen plays a crucial role in the primary and secondary events leading to neuronal cell death and damage [1–3]. Currently, systemic thrombolysis with tissue-type plasminogen activator remains the only FDA-approved reperfusion strategy for acute ischemic stroke [4], which must be given during the early onset of stroke and delayed therapy significantly increases the risks of intracranial hemorrhage [5]. For this reason, improving tissue oxygenation with oxygen therapy has been considered a logical and potentially important therapy for acute ischemic stroke. As adequate cerebral oxygen supply is crucial to neuron survival, the rational for this approach is to restore cellular oxygenation of damaged neurons, particularly in the penumbra, compromised after ischemic stroke, and stimulate those damaged neurons to function normally. Specifically, normobaric hyperoxia (NBO) may be especially useful for improving brain tissue oxygen [6–9]. NBO is considered a good approach because of its potential clinical advantages. It is inexpensive, non-invasive, can be widely available and simple to administer to acute stroke patients by medical staff in a broad range of conditions, including by paramedics or at home. In accord, there are many studies that suggest that NBO is neuroprotective and can reduce acute ischemic brain injury, reduce infarct volume and improve pathologic and neurobehavorial outcomes [10–26].
Electron paramagnetic resonance (EPR) oximetry studies highlight the importance of monitoring tissue oxygen during acute ischemic stroke and NBO treatment [13, 17, 27]. EPR studies have shown that NBO treatment applied during ischemia restores penumbra tissue back to or above preischemic levels [13, 17, 27], while NBO during reperfusion increases penumbra tissue oxygen to twice preischemic levels [17]. As the ischemic penumbra is considered the main target of oxygen therapy, increased oxygen status in the penumbra can improve metabolic status and survival of tissue, but more importantly may also trigger the generation of reactive oxygen species (ROS) and oxidative damage when brain tissue oxygen levels are too high or too low [28, 29]. Thus, a major concern with NBO in acute ischemic stroke is the potential increase in the generation of ROS, thereby exacerbating brain injury [7]. In this review, we will discuss the history of oxidative stress with respect to ischemia and hyperoxia, and the existing controversy surrounding NBO and oxidative stress, particularly from relevant experimental models. We will also summarize the current literature on the impact of NBO treatment on oxidative stress in acute focal ischemia stroke animal models, and if potential NBO induced oxidative stress offset the suggested neuroprotective properties of NBO.
Oxidative stress in ischemia/reperfusion injury and the evolution of oxygen treatment
The concept of oxidative damage results from the production of ROS and byproducts at rates which exceed the ability of natural antioxidant defense mechanisms to detoxify these deleterious products, likely promoting damage. It has been suggested that ROS and oxidative stress is an important mechanism in ischemic/reperfusion injury when ischemia is long and when conditions allow for oxygen to be restored to the ischemic tissue [30–34]. Reintroduction of oxygen to ischemic tissue may trigger a series of harmful reactions leading to the production of ROS and as a consequence inflammation and oxidative damage rather than the restoration to normal function. Accordingly, oxygen therapy or hyperoxia may contribute to oxidative ischemic/reperfusion injury by introducing a compliment of ROS and oxidative stress.
The idea that hyperoxia causes oxidative stress dates back to a novel study hypothesizing that the similarities between the toxic effects of oxygen toxicity and gamma radiation in mice were associated with the production of ROS [35]. Many brain specific studies, beginning in the early 1960s, would then provide supporting evidence of the involvement of ROS in oxygen toxicity by hyperoxia either by an increase in ROS, increase in lipid peroxidation, or circumstantial evidence from studies using agents to effect antioxidant mechanisms and pretreatments to make animals tolerant to hyperoxia [36–53]. However, in a number of these studies, hyperbaric oxygen (HBO) treatment, 100 % oxygen at a pressure greater than 1 atm absolute, was used as the model for hyperoxia and oxygen toxicity. HBO will not be discussed in detail herein, but in clinical trials for ischemia, HBO has shown mixed outcomes [54–58]. Subsequently, recent review articles have provided detailed findings on the effects of HBO on ischemia and oxidative stress in animal studies [7, 59–61]. While controversial, these reviews have emphasized that HBO is associated with increased oxidative stress in several studies, mainly linked to excessive duration of exposure along with high concentrations and pressures. Likewise, differences in experimental conditions and trial design in clinical trials have been recognized as a contributing factor for the mixed results and more controlled studies are needed to determine the effectiveness of HBO treatment.
The risks of NBO induced oxidative stress are also suggested to be associated with duration or prolonged exposure [21, 62, 63]. In addition, the degree of hyperoxia, composition of the inhaled gas mixture, age (especially neonatal vs adult), health and species of animal may play a significant role in determining the therapeutic potential of NBO [37, 48, 64–67]. Thus far, there are a small number of clinical trials using NBO as a potential treatment for ischemic stroke, and mostly all have shown either a potential benefit or no effect [15, 16, 20, 22, 24] with one trial cautioning the use of NBO when given for 24 h [68]. To date, no trials have provided evidence that NBO was detrimental. One clinical study in particular has shown NBO to be neuroprotective when applied within 12 h after the onset of stroke, and these positive results, despite the small sample size, call for further studies to investigate the optimal time window for NBO duration and effects in different types of stroke [15]. Even though neurological outcomes are promising in clinical trials of NBO, the uncertainty of potential damage from NBO induced oxidative stress which may aggravate ischemia/reperfusion injury remains a concern. However, many animal studies have presented findings on the use of NBO treatment in focal acute ischemia/reperfusion injury models, and its effects on the formation of ROS, reactive nitrogen species (RNS) or increased oxidative stress, if any, were reported. These studies are summarized in the following sections (Table 1).
The Effect of NBO on oxidative stress in focal acute ischemia animal models
Initially, we will review a series of studies in which NBO is given shortly after the onset of focal acute ischemia and the impact NBO has on oxidative stress under these conditions. In 1991, a study by Agardh CD et al. found that NBO following brief 15 min periods of ischemia, by bilateral carotid artery occlusion, did not lead to enhanced H2O2 production and that there was no indication that postischemic oxygen supply altered the productions of ROS [32]. Singhal AB et al. indicated that NBO treatment during a filament model of transient middle cerebral artery occlusion (MCAO) ischemia/reperfusion did not increase oxidative stress as measured by heme oxygenase-1 induction and protein carbonyl formation [12]. The same group also indicated that NBO did not increase markers for superoxide (O2·−) generation, measured by dihydroethidium (Het) fluorescence [14]. In these studies, no significant difference was observed between NBO and non-treatment animals suggesting that NBO neither increased nor decreased oxidative stress or ROS formation.
In a series of studies by the Liu group, it was shown that NBO treatment, given intermittently or continuously, during MCAO (90 min) either had no effect or reduced O2·− generation as measured by the Het fluorescence [17, 69]. The generation of 8-hydroxy-2′–deoxyguanosine (8-OHdG), a biomarker for oxidative DNA damage, was also reduced compared to non-treatment animals [17, 70]. Alongside, an increase in ROS and 8-OHdG was not observed in the contralateral hemispshere of the ischemic animals given NBO suggesting that the brain is more tolerable to hyperoxia [17]. Follow up studies indicated that NBO inhibited the upregulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase catalytic subunit, gp91phox, along with the expression of mRNA and protein of gp91phox. It was concluded that NBO reduced NADPH activity, a principal enzyme responsible for O2·− generation [67, 69, 71], and a subsequent study would then suggest that gp91phox containing NADPH oxidase was an important mechanism in NBO-afforded neuroprotection in ischemia using gp91phox knock-out mice [23]. Of note, Yaun Z et al. also demonstrated that NBO for 8 h starting during ischemia and continued during reperfusion was the most effective at decreasing oxidative stress compared to shorter durations [70]. Likewise, a study by the Veltkamp group indicated that O2·− formation, again measured by Het, significantly decreased in mice treated with NBO during ischemia compared to room air controls, while superoxide dismutase (SOD) 2 knock-out mice, genetically susceptible to ROS-induced brain injury, were unaffected by NBO suggesting that NBO does not appear to promote damage associated with ROS [72].
Similar reports also support the hypothesis that NBO does not increase ROS production or oxidative stress when given during reperfusion. In the Liu S et al. study reviewed above, NBO given during reperfusion (90 min) instead of ischemia exhibited no difference in ROS and 8-OHdG production compared to control animals [17]. Rink C et al. showed that NBO given during reperfusion (90 min) did not significantly increase histochemical markers of oxidative stress, 4-hydroxynonenal (4HNE) (a measure of lipid peroxidation), the protein nitrotyrosine (NT) and 8-OHdG oxidation, in the ischemic hemisphere compared to room air controls [27]. Of note, in the same study, Rink C et al. showed that NBO given during MCAO decreased levels of 4HNE, NT and 8-OHdG oxidation in the ischemic hemisphere compared to room air controls [27] similar to previous studies [17, 70].
The effect of NBO on RNS has also been investigated in an acute ischemia small animal model. In an earlier study by Yuan et al., they showed that NBO treatment during MCAO delayed and attenuated nitric oxide (·NO) production via the measurement of nitrite plus nitrate (NOx−), presumably by inhibiting neuronal nitric oxide synthase (nNOS) [73]. Specifically, ischemia caused a rapid production of ·NO 10 min after stroke onset and NBO delayed this up to 30 min and decreased the total amount of NOx− produced. NBO also inhibited the oxidative stress marker, 3-NT, similar to the nNOS inhibitor, 7-nitroxindazole, compared to untreated rats [73].
Collectively, all of these studies suggest that short duration of NBO treatment does not promote additional oxidative stress in acute ischemic animal models and may potentially decrease oxidative stress or ROS/RNS depending on experimental conditions (Table 1). As a majority of acute ischemia experimental studies mostly focus on acute 24 or 48 h outcomes, the question can be raised that more time may be needed for secondary events to develop with NBO treatment and thus we may be monitoring different aspects of oxidative stress mechanisms [38]. For clinical translation, it may be advantageous to show that NBO does not promote oxidative stress outside the acute 48 h window or at least that any effect is extraneous, which current studies, herein, support and the long term benefits of NBO treatment suggests [26]. That said, the mechanisms underlying the effect of NBO on oxidative stress are still not fully understood and further investigations are needed.
NBO afforded neuroprotection in focal acute ischemia animal models
Seeing that a large pool of evidence suggests that NBO does not increase oxidative stress in acute ischemic stroke or may possibly decrease oxidative stress dependent on experimental condition, many of these studies also insinuate that NBO affords neuroprotection or does not worsen neurological outcomes in acute ischemic stroke. Uninterrupted or continuous NBO given shortly after ischemia decreased infarct volume significantly in many of the studies [12, 17, 27, 70, 72, 73]. It was even demonstrated in a novel study by Liu et al. that intermittent NBO and a combination of intermittent and continuous NBO, given during MCAO, significantly reduced infarct volume [68]. Similarly, measurements of lesion volume, a compliment to infarct volume, by MRI apparent diffusion coefficients was also significantly reduced in a study wherein NBO was given during MCAO [67]. This study was in agreement with several clinical studies which demonstrate improvement of clinical deficits and MRI abnormalities [15, 16, 20, 24]. Additionally, NBO given during MCAO was shown to reduce BBB damage and brain edema, a measure of oxidative damage [70], and more recently, Sun et al. indicated that NBO therapy had no effect on infarct volume in SOD2 knockout mice, again suggesting that NBO did not appear to promote damage inflicted by ROS [72].
The neuroprotective benefits of NBO have also been investigated when given during reperfusion. In the study conducted by Liu S et al., in which NBO given during ischemia reduced infarct volumes, it was determined that NBO given during reperfusion had no significant effect on infarct volume [17]. However, in a similar study, Rink et al. indicated that while NBO given during ischemia reduced infarct volume, NBO given during reperfusion significantly exacerbated the ischemic lesion. [27]. In the same study NBO given during reperfusion had no significant effect on sensorimotor assessment and did not exhibit elevated levels of oxidative stress suggesting other factors may be involved. The authors commented that timing of NBO treatment is crucial in determining the benefits of NBO, and the mixed outcomes of these studies warrants further investigation. Nonetheless, in a study investigating the relationship between outcome and NBO duration, Yuan Z et al. showed that NBO given for 8 h, started during MCAO and continued during reperfusion, offered greater efficacy at reducing infarct volume compared to NBO given for 2 and 4 h [71]. In this study, NBO treatment started during ischemia and extended into the reperfusion window seemingly improved the benefits of NBO.
In addition to infarct volume, neurological score assessments have been used to address NBO afforded neuroprotection or damage in studies investigating NBO treatment and acute ischemic stroke. In a study wherein NBO was given during ischemia, Kim et al. showed that NBO did improve neurological scores [14], while Agardh CD et al. indicated that NBO given following brief ischemia (15 min) did not enhance or decrease damage as measured by neurological damage scores [32]. Conversely, two studies have demonstrated that NBO preconditioning, although unbefitting for ischemic stroke patients, could prevent ischemic/reperfusion injury or increase ischemic tolerance [21, 25]. Both studies indicated that NBO preconditioning decreased infarct volume, significantly, while Chen et al. [25] showed that NBO also significantly improved neurological scores compared to controls. It was suggested that overactivation of antioxidant activities [21] and the hypoxia-inducible factor signaling pathway [25] may play a role in ischemic tolerance or protection. Given that NBO preconditioning was neuroprotective, it was suggested that preconditioning may be beneficial for those at risk of stroke or in surgeries involving selective cerebral perfusion [21, 25, 74, 75], but the potential neuroprotective effects of NBO preconditioning need to be investigated further.
Worthy of note, a study conducted by Shin HK et al. indicated that NBO treatment, in a distal MCAO model, reduced infarct volume and improved neurological function in normal wild-type mice compared to mice with endothelial dysfunction [76]. It was suggested that the neuroprotective effects of NBO are critically dependent on normal endothelial function possibly linked to RNS production from endothelial NOS, but production of RNS was not investigated. Notwithstanding, a majority of the studies in this section support the hypothesis that NBO treatment is neuroprotective or does not stimulate additional damage with respect to acute ischemia/reperfusion injury. One study, by Esposito et al., even supports the neuroprotective effects of NBO long term (up to 2 weeks) in an animal model [26], but if these effects last for months or years in human patients warrants further examination. It is also suggested that NBO may widen the time window for reperfusion [14] and for combination treatment with other neuroprotective therapies [9], but clearly many investigators agree that because of considerable differences in clinical studies, firm conclusions are difficult to make and more studies are needed to confirm neuroprotection in humans as those observed in animal studies, herein. The protective mechanisms of NBO remain misunderstood and controversial [77], but it seems well documented that NBO, if applied shortly after acute ischemia, may provide neuroprotection particularly when NBO is shown to reduce or not influence oxidative stress.
The effect of NBO on oxidative stress in other experimental models
Although the above studies suggest that NBO may be neuroprotective in acute ischemic models with no adverse effects related to oxidative stress, the present controversy surrounding the association between NBO and oxidative stress may result from conflicting results in various in vitro, ex vivo and in vivo experimental models and diseases states. We will first review the relationship between NBO and oxidative stress in in vitro and ex vivo models. Cell culture reoxygenation studies which mimic hyperoxia conditions similar to NBO show an increase in intracellular ROS (O2·−, hydrogen peroxide (H2O2), hydroxyl radicals, and potent oxidants such as peroxynitrite) and increased oxidative stress compared to controls [78–80]. Ex vivo studies of tissue from the lungs, brain, liver, and kidney of NBO exposed animals also indicate an increase in oxidative stress with each organ responding differently to NBO and severity of injury being time, dose and exposure dependent [62–64, 80–82]. Taken together, these in vitro and ex vivo studies strongly associate NBO with increased oxidative stress, but care should be taken when interpreting these results as the duration of hyperoxia or NBO exposure is significant compared to suggested treatment conditions for acute ischemia in vivo [11]. Furthermore, hyperoxia conditions in in vitro studies exposes the cells to much higher oxygen concentration (~100 % directly, or 760 mmHg) than in vivo conditions where actual tissue oxygen level is only about 30–40 mmHg [13, 17, 27].
The effect of NBO on oxidative stress in permanent ischemic models has also added to the controversy surrounding NBO treatment. A study using Mongolian gerbils indicated that NBO (as short as 60 min) given 15 min after bilateral carotid occlusion resulted in significant production of lipid peroxidation measured by pentane production and that animals given NBO, for 3–6 h, displayed significantly higher mortality compared to control animals [83]. It was suggested that the brain was the likely site of pentane production, but not confirmed. In a more recent study, Geng et al. indicated that NBO (given for 6 h) after permanent MCAO had no significant effect on infarct volume, neurological deficit or ROS production measured via H2O2 formation. [84]. In addition, infarct volume was significantly reduced when NBO was given 45 min after MCA coagulation, while NBO (given for 60 or 120 min) had no significant effect on infarct volume in a permanent MCAO model [85]. The same study indicated that the benefits of NBO were dependent on the extent and location of permanent ischemia, which may explain the differences in results between studies [85]. Consequently, more controlled studies are needed to evaluate the effects of NBO in permanent ischemia.
Moreover, several studies have investigated the effect of NBO and oxidative stress in brain diseases such as traumatic brain injury (TBI) and multiple sclerosis. Thus far, no studies have shown preliminary evidence of increased oxidative stress from NBO in TBI models; alongside, there is supporting evidence of possible benefits in animal models [86, 87]. It is emphasized that results are somewhat controversial, but clinical TBI studies have shown no impairment from NBO as well [86, 87]. Similarly, a recent study indicated that NBO did not exacerbate oxidative stress injury or neurological outcomes in a collagenase-induced intracerebral hemorrhage model [66]. Given that NBO did not worsen outcomes related to TBI, the study suggests that NBO, if proven to be beneficial in ischemic stroke, could be started before a definitive diagnosis is made to distinguish between ischemic versus hermorrhagic stroke [88]. Finally, a novel study on multiple sclerosis showed that NBO, whether brief (1 h) or continued for 7 days did not reveal evidence of increased O2·− production or oxidative damage when compared to animals breathing room air, and that NBO therapy may be surprisingly beneficial as a treatment for multiple sclerosis that had not been noticed previously [89].
The results of the studies presented in this section highlight the mixed results and controversy surrounding NBO, oxidative stress and its effectiveness as treatment in several disease models. Differing experimental conditions, mainly duration of exposure is attributed to the observed increases in oxidative stress and varying results, further supporting the hypothesis that long duration of NBO would likely produce ROS. However, these studies are dissimilar to acute ischemia models which theorized that early timing and short durations of NBO may minimize deleterious effects as shown in many acute ischemia animal studies, herein; thus, making NBO a potential neuroprotective treatment for several brain diseases including acute ischemic stroke.
Conclusions
As a potential treatment for acute ischemic stroke, NBO has distinct advantages over pharmaceutical drugs such as the ease of diffusibility across the blood–brain barrier and the mechanism of action may be via multiple pathways. It is suggested that high concentrations of oxygen are well tolerated; however, decades of research have cautioned the possible harmful effects of oxygen treatment and oxidative damage. The claim is reasonable considering the oxidant radical damage hypothesis which states that increased oxygen availability produces increased ROS and oxidative stress. Nonetheless, considering the available data from relevant animal models that NBO does not increase ROS or oxidative stress if applied for a short duration, the potential that NBO is a viable neuroprotective strategy for acute ischemic stroke is compelling. The benefits of NBO may outweigh the risks of potentially enhanced ROS generation even if deemed clinically relevant. If we can manage the variables controlling oxygen delivery for the best possible clinical outcome, patients could benefit from receiving NBO at the earliest possible time. The risk to benefit ratio of oxygen therapy must be balanced to determine the safest plan for treatment of acute ischemic stroke.
Abbreviations
- NBO:
-
Normobaric hyperoxia
- EPR:
-
Electron paramagnetic resonance
- ROS:
-
Reactive oxygen species
- HBO:
-
Hyperbaric oxygen
- RNS:
-
Reactive nitrogen species
- MCAO:
-
Middle cerebral artery occlusion
- O2·- :
-
Superoxide
- Het:
-
Dihydroethidium
- 8-OHdG:
-
8-hydroxy-2′–deoxyguanosine
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- SOD:
-
Superoxide dismutase
- 4HNE:
-
4-hydroxynonenal
- NT:
-
Nitrotyrosine
- ·NO:
-
Nitric oxide
- NOx− :
-
Nitrite plus nitrate
- NOS:
-
Nitric oxide synthase
- H2O2 :
-
Hydrogen peroxide
- TBI:
-
Traumatic brain injury
References
Zauner A, Daugherty WP, Bullock MR, Warner DS. Brain oxygenation and energy metabolism: part I biological function and pathophysiology. Neurosurgery. 2002;51(2):289–301. discussion 302.
Mathers CD, Boerma T, Ma FD. Global and regional causes of death. Br Med Bull. 2009;92:7–32.
Stinear C. Prediction of recovery of motor function after stroke. Lancet Neurol. 2010;9(12):1228–32.
Furlan A, Higashida R, Wechsler L, Gent M, Rowley H, Kase C, et al. Intra-arterial prourokinase for acute ischemic stroke. The PROACT II study: a randomized controlled trial. Prolyse in Acute Cerebral Thromboembolism. JAMA. 1999;282(21):2003–11.
Hatcher MA, Starr JA. Role of tissue plasminogen activator in acute ischemic stroke. Ann Pharmacother. 2011;45(3):364–71.
Calvert JW, Cahill J, Zhang JH. Hyperbaric oxygen and cerebral physiology. Neurol Res. 2007;29(2):132–41.
Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29(2):173–83.
Singhal AB, Lo EH. Advances in emerging nondrug therapies for acute stroke. Stroke. 2008;39(2):289–91.
Qi Z, Liu W, Luo Y, Ji X, Liu KJ. Normobaric hyperoxia-based neuroprotective therapies in ischemic stroke. Med Gas Res. 2013;3(1):2.
Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52(5):566–72.
Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002;58(6):945–52.
Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2002;22(7):861–8.
Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24(3):343–9.
Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neurol. 2005;57(4):571–5.
Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, et al. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36(4):797–802.
Chiu EH, Liu CS, Tan TY, Chang KC. Venturi mask adjuvant oxygen therapy in severe acute ischemic stroke. Arch Neurol. 2006;63(5):741–4.
Liu S, Liu W, Ding W, Miyake M, Rosenberg GA, Liu KJ. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26(10):1274–84.
Shin HK, Dunn AK, Jones PB, Boas DA, Lo EH, Moskowitz MA, et al. Normobaric hyperoxia improves cerebral blood flow and oxygenation, and inhibits peri-infarct depolarizations in experimental focal ischaemia. Brain. 2007;130(pt 6):1631–42.
Henninger N, Bouley J, Nelligan JM, Sicard KM, Fisher M. Normobaric hyperoxia delays perfusion/diffusion mismatch evolution, reduces infarct volume, and differentially affects neuronal cell death pathways after suture middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2007;27(9):1632–42.
Singhal AB, Ratai E, Benner T, Vangel M, Lee V, Koroshetz WJ, et al. Magnetic resonance spectroscopy study of oxygen therapy in ischemic stroke. Stroke. 2007;38(10):2851–4.
Bigdeli MR, Rasoulian B. Ali Akbar Meratan AA. In vivo normobaric hyperoxia preconditioning induces different degrees of antioxidant enzymes activities in rat brain tissue. Eur J Pharmacol. 2009;611(1–3):22–9.
Padma MV, Bhasin A, Bhatia R, Garg A, Singh MB, Tripathi M, et al. Normobaric oxygen therapy in acute ischemic stroke: a pilot study in Indian patients. Ann Indian Acad Neurol. 2010;13(4):284–8.
Liu W, Chen Q, Liu J, Liu KJ. Normobaric hyperoxia protects the blood brain barrier through inhibiting Nox2 containing NADPH oxidase in ischemic stroke. Med Gas Res. 2011;1(1):22.
Wu O, Benner T, Roccatagliate L, Zhu M, Schaefer PW, Sorensen AG, et al. Evaluating effects of normobaric oxygen therapy in acute stroke with MRI-based predictive models. Med Gas Res. 2012;2(1):5.
Chen C, Cui H, Li Z, Wang R, Zhou C. Normobaric oxygen for cerebral ischemic injury. Neural Regen Res. 2013;8(31):2885–94.
Esposito E, Mandeville ET, Hayakawa K, Singhal AB, Lo EH. Effects of normobaric oxygen on the progression of focal cerebral ischemia in rats. Exp Neurol. 2013;249:33–8.
Rink C, Roy S, Khan M, Ananth P, Kuppusamy P, Sen CK, et al. Oxygen-sensitive outcomes and gene expression in acute ischemic stroke. J Cereb Blood Flow Metab. 2010;30(7):1275–87.
Kulkarni AC, Kuppusamy P, Parinandi N. Oxygen, the lead actor in the pathophysiologic drama: enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid Redox Signal. 2007;9(10):1717–30.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–90.
Kontos HA, George E. Brown memorial lecture. Oxygen radicals in cerebralvascular injury. Circ Res. 1985;57(4):508–16.
Siesjo BK, Agardh CD, Bengtsson F. Free radicals and brain damage. Cerebrovasc Brain Metab Rev. 1989;1(3):165–211.
Agardh CD, Zhang H, Smith ML, Siesjo BK. Free radical production and ischemic brain damage: influence of postischemic oxygen tension. Int J Dev Neurosci. 1991;9(2):127–38.
Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14.
Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–70.
Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn WO. Oxygen poisoning and X-irradiation: a mechanism in common. Science. 1954;119(3097):623–6.
Jamieson D, Chance B, Cadenas E, Boveris A. The relation of free radical production to hyperoxia. Annu Rev Physiol. 1986;48:703–19.
Bostek CC. Oxygen toxicity an introduction. AANA J. 1989;57(3):231–7.
Jamieson D. Oxygen toxicity and reactive oxygen metabolites in mammals. Free Radic Bio Med. 1989;7(1):87–108.
Becker NH, Galvin JF. Effect of oxygen-rich atmospheres on cerebral lipid peroxides. Aerosp Med. 1963;33:985–7.
Jamieson D, van den Brenk HAS. The effects of antioxidants on high pressure oxygen toxicity. Biochem Pharmacol. 1964;13:159–64.
Zirkle LG, Mengel CE, Horton BD, Duffy EJ. Studies of oxygen toxicity in the central nervous system. Aerosp Med. 1965;36(11):1027–32.
Jerrett SA, Jefferson D, Mengel CE. Seizures, H202 formation and lipid peroxides in brain during exposure to oxygen under high pressure. Aerosp Med. 1973;44(1):40–4.
Hilton JG, Brown GL, Proctor PH. Effects of superoxide dismutase and catalase on central nervous system toxicity of hyperbaric oxygen. Toxicol Appl Pharmacol. 1980;53(1):50–3.
Cadenas E, Varsavasky AI, Boveris A, Chance B. Oxygen- and organic hydroperoxide-induced chemiluminescence of brain and liver homogenates. Biochem J. 1981;198(3):645–54.
Gabivov MM, Karagezian KG. Lipid oxidation processes in different rat tissues in the presence of hyperoxia and in the posthyperoxic period. Bull Exp Med Biol. 1981;91(6):682–5.
Kovachich GB, Mishra OP. Partial inactivation of Na+, K + −ATPase in cortical brain slices incubated in normal Krebs-Ringer phosphate medium at 1 and 10 atm oxygen pressures. J Neurochem. 1981;36(1):333–5.
Dirks CD, Faiman MD. Free radical formation and lipid peroxidation in rat and mouse cerebral cortex slices exposed to high oxygen pressure. Brain Res. 1982;248(2):355–60.
Noda Y, McGeer PL, McGeer EG. Lipid peroxide distribution in brain and the effect of hyperbaric oxygen. J Neurochem. 1983;40(5):1329–32.
Clark JM. Interacting effects of hyperoxia adaptation and acute hypercapnia on oxygen tolerance in rats. J Appl Physiol. 1984;56(5):1191–8.
Yusa T, Crapo JD, Freeman BA. Liposome-mediated augmentation of brain SOD and catalase inhibits CNS 02 toxicity. J Appl Physiol. 1984;57(6):1674–81.
Yusa T, Beckman JS, Crapo JD, Freeman BA. Hyperoxia increases H2O2 production by brain in vivo. J Appl Physiol (1985). 1987;63(1):353–8.
Bickford PC, Chadman K, Williams B, Shukitt-Hale B, Holmes D, Taglialata G, et al. Effect of normobaric hyperoxia on two indexes of synaptic function in fisher 344 rats. Free Radic Biol Med. 1999;26(7–8):817–24.
Demchenko IT, Boso AE, Whorton AR, Piantadosi CA. Nitric oxide production is enhanced in rat brain before oxygen-induced convulsions. Brain Res. 2001;917(2):253–61.
Neubauer RA, End E. Hyperbaric oxygenation as an adjunct therapy in strokes due to thrombosis. A review of 122 patients. Stroke. 1980;11(3):297–300.
Anderson DC, Bottini AG, Jagiella WM, Westphal B, Ford S, Rockswold GL, et al. A pilot study of hyperbaric oxygen in the treatment of human stroke. Stroke. 1991;22(9):1137–42.
Nighoghossian N, Trouillas P, Adeleine P, Salord F. Hyperbaric oxygen in the treatment of acute ischemic stroke. A double-blind pilot study. Stroke. 1995;26(8):1369–72.
Rusyniak DE, Kirk MA, May JD, Kao LW, Brizendine EJ, Welch JL, et al. Hyperbaric oxygen therapy in acute ischemic stroke: results of the Hyperbaric Oxygen in Acute Ischemic Stroke Trial Pilot Study. Stroke. 2003;34(2):571–4.
Bennett MH, Weibel S, Wasiak J, Schnabel A, French C, Kranke P. Hyperbaric oxygen therapy for acute ischaemic stroke (Review). Cochrane Database Syst Rev. 2014;12(11):CD004954.
Nemoto EM, Betterman K. Basic physiology of hyperbaric oxygen in brain. Neurol Res. 2007;29(2):116–26.
Ding Z, Tong WC, Lu X-X, Peng H-P. Hypebaric oxygen therapy in acute ischemic stroke: a review. Interv Neuro. 2014;2(4):201–11.
Liu W, Khatibi N, Sridharan A, Zhang JH. Application of medical gases in the field of neurobiology. Med Gas Res. 2011;1(1):13.
Tatarkova Z, Engler I, Calkovska A, Mokra D, Drgova A, Hodas P, et al. Effect of long-term normobaric hyperoxia on oxidative stress in mitochondria of the guinea pig brain. Neurochem Res. 2011;36(8):1475–81.
Tatarkova Z, Engler I, Calkovska A, Mokra D, Drgova A, Kuka S, et al. Effect of normobaric oxygen treatment on oxidative stress and enzyme activities in guinea pig heart. Gen Physiol Biophys. 2012;31(2):179–84.
Ahotupa M, Mäntylä E, Peltola V, Puntala A, Toivonen H. Pro-oxidant effects of normobaric hyperoxia in rat tissues. Acta Physiol Scand. 1992;145(2):151–7.
Oter S, Korkmaz A, Topal T, Ozcan O, Sadir S, Ozler M, et al. Correlation between hyperbaric oxygen exposure pressures and oxidative parameters in rat lung, brain, and erythrocytes. Clin Biochem. 2005;38(8):706–11.
Hensley K, Howard BJ, Carney JM, Butterfield DA. Membrane protein alterations in rodent erythrocytes and synaptosomes due to aging and hyperoxia. Biochim Biophys Acta. 1995;1270(2–3):203–6.
Tang X, Liu KJ, Ramu J, Chen Q, Li T, Liu W. Inhibition of gp91(phox) contributes towards normobaric hyperoxia afforded neuroprotection in focal cerebral ischemia. Brain Res. 2010;1348:174–80.
Rønning OM, Guldvog B. Should stroke victims rountinely receive supplemental oxygen. Stroke. 1999;30(Rønning OM and Guldvog B):2033–7. renumber references now after 68.
Liu C, Weaver J, Liu KJ. Rapid conditioning with oxygen oscillation: neuroprotection by intermittent normobaric hyperoxia after transient focal cerebral ischemia in rats. Stroke. 2012;43(1):220–6.
Yuan Z, Pan R, Liu W, Liu KJ. Extended normobaric hyperoxia therapy yields greater neuroprotection for focal transient ischemia-reperfusion in rats. Med Gas Res. 2014;4:14.
Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, et al. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107(5):1196–205.
Sun L, Wolferts G, Veltkamp R. Oxygen therapy does not increase production and damage induced by reactive oxygen species in focal cerebral ischemia. Neurosci Lett. 2014;577:1–5.
Yuan Z, Liu W, Liu B, Schnell A, Liu KJ. Normobaric hyperoxia delays and attenuates early nitric oxide production in focal cerebral ischemic rats. Brain Res. 2010;1352:248–54.
Bigdeli MR, Khoshbaten A. In vivo preconditioning with normobaric hyperoxia induces ischemic tolerance partly by triggering tumor necrosis factor-alpha converting enzyme/tumor necrosis factor-alpha/nuclear factor-kappaB. Neuroscience. 2008;153(3):671–8.
Bigdeli MR, Hajizadeh S, Froozandeh M, Heidarianpour A, Rasoulian B, Asgari AR, et al. Normobaric hyperoxia induces ischemic tolerance and upregulation of glutamate transporters in the rat brain and serum TNF-α level. Exp Neurol. 2008;212(2):298–306.
Shin HK, Oka F, Kim JH, Atochin D, Huang PL, Ayata C. Endothelial dysfunction abrogates the efficacy of normobaric hyperoxia in stroke. J Neurosci. 2014;34(46):15200–7.
Haelewyn B, Chazalviel L, Nicole O, Lecocq M, Risso JJ, Abraini JH. Moderately delayed post-insult treatment with normobaric hyperoxia reduces excitotoxin-induced neuronal degeneration but increases ischemia-induced brain damage. Med Gas Res. 2011;1(1):2.
Zweier JL, Duke SS, Kuppusamy P, Sylvester JT, Gabrielson EW. Electron paramagnetic resonance evidence that cellular oxygen toxicity is caused by the generation of superoxide and hydroxyl free radicals. FEBS Lett. 1989;252(1–2):12–6.
Gille JJ, Joenje H. Cell culture models for oxidative stress: superoxide and hydrogen peroxide versus normobaric hyperoxia. Mutat Res. 1992;275(3–6):405–14.
Flandin P, Donati Y, Barazzone-Argiroffo C, Muzzin P. Hyperoxia-mediated oxidative stress increases expression of UCP3 mRNA and protein in skeletal muscle. FEBS let. 2005;579(16):3411–5.
Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis. 1980;122(1):123–43.
Kaplan P, Tatarkova Z, Engler I, Calkovska A, Mokra D, Drgova A, et al. Effects of long-term oxygen treatment on alpha-ketoglutarate dehydrogenase activity and oxidative modifications in mitochondria of the guinea pig heart. Eur J Med Res. 2009;14 Suppl 4:116–20.
Mickel HS, Vaishnav YN, Kempski O, von Lubitz D, Weiss JF, Feuerstein G. Breathing 100 % oxygen after global brain ischemia in Mongolian Gerbils results in increased lipid peroxidation and increased mortality. Stroke. 1987;18(2):426–30.
Geng X, Elmadhoun O, Peng C, Ji X, Hafeez A, Liu Z, et al. Ethanol and normobaric oxygen: novel approach in modulating pyruvate dehydrogenase complex after severe transient and permanent ischemic stroke. Stroke. 2015;46(2):492–9.
Veltkamp R, Sun L, Herrmann O, Wolferts G, Hagmann S, Siebing DA, et al. Oxygen therapy in permanent brain ischemia: potential and limitations. Brain Res. 2006;1107(1):185–91.
Kumaria A, Tolias CM. Normobaric hyperoxia therapy for traumatic brain injury and stroke: a review. Br J Neurosurg. 2009;23(6):576–84.
Diringer MN. Hyperoxia: good or bad for the injured brain? Curr Opin Crit Care. 2008;14(2):167–71.
Fujiwara N, Mandeville ET, Geng X, Luo Y, Arai K, Wang X, et al. Effect of normobaric oxygen therapy in a rat model of intracerebral hemorrhage. Stroke. 2011;42(5):1469–72.
Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. 2013;74(6):815–25.
Acknowledgements
This work was supported in part by grants from National Institute of Health [P30GM103400, R01AG031725] and Dedicated Health Research Funds from the University of New Mexico School of Medicine [UNM HSC RAC grant].
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
JMW reviewed the NBO studies, participated in the overall design of the review and wrote the manuscript. KJL edited, improved, participated in the overall design of the review and obtained funding. All authors have read and approved the final review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Weaver, J., Liu, K.J. Does normobaric hyperoxia increase oxidative stress in acute ischemic stroke? A critical review of the literature. Med Gas Res 5, 11 (2015). https://doi.org/10.1186/s13618-015-0032-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13618-015-0032-4