
Abstract
Background
Observational studies have demonstrated an association between gut microbiota and myasthenia gravis; however, the causal relationship between the two still lacks clarity. Our goals are to ascertain the existence of a bidirectional causal relationship between gut microbiota composition and myasthenia gravis, and to investigate how gut microbiota plays a role in reducing the risk of myasthenia gravis.
Methods
We acquired gut microbiota data at the phylum, class, order, family, and genus levels from the MiBioGen consortium (N = 18,340) and myasthenia gravis data from the FinnGen Research Project (426 cases and 373,848 controls). In the two-sample Mendelian randomization analysis, we assessed the causal relationship between the gut microbiota and myasthenia gravis. We also conducted bidirectional MR analysis to determine the direction of causality. The inverse variance weighted, mendelian randomization-Egger, weighted median, simple mode, and weighted mode were used to test the causal relationship between the gut microbiota and severe myasthenia gravis. We used MR-Egger intercept and Cochran's Q test to assess for pleiotropy and heterogeneity, respectively. Furthermore, we utilized the MR-PRESSO method to evaluate horizontal pleiotropy and detect outliers.
Results
In the forward analysis, the inverse-variance weighted method revealed that there is a positive correlation between the genus Lachnoclostridium (OR = 2.431,95%CI 1.047–5.647, p = 0.039) and the risk of myasthenia gravis. Additionally, the family Clostridiaceae1 (OR = 0.424,95%CI 0.202–0.889, p = 0.023), family Defluviitaleaceae (OR = 0.537,95%CI 0.290–0.995, p = 0.048), family Enterobacteriaceae (OR = 0.341,95%CI 0.135–0.865, p = 0.023), and an unknown genus (OR = 0.407,95%CI 0.209–0.793, p = 0.008) all demonstrated negative correlation with the risk of developing myasthenia gravis. Futhermore, reversed Mendelian randomization analysis proved a negative correlation between the risk of myasthenia gravis and genus Barnesiella (OR = 0.945,95%CI 0.906–0.985, p = 0.008).
Conclusion
Our research yielded evidence of a causality connection in both directions between gut microbiota and myasthenia gravis. We identified specific types of microbes associated with myasthenia gravis, which offers a fresh window into the pathogenesis of this disease and the possibility of developing treatment strategies. Nonetheless, more studies, both basic and clinical, are necessary to elucidate the precise role and therapeutic potential of the gut microbiota in the pathogenesis of myasthenia gravis.
Similar content being viewed by others

Introduction
Myasthenia gravis (MG) is an autoimmune disease characterized by the attack of antibodies on the neuromuscular junction, leading to muscle weakness. The most common symptom is ocular muscle weakness, which can also affect bulbar, limb, axial, and ventilator muscles, progressing to generalized MG [1]. Acute respiratory failure requiring mechanical ventilation occurs in 20% of patients with MG in clinical practice, resulting in significantly increased mortality [2]. The etiology of MG is multifaceted, resulting from genetic and several environmental risk factors [3,4,5]. However, the exact factors that result in an individual's susceptibility to MG are unknown. The gut microbiota is critical for the development and maintenance of host metabolism and immune homeostasis in the human intestine, which affects human nutrition as well as gastrointestinal function and integrity [6, 7]. Mounting scientific evidence indicates that microbial-host interactions affect not only the gut environment but also remote organs [8, 9]. Evidence suggests that gut microbiota can significantly affect both the innate and adaptive immune systems and have a dual role in either promoting or protecting against disease development [10]. Studies suggest that disturbances in the composition of the gut microbiota can be associated with several autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis [11].
Alterations in the composition of the gut microbiota have been observed in both myasthenia gravis patients and animal models. A case–control study demonstrated that the gut microbiota diversity and abundance differed between the myasthenia gravis (MG) group and the healthy control group. Specifically, the MG group exhibited reduced levels of Firmicutes, Clostridium, Eubacterium, and F. prausnitzii. Conversely, higher levels of Proteobacteria, Bacteroidetes, Streptococcus, and Parasutterella were noted in the MG group. The healthy control group displayed Clostridium levels approximately three times that of the MG group [12]. Based on German Moris et al.'s research, it was found that Verrucomicrobiaceae and Bifidobacteriaceae were less abundant in patients with MG than in the healthy control group, while the abundance of Bacteroidetes and Desulfovibrionaceae was greater [1]. An animal experiment showed that transplanting microbiota from MG mice to germ-free mice resulted in reduced motor function, which could be restored through a mixed microbiota (made up of both the MG and healthy microbiota) [2]. This suggests that the gut microbiota may play a role in MG development.
However, observational studies dominate most current research; while suggesting an association between gut microbiota and MG, the conclusions drawn in observational studies tend to be based on “association” rather than “causation”. Additionally, confounding factors like demographics, comorbidities, medications, and diet cannot be ruled out. And, experiments conducted on animals do not guarantee the same results in humans. The Mendelian Randomization method aims to alleviate these limitations.
Mendelian randomization (MR) is a method of data analysis widely used recently to infer causality in epidemiology. Traditional epidemiological causal inference is hampered by reverse causation and confounding factors. Randomized controlled trials are difficult to implement due to ethical limitations in human medicine and trial design. MR complements these limitations by using gene variation as instrumental variables (IVs), normally single nucleotide polymorphisms (SNPs), to infer the causal effect of treatment exposure on outcomes [13]. Mendel’s laws of inheritance ensure a random distribution of parental alleles to offspring during gamete formation, thereby minimizing interference from common confounding factors such as postnatal environment, socioeconomic status, and behavioral factors [13, 14]. This reasonable causal sequence results in a more accurate estimation of true causal effects. The present study aims to shed light on the association between changes in gut microbiota and MG using MR method.
Materials and methods
Study design
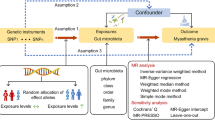
We conducted a bidirectional MR analysis to investigate the causal relationship between the gut microbiota and MG; the flowchart and design for this analysis is presented in Fig. 1A. First, the genome-wide association study (GWAS) data for the gut microbiota and MG were obtained from the Mibiogen Consortium [15] and FinnGen Research Project, respectively. Genetic variants from the GWAS data were retrieved and used as IVs. Then, a Two-Sample MR was performed using the R package “TwoSampleMR” (0.5.6), which included five MR methods. Sensitivity analyses, including pleiotropy [16] and heterogeneity tests [17], and leave-one-out analyses were conducted.MR-PRESSO was used to detect and correct outliers [18]. Finally, reversed analysis was conducted to obtain a comprehensive conclusion regarding causation.

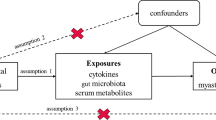
Study design and Mendelian randomization core assumption. A Data sources and study design of bidirectional Mendelian randomization. B Three core assumptions in the Mendelian randomization
For results with minimal bias, adhering to three key assumptions is crucial in utilizing the MR method: (1) IVs were significantly correlated with exposure; (2) IVs did not have any confounding factors associated with exposure-outcome associations, and (3) IVs only affect outcomes through exposure [19] (Fig. 1B).
Selection of data sources and instrumental variables
Data sources
The summary data on the gut microbiota utilized in this research was derived from a recently conducted genome-wide association study by the global consortium, MiBioGen [15]. The study’s vast database collected 16S ribosomal RNA gene sequencing and genotype data from 18,340 predominantly European participants (N = 13,266). The researchers amassed records of 211 bacterial characteristics belonging to 131 genera in nine phyla with sixteen classes, 20 orders, and 35 families. The data on MG was collected from the FinnGen Research Project, consisting of a group of 426 MG diagnosed cases and 373,848 European controls. The data used in this study was obtained from publicly available repositories and did not need any further ethics approval or patient consent.
Instrumental variables selection
In the forward analysis, the exposure variable was gut microbiota, while the outcome variable was MG. The process for selecting IVs is illustrated in Fig. 1A.Firstly, a set of SNPs was selected as IVs which fell below the genome-wide significance threshold of 1 × 10−5 in line with previous studies[20]. Secondly, to ensure that each IV was independent, we clumped together SNPs utilizing the European 1000 Genomes Project reference dataset with an r2 value of 0.001 and a clumping window of 10,000 kb [21]. Third, we harmonized the effects of the SNP on outcome and exposure by ensuring that they referred to the same allele, correcting the strand for non-palindromic SNPs, and removing all palindromic sequences. The F-statistic calculated the potency of each SNP as an instrumental variable with a value exceeding 10 indicating a strong instrument.
During reverse causality studies with MG utilized as an exposure variable, a set of SNPs below the genome-wide threshold of statistical significance (5 × 10−8) was used as IVs; however, only a SNP reached this threshold. Thus, SNPs with a second p-value less than the genome-wide significance level (5 × 10−6) were selected to identify the underlying causal links. The remaining flow and parameters remained identical to the forward MR.
Statistical analysis
To evaluate the causal link between gut microbiota and MG, multiple methods, including inverse variance weighted (IVW), MR-Egger regression, weighted median, simple mode, and weighted mode were employed. The primary analysis was conducted using the IVW method, while the other four methods supplemented the IVW [22]. The IVW method uses a meta-analytical approach to combine the Wald ratios for each SNP, and it provides the most precise estimates when all IVs are valid [23]. The MR-Egger regression relies on the “instrument strength independent of the direct effect” assumption that exposure and outcome are independent. It has lower precision and statistical power but can be used to correct for horizontal pleiotropy [16]. When up to 50% of the information originates from invalid genetic variants, the weighted median method provides the most unbiased estimate of the causal effects [24]. As an additional step, we used weighted mode and simple mode to enhance accuracy and stability [25].
Various sensitivity analyses were conducted to evaluate the strength of the results. MR-Egger intercept was employed to identify horizontal pleiotropy based on the distance of the regression intercept line from zero [26]. Cochran’s Q test is a method used to assess heterogeneity among different IVs. If the p-value of the Cochran's Q test is less than the pre-defined significance level (0.05), significant heterogeneity is considered to be present. Conversely, if the p-value is greater than the significance level (0.05), it is concluded that there is no significant heterogeneity among the IVs [27]. The leave-one-out sensitivity method was used to assess if one SNP significantly influenced causality estimates [28]. The MR-PRESSO test was used to detect and remove possible outliers, then provide estimates thereby correcting for horizontal pleiotropy [18]. Finally, the F-statistic for each SNP was computed using the formula β2/SE2 [28], with β and SE expressing the estimated and standard errors of the effect allele, respectively [29]. In this study, SNP with F-statistics less than or equal to 10 (defined as weak IVs) were excluded from the MR analysis. To adjust for multiple comparisons, we applied a Bonferroni correction, giving a cutoff of p = 2.37 × 10−4 (0.05/211) for gut microbiota and MG, and the corrected p > 0.05 was considered to suggest an association [30].
All statistical analyses were performed using R version 4.2.2. MR analyses were performed using the two-sample MR (version 0.5.6) [28] and MR-PRESSO (version 1.0)[31] R packages.
Results
The results of IVW analyses demonstrated that the family Clostridiaceae1 (OR = 0.424,95%CI 0.202–0.889,p = 0.023), family Defluviitaleaceae (OR = 0.537,95%CI 0.290–0.995, p = 0.048), family Enterobacteriaceae (OR = 0.341,95%CI 0.135–0.865, p = 0.023), and an unknown genus (OR = 0.407,95%CI 0.209–0.793, p = 0.008) were negatively correlated with the risk of MG (Table 1 and Fig. 2A–D), whereas the genus Lachnoclostridium (OR = 2.431,95%CI 1.047–5.647, p = 0.039) was positively correlated with the risk of MG (Table 1 and Fig. 2E). The same results were obtained using the four additional methods.

Scatter plots for the causal association between gut microbiota and MG
Reverse IVW analysis indicated that MG was associated with a lower abundance of the genus Barnesiella (OR = 0.945,95%CI 0.906–0.985, p = 0.008). The same results were obtained using the four additional methods (Table 2 and Fig. 2F).
No potential heterogeneity or pleiotropy was found in the sensitivity analysis (p > 0.05), as demonstrated in Table 1 and Table 2. Moreover, no notable outliers were identified in the MR-PRESSO or leave-one-out analyses (Table 1 and Figs. 3). The estimated F-statistics were all higher than 10, signifying that there were no weak IVs. More details regarding the final SNPs and the corresponding funnel plots and forest plots are summarized in Additional file 1.

Leave-one-out analysis between gut microbiota and MG
Discussion
Several hypotheses have been postulated to explain the pathogenesis of MG; these include antibodies against acetylcholine receptors (AchR), the role of CD4 + T cells in the pathogenesis of MG, and the effects of CD4 + T cell subtypes and cytokines in MG and experimental autoimmune yasthenia gravis [32,33,34]. Additionally, other autoantibodies such as anti-musk antibodies are observed in MG patients who lack anti-AchR antibodies [35]. It is worth noting that there is growing attention to the role of intestinal flora in the disease. Changes in either a single microbial species or the global commensal communities could play a therapeutic role in autoimmune diseases by altering the balance between pathogenic and protective immune responses [36]. Improvement in this regard has been observed in cases of rheumatoid arthritis as well as inflammatory bowel disease [37, 38]. Our study highlights the effects of alterations in gut microbiota on MG, as well as the potential for MG to cause changes in specific gut microbiota levels. Currently, this is the first two-sample bidirectional MR study to demonstrate the association between the gut microbiota and MG. Specifically, Clostridiaceae1, Defluviitaleaceae, Enterobacteriaceae, and an unknown genus were observed as having a negative relationship, while Lachnoclostridium was identified as a potential risk factor for MG. Additionally, reverse analysis revealed a negative correlation between the presence of MG and the level of Barnesiella.
Effect of gut microbiota on MG
Our research indicates that multiple bacteria serve as inhibitory factors in the onset of MG, and the specific mechanisms may be diverse.
Foxp3 + CD4 + Treg cells play a critical role in maintaining self-tolerance and immune homeostasis by regulating the production of pathogenic antibodies through the modulation of the quantity of autoreactive T cells and the suppression of the activity of autoreactive B cells, thereby reducing the severity and progression of diseases[39]. The frequency of Foxp3 + CD4 + Treg cells in peripheral blood lymphocytes of MG patients is significantly insufficient [12]. Therefore, the abundance of Foxp3 + CD4 + Treg cells is vital for preventing and treating MG, and it has become the main focus of current research on the pathogenesis of MG. Studies have shown that gut microbiota, especially Clostridium, can affect the number of Foxp3 + CD4 + Treg cells and the surface T cell receptor (TCR). The TCR on Foxp3 + CD4 + Treg cells can recognize commensal bacterial subsets, inducing naive CD4 + T cells to differentiate into antigen-specific Foxp3 + CD4 + Treg cells, and within this way, increase their numbers. An example that highlights this is Clostridium. The strains of Clostridium are able to colonize the mucous layer that is in close proximity to the epithelial, increasing the expression of 2,3-dioxygenase and TGF-β1 [40, 41]. These changes may promote the differentiation of immature T cells, resulting in the formation of Foxp3 + CD4 + Treg cells. This biological mechanism provides a protective effect, which has been observed in germ-free mice and in the colon of humans [42, 43]. The proliferator-activated receptor (PPARγ) participates in regulating the proliferation and differentiation of immune cells and can increase the number of Foxp3 + CD4 + Treg cells through inducing differentiation [44]. Streptococcus has been proven to regulate PPARγ and its ligand 15d-PGJ2 by activating PPARγ through inhibiting certain pathways or immune cell functions [45]. Its efficacy has been confirmed in treating other immune diseases such as rheumatoid arthritis and inflammatory bowel disease [46, 47]. These findings highlight the fact that gut microbiota can affect transcriptional regulation through factors such as PPARγ, thus resulting in a tightly coordinated balance of immune system reactions.
Short-chain fatty acids (SCFAs) are non-nutritional substances produced by gut microbiota, with significant physiological regulatory functions. They provide some of the energy needed by the human body, protect the intestinal mucosal barrier, inhibit intestinal inflammation, and regulate immune responses [48, 49]. SCFAs are another metabolite that can regulate Foxp3 + CD4 + Treg cells. They have a profound influence on T cells and directly regulate the differentiation of T cells into Foxp3 + CD4 + Treg cells [43, 50]. Therefore, the gut microbiota may increase the number of Foxp3 + CD4 + Treg cells by increasing the microbial metabolites (SCFAs), indirectly exerting a protective effect.
Adjusting the gut microbiota to increase the number of Foxp3 + CD4 + Treg cells may become a new strategy for treating MG. The use of probiotics to regulate the course of MG has already been validated in animal models, with a mixture of five probiotic strains (Streptococcus thermophilus, Lactobacillus reuteri, Bifidobacterium bifidum, Lactobacillus acidophilus, and Lactobacillus casei) observed to suppress pro-inflammatory lymphocyte reactions and reduce AchR antibody levels in MG model rats, and this effect is also achieved by increasing the number of Foxp3 + CD4 + Treg cells. However, the specific strains of probiotics applicable to the human body, as well as the required dosage for maximal efficacy against different types of MG, need further investigation. Our study did not show positive results for protective bacteria mentioned above, namely Clostridium and Streptococcus, which may be due to the small sample size. However, the protective bacteria with suggestive results obtained in this study, such as Clostridiaceae1, Defluviitaleaceae, Enterobacteriaceae, and an unknown type of bacteria, broaden the scope of bacteria to be studied for future research.
In previous animal experiments, it was demonstrated that transplanting the gut microbiota of mice with MG into germ-free mice can cause movement impairments, yet the specific bacterial strain(s) responsible remain unidentified. This study represents the first evidence that Lachnoclostridium is a risk factor for the development of MG. Lachnoclostridium is an important bacteria that produces SCFAs and can exhibit anti-inflammatory effects in the body [51, 52]. While previous research has shown that SCFAs have a beneficial effect on foxp3 + CD4 + Tregs, the association of Lachnoclostridium with MG is paradoxical. The reasons for this phenomenon are complex. On the one hand, SCFAs do not always have a neuroprotective effect. For example, oral administration of SCFAs can exacerbate movement impairments in mouse models that overexpress alpha-synuclein [53], suggesting that SCFAs may produce different effects in diverse pathological contexts. On the other hand, the 16S rRNA gene sequencing of Lachnoclostridium was performed at the genus level, it cannot be determined whether specific strains or species of bacteria are associated with this inconsistent result. In addition, fecal metabolites are currently considered an external manifestation of the function of the gut microbiota [2]. Besides SCFA aforementioned, studies have demonstrated a significant correlation between the microbiota of the gut and a range of metabolic biomarkers, such as valine, leucine, xanthine, cytosine, naphthalene, and catechol [2]. These metabolic products may support the view that gut microbiota disorders are related to MG, as gut microbiota may affect the occurrence of MG through metabolic pathways such as amino acids, nucleotides, and microbial metabolism[54, 55]. This is a potential new supplement to the previously proposed antibody-mediated mechanism for MG pathogenesis. However, it should be acknowledged that it is still unclear whether Lachnoclostridium, which was identified in this study, participates in these metabolic pathways and how it contributes to the pathogenesis of MG- a point that warrants further research.
Effect of MG on gut microbiota
In the reverse MR analysis, it was discovered that MG led to a decrease in Barnesiella. Previous studies have shown that MG can alter the relative abundance of bacterial groups in the gut microbiota. Specifically, it results in a decrease in Clostridium [56]. In contrast, this study only found a negative correlation between MG and Barnesiella, with no observed correlation with Clostridium. According to a case–control study, bacterial diversity and abundance were observed to be lower in the MG group than the healthy control group [12]. As such, the microbial diversity index may serve as a new clinical tool for evaluating the severity of MG [2]. Additionally, changes in gut microbiota that occur in MG patients are slightly associated with some clinical parameters. Combining gut microbiota with intestinal metabolites can help distinguish MG subjects from healthy controls [2]. Given the current lack of detailed data on Barnesiella, it is difficult to assess the efficacy of its level changes in evaluating diseases. Nevertheless, it is important to note that the findings of this study may offer important insights for future research.
Limitations
There are several limitations to our study that need to be considered. Firstly, the sample population we analyzed only comprised European individuals. Consequently, the extent to which findings can be generalized to non-European populations is unclear. Secondly, due to dataset constraints, it was not possible to explore the causal relationship between gut microbiota and MG at the genus level. Thirdly, the associations we identified in our study, after multiple comparisons, were only suggestive of causal relationships, not certain. Finally, the summary statistics we used instead of raw data made it challenging to carry out subgroup analyses for MG. Therefore, in the future, more comprehensive data is needed to confirm our findings.
Conclusion
Our research yielded evidence of a causality connection in both directions between gut microbiota and MG. We identified specific types of microbes associated with MG, which offers a fresh window into the pathogenesis of this disease and the possibility of developing treatment strategies. Nonetheless, more studies, both basic and clinical, are necessary to elucidate the precise role and therapeutic potential of the gut microbiota in the pathogenesis of MG.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files.
Abbreviations
- GWAS:
-
Genome-wide association studies
- IVW:
-
Inverse variance weighted
- IVs:
-
Instrumental variables
- MG:
-
Myasthenia gravis
- MR:
-
Mendelian randomization
- PPARγ:
-
Proliferator-activated receptor
- SCFAs:
-
Short-chain fatty acids
- SNPs:
-
Single nucleotide polymorphisms
- TCR:
-
T cell receptor
References
Moris G, Arboleya S, Mancabelli L, Milani C, Ventura M, de Los Reyes-Gavilán CG, et al. Fecal microbiota profile in a group of myasthenia gravis patients. Sci Rep. 2018;8(1):14384.
Zheng P, Li Y, Wu J, Zhang H, Huang Y, Tan X, et al. Perturbed microbial ecology in myasthenia gravis: evidence from the Gut microbiome and fecal metabolome. Adv Sci. 2019;6(18):1901441.
Selmi C, Leung PSC, Sherr DH, Diaz M, Nyland JF, Monestier M, et al. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun. 2012;39(4):272–84.
Germolec D, Kono DH, Pfau JC, Pollard KM. Animal models used to examine the role of the environment in the development of autoimmune disease: findings from an NIEHS Expert Panel Workshop. J Autoimmun. 2012;39(4):285–93.
Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun. 2012;39(4):259–71.
Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904.
Sommer F, Bäckhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–38.
Diaz Heijtz R, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA. 2011;108(7):3047–52.
Clarke G, O’Mahony SM, Dinan TG, Cryan JF. Priming for health: gut microbiota acquired in early life regulates physiology, brain and behaviour. Acta Paediatr. 2014;103(8):812–9.
Berrih-Aknin S. Myasthenia gravis: paradox versus paradigm in autoimmunity. J Autoimmun. 2014. https://doi.org/10.1016/j.jaut.2014.05.001.
Xu Q, Ni J-J, Han B-X, Yan S-S, Wei X-T, Feng G-J, et al. Causal relationship between Gut microbiota and autoimmune diseases: a two-sample mendelian randomization study. Front Immunol. 2021;12: 746998.
Qiu D, Xia Z, Jiao X, Deng J, Zhang L, Li J. Altered Gut microbiota in myasthenia gravis. Front Microbiol. 2018;9:2627.
Emdin CA, Khera AV, Kathiresan S. Mendelian Randomization. JAMA. 2017;318(19):1925–6.
Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10(4):486–96.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Greco MFD, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–40.
Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Lu Y, Tang H, Huang P, Wang J, Deng P, Li Y, et al. Assessment of causal effects of visceral adipose tissue on risk of cancers: a Mendelian randomization study. Int J Epidemiol. 2022;51(4):1204–18.
Ni J-J, Xu Q, Yan S-S, Han B-X, Zhang H, Wei X-T, et al. Gut Microbiota and Psychiatric Disorders: A Two-Sample Mendelian Randomization Study. Front Microbiol. 2021;12: 737197.
Li P, Wang H, Guo L, Gou X, Chen G, Lin D, et al. Association between gut microbiota and preeclampsia-eclampsia: a two-sample Mendelian randomization study. BMC Med. 2022;20(1):443.
Long Y, Tang L, Zhou Y, Zhao S, Zhu H. Causal relationship between gut microbiota and cancers: a two-sample Mendelian randomisation study. BMC Med. 2023;21(1):66.
Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98.
Yang Y, Ma X, Pang W, Jiang C. Causal associations of PM2.5 and GDM: a two-sample mendelian randomization study. Toxics. 2023. https://doi.org/10.3390/toxics11020171.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018. https://doi.org/10.7554/eLife.34408.
Burgess S, Thompson SG. Mendelian Randomization: Methods for Causal Inference Using Genetic Variants. Boca Raton: CRC Press; 2021.
Hemani G, Tilling K, Davey SG. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11): e1007081.
Xu H, Wu Z, Feng F, Li Y, Zhang S. Low vitamin D concentrations and BMI are causal factors for primary biliary cholangitis: a mendelian randomization study. Front Immunol. 2022;13:1055953.
Jiang L, Li J-C, Tang B-S, Guo J-F. Associations between gut microbiota and Parkinson disease: a bidirectional Mendelian randomization analysis. Eur J Neurol. 2023. https://doi.org/10.1111/ene.15848.
Xue H, Shen X, Pan W. Constrained maximum likelihood-based Mendelian randomization robust to both correlated and uncorrelated pleiotropic effects. Am J Hum Genet. 2021;108(7):1251–69.
Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116(11):2843–54.
Feferman T, Maiti PK, Berrih-Aknin S, Bismuth J, Bidault J, Fuchs S, et al. Overexpression of IFN-induced protein 10 and its receptor CXCR3 in myasthenia gravis. J Immunol. 2005;174(9):5324–31.
Christadoss P, Goluszko E. Treatment of experimental autoimmune myasthenia gravis with recombinant human tumor necrosis factor receptor Fc protein. J Neuroimmunol. 2002;122(1–2):186–90.
Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J. Myasthenia gravis: a review. Autoimmune Dis. 2012;2012: 874680.
Hemarajata P, Versalovic J. Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therap Adv Gastroenterol. 2013;6(1):39–51.
So J-S, Kwon H-K, Lee C-G, Yi H-J, Park J-A, Lim S-Y, et al. Lactobacillus casei suppresses experimental arthritis by down-regulating T helper 1 effector functions. Mol Immunol. 2008;45(9):2690–9.
Kim N, Kunisawa J, Kweon M-N, Eog Ji G, Kiyono H. Oral feeding of Bifidobacterium bifidum (BGN4) prevents CD4(+) CD45RB(high) T cell-mediated inflammatory bowel disease by inhibition of disordered T cell activation. Clin Immunol. 2007;123(1):30–9.
Shin DS, Jordan A, Basu S, Thomas RM, Bandyopadhyay S, de Zoeten EF, et al. Regulatory T cells suppress CD4+ T cells through NFAT-dependent transcriptional mechanisms. EMBO Rep. 2014;15(9):991–9.
Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci USA. 2009;106(46):19256–61.
Kuhn KA, Stappenbeck TS. Peripheral education of the immune system by the colonic microbiota. Semin Immunol. 2013;25(5):364–9.
Nagano Y, Itoh K, Honda K. The induction of Treg cells by gut-indigenous Clostridium. Curr Opin Immunol. 2012;24(4):392–7.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500(7461):232–6.
Couvigny B, de Wouters T, Kaci G, Jacouton E, Delorme C, Doré J, et al. Commensal Streptococcus salivarius modulates PPARγ transcriptional activity in human intestinal epithelial cells. PLoS ONE. 2015;10(5): e0125371.
Nettleford SK, Prabhu KS. Selenium and selenoproteins in Gut inflammation—a review. Antioxidants. 2018. https://doi.org/10.3390/antiox7030036.
Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106(2):189–97.
Hammad H, de Heer HJ, Soullié T, Angeli V, Trottein F, Hoogsteden HC, et al. Activation of peroxisome proliferator-activated receptor-gamma in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol. 2004;164(1):263–71.
Asadpoor M, Ithakisiou G-N, Henricks PAJ, Pieters R, Folkerts G, Braber S. Non-Digestible oligosaccharides and short chain fatty acids as therapeutic targets against enterotoxin-producing bacteria and their toxins. Toxins. 2021. https://doi.org/10.3390/toxins13030175.
Tran SM-S, Mohajeri MH. The role of gut bacterial metabolites in brain development, aging and disease. Nutrients. 2021. https://doi.org/10.3390/nu13030732.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6.
Wu W, Zhang L, Xia B, Tang S, Xie J, Zhang H. Modulation of pectin on mucosal innate immune function in pigs mediated by Gut microbiota. Microorganisms. 2020. https://doi.org/10.3390/microorganisms8040535.
Dandachi I, Anani H, Hadjadj L, Brahimi S, Lagier JC, Daoud Z, et al. Genome analysis of Lachnoclostridium phocaeense isolated from a patient after kidney transplantation in Marseille. New Microbes New Infect. 2021;41: 100863.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016. https://doi.org/10.1016/j.cell.2016.11.018.
Yang D, Su Z, Wu S, Bi Y, Li X, Li J, et al. Low antioxidant status of serum bilirubin, uric acid, albumin and creatinine in patients with myasthenia gravis. Int J Neurosci. 2016;126(12):1120–6.
Fuhua P, Xuhui D, Zhiyang Z, Ying J, Yu Y, Feng T, et al. Antioxidant status of bilirubin and uric acid in patients with myasthenia gravis. NeuroImmunoModulation. 2012;19(1):43–9.
Thye AY-K, Law JW-F, Tan LT-H, Thurairajasingam S, Chan K-G, Letchumanan V, et al. Exploring the Gut microbiome in myasthenia gravis. Nutrients. 2022. https://doi.org/10.3390/nu14081647.
Acknowledgements
We express our gratitude to the Core facility, and Bioinformatics Laboratory of The First Hospital of Jilin University for the training and generous sharing of experiences and codes.
Funding
This research was funded by the National Natural Science Foundation Committee of China Grant Number 82071351.
Author information
Authors and Affiliations
Contributions
All the authors contributed substantially to the manuscript. TF S drafted the manuscript and made substantial contributions to the conception and design of this study. XY, JX R, YL, and WGL Z made substantial, direct, and intellectual contributions to this study. LC agrees to be accountable for all aspects of the work and ensures that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics statement and consent to participate
Not applicable. The GWAS summary data on gut microbiota and myasthenia gravis were derived from published studies; therefore, no additional ethical approval was required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Original data used in this study, as well as the results of Mendelian randomization analysis, heterogeneity and pleiotropy tests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Su, T., Yin, X., Ren, J. et al. Causal relationship between gut microbiota and myasthenia gravis: a bidirectional mendelian randomization study. Cell Biosci 13, 204 (2023). https://doi.org/10.1186/s13578-023-01163-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01163-8