
Abstract
Background
Myasthenia gravis (MG) is an autoimmune disease that affects neuromuscular junction. The literature suggests the involvement of circulating cytokines (CK), gut microbiota (GM), and serum metabolites (SM) with MG. However, this research is limited to observational trials, and comprehensive causal relationship studies have not been conducted. Based on published datasets, this investigation employed Mendelian Randomization (MR) to analyze the known and suspected risk factors and biomarkers causal association of MG and its subtypes.
Methods
This research used two-sample MR and linkage disequilibrium score (LDSC) regression of multiple datasets to aggregate datasets acquired from the genome-wide association studies (GWAS) to assess the association of MG with 41-CK, 221-GM, and 486-SM. For sensitivity analysis and to validate the robustness of the acquired data, six methods were utilized, including MR-Egger regression, inverse variance weighting (IVW), weighted median, and MR-PRESSO.
Results
The MR method identified 20 factors significantly associated with MG, including 2 CKs, 6 GMs, and 9 SMs. Further analysis of the factors related to the two MG subtypes, early-onset MG (EOMG) and late-onset MG (LOMG), showed that EOMG had a high overlap with MG in the intestinal flora, while LOMG had a greater similarity in CKs and SMs. Furthermore, LDSC regression analysis indicated that Peptococcaceae, oxidized biliverdin, and Kynurenine had significant genetic correlations with general MG, whereas EOMG was highly correlated with Intestinibacter, while LOMG had significant genetic associations with Kynurenine and Glucose.
Conclusion
This research furnishes evidence for the potential causal associations of various risk factors with MG and indicates a heterogeneous relationship between CKs, GMs, and SMs with MG subtypes.
Similar content being viewed by others

Introduction
Myasthenia Gravis (MG), a chronic autoimmune disease, is manifested with serum auto-antibodies against the nicotinic acetylcholine receptor (AChR) at the neuromuscular junction (NMJ) in most patients. The main clinical symptoms of MG are the changes in the NMJ and damage caused by antibodies, leading to partial or systemic abnormal muscle fatigue and weakness [1, 2]. Its incidence per annum is about 4–12 per million, whereas the global prevalence is 40‒180 per million people [3, 4]. Depending on whether the patient is over 50 years old at the time of onset, MG can be divided into late-onset MG (LOMG) and early-onset MG (EOMG) subtypes [5]. With the rising prevalence and morbidity in the elderly population, especially LOMG, the mortality rate of MG has markedly increased [6]. Adjuvant antibody testing is time-consuming, relatively expensive, not readily available, and has a high false-negative result rate [1]. Furthermore, although immunosuppressive and conventional hormonal therapies can relieve symptoms, the MG recurrence rate is still quite high, and no consensus has been reached on the ideal therapeutic algorithm for MG [7]. Therefore, early identification and development of more specific therapies are critical for MG patient’s better quality of life.
A thorough understanding of the pathogenesis of MG is helpful for the diagnosis and treatment. Some studies have found genes related to genetic susceptibility to MG, such as HLA-DQ5, CTLA-4, etc., but the etiology of MG immune-related needs further study. The epidemiological literature has indicated the associations of MG with cytokines (CK) [8, 9], gut microbiotas (MG) [10, 11], serum metabolites (SM) [12, 13], circulating inflammatory proteins (CIF), and immune cell signatures (ICS). The causal associations of many of these remain undetermined, as most evidence was acquired from observational studies, which are often limited by recall, selection, confounding, reverse causation biases, and measurement errors.
In epidemiologic research, Mendelian randomization (MR) assessment has widely been used for assessing causal inference [14]. The MR analysis employs genetic variants as instrumental variables (IVs) to calculate exposures of interest for association analysis of disease outcomes. Since during gametogenesis, genetic alleles are classified randomly, MR resembles randomized clinical trials, minimizing or eliminating the potential for confusion and reverse causality bias common in traditional epidemiological research [15]. The associations of genetic variants with MG have been assessed in various genome-wide association studies (GWAS). The molecular analyses of peripheral T-cell traits have determined three T-cell traits as causally protective factors against MG [16]. Currently, studies for the evaluation of a large number of known and suspected MG risk factors are lacking. MR analysis could serve as an efficient tool for exploring causal relationships between new biomarkers and MG risk using the existing large data sets, which is rarely done in the field. Using the same methodology to assess these risk factors in one study will help compare the association strength and furnish a comprehensive understanding of MG etiology. Understanding the relationship between MG’s intrinsic subtypes and these risk factors may provide additional insights into its etiology and biology.
Therefore, this large MR research comprehensively elucidated the associations of 41-CK, 221-GM, and 486-SM with MG risk and assessed their associations with the EOMG and LOMG subtypes.
Materials and methods
Study design
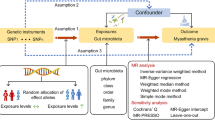
Figure 1 indicates the assumptions of this MR study. This research followed three basic assumptions: (1) genetic variants were related to exposure, (2) confounders and genetic variants are not associated, and (3) outcomes were only linked with genetic variants through exposure. The inverse variance-weighted (IVW) method was applied as the primary method, which equals a weighted regression of the SNP outcome effect on SNP exposure with a fixed y-intercept of zero [17]. The simple mode, weighted median, and MR-Egger test were determined to enhance the robustness of the data. Furthermore, to assess the robustness or potential bias of results, sensitivity analyses were carried out. These included the MR-Egger intercept test for pleiotropy, Cochrane’s Q test for heterogeneity, and leave-one-out analyses [18]. The heterogeneity analysis determined the differences between each IV, whereas the pleiotropy assessed the presence of horizontal pleiotropy.

The primary design and assumptions of the bidirectional Mendelian randomization research: (1) instrumental variables are closely linked with exposure, (2) instrumental variables and any confounding factors were independent, and (3) instrumental variables could influence outcomes only via exposure. SNP: single-nucleotide polymorphism
Data sources
The data were sourced from GWAS. Table 1 depicts the detailed information of various GWAS datasets. This investigation employed a two-sample MR method to elucidate the causal relationship of MG with 41 CK, 221 gut microbiotas (GM) (genus level), and 486 SM. In detail, the CK dataset comprised the meta-analysis summary statistics of GWAS on inflammatory CKs performed on 3 Finnish cohorts (FINRISK 1997, YFS, and 2002) [19]. For gut microbial taxa, the summary statistics were acquired from a multi-ethnic large-scale GWAS meta-analysis carried out by the MiBioGen consortium and included 19,790 participants from 18 cohorts [20]. MiBioGen is the most comprehensive effort for elucidating host-genetics vs. microbiome correlation on a population scale and comprises 211 taxa (131 genera, 35 families, 20 orders, 16 classes, and 9 phyla). The 486 human SM genetic datasets were acquired from the Metabolomics GWAS Server (http://metabolomics.helmholtz-muenchen.de/gwas/), which included 7824 European participants (KORA and TwinsUK cohorts) [21]. The MG-related genetic variants were identified from 3 datasets. The primary analysis MG dataset was obtained from the current largest meta-GWAS performed in Italy and the US (1,873 patients vs. 36,370 controls) [22]. Only MG patients who were anti-AChR antibody-positive (AChR+) were selected for this study, and those who were positive for muscle-specific kinase (MuSK+) antibodies were excluded. To specify the age-dependent genetic heterogeneity, summary statistics of LOMG (1,278 patients vs. 33,652 controls) and EOMG (595 patients vs. 2,718 controls, aged ≤ 40 years) were separately analyzed. The validation datasets included UK (http://www.nealelab.is/uk-biobank) (234 patients vs. 324,074 controls) and FIN (https://gwas.mrcieu.ac.uk/datasets/finn-b-G6 MYASTHENIA/) (232 patients vs. 217,056 controls) Biobanks [23]. The MG phenotype was identified using questionnaires completed by the participants; data on MG subtypes were not applicable. All original articles had received ethical approval and the participants had provided informed consent. This investigation included individuals of European ancestry to minimize population stratification bias.
SNP selection criteria
To identify the most representative, unbiased, and key genetic variables, various quality control procedures were carried out to assess eligible crucial single nucleotide polymorphisms (SNPs). (1) Based on minor allele frequency (MAF) > 0.01 and genome-wide significance (P < 5 × 108), SNPs related to the exposures were identified. (2) Considering that multiple SNPs might be present adjacent in linkage disequilibrium status, the Clumping step was carried out with \({r}^{2}\) < 0.001, and the window size = 10,000 kb based on the European 1000 Genome Project, and those with the lowest p-value were retained [24]. (3) The exposure SNPs were isolated from the outcome GWAS summary data. In case a specific exposure SNP was missing in the outcome GWAS, a proxy SNP in linkage disequilibrium was employed with the exposure SNP (minimum LD \({r}^{2}\) = 0.8). (4) Ambiguous SNPs, from which effect allele could not be assessed, were eliminated by harmonizing the outcome SNPs and exposure. Palindromic SNPs in the original datasets were reviewed separately to avoid undesired reverse effects. F-statistics evaluated the strength of the genetic instrument and a genetic variant with < 10 F-statistics for a specific IV was termed weak and not included in the MR analysis. The F-statistic was measured as follows:
where \(N\) = the sample size, \(k\) = the number of contained SNPs, and \({R}^{2}\) = the proportion of exposure variance explained by a particular genetic variant. R2 was assessed as follows:
where \(\beta\) = the estimated effect of the genetic variant, and EAF = the effect allele frequency [25]. Table 1 and S1 provide details of the genetic instruments used.
Two-sample mendelian randomization analysis
Figure 2 depicts the workflow of a two-sample MR analysis. Various MR methods were employed to elucidate the causal impact of identified exposure variables on the outcome. The primary analysis method was Inverse variance weighting (IVW), and all causality estimates were converted into odds ratios (OR) of dichotomous phenotypic outcomes. MR Egger, weight median [26], simple model, and MR-PRESSO [27] were performed at the same time. Sensitivity analysis was performed, which included the MR-Egger intercept test for pleiotropy [18], Cochrane’s Q test for heterogeneity, and leave-one-out analyses to assess the robustness and potential bias in the results. Heterogeneity tests were carried out to examine the differences between each IV, and pleiotropy tests examined the presence of horizontal pleiotropy. Finally, each exposure’s statistical power was elucidated using a two-sided type-I error rate α = 0.05 [28].

The Mendelian randomized study workflow revealed causal relationships between cytokines, gut microbiotas, and serum metabolites and the risk of myasthenia gravis. MR-PRESSO: MR pleiotropy residual sum and outliers, MR: Mendelian randomization, IVW: inverse variance weighting
Evaluation of generation of genetic correlation and directionality
Linkage Disequilibrium Score (LDSC) regression was implemented to elucidate the genetic association across exposures to explore the probability of a shared genetic architecture [29]. Pre-calculated LD scores of European participants in the 1000 Genomes project were used for high-quality HapMap3 SNPs. Through LDSC regression, the genetic correlation (rg) between the two traits was evaluated by combining GWAS summary statistics and the LD scores with a regression model. Furthermore, the Steiger test was performed to avoid the resulting bias caused by reverse causality [30].
Metabolic pathway analysis
For metabolic pathways analysis, web-based Metaconflict 5.0 (https://www.metaboanalyst.ca/) [31] was used. With the help of functional enrichment and pathway analysis modules, potential groups of metabolites or pathways linked with outcome biological processes were identified. This investigation used the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and for pathway analysis, the significance level was set at 0.10.
Statistical analysis
Two Sample MR package [17] was used for MR analysis in R version 4.2.2. Other packages for data processing and graph generation included ieugwasr, tidyverse [32], readr, ldscr, and forestplot.
Results
The detailed MR analysis results on the association of 41 CKs, 221 GMs, and 486 SMs exposures with overall MG, EOMG, and LOMG are presented in Supplement File 1–3. Furthermore, the MR analysis results of replicate data of exposure factors and overall MG are displayed in Supplement File 4–5.
Associations with general myasthenia gravis risk
Figure 3 shows the CKs, GMs, and SMs significantly associated with general MG risk. MR analysis revealed that the quantity of SNPs for all exposures ranged from 6 to 33, the proportion of variance explained (PVE, %) ranging from 3.3 to 25.9, and F-statistics for all exposures were > 10. IVW analysis showed that among the CK exposure factors, IL-2 RA and MCP 1 MCAF were substantially related to a reduced risk of general MG. In the GMs, six types of bacteria were significantly correlated with general MG. An increased level in Faecalibacterium and MollicutesRF9 were markedly linked with an increased risk of general MG, while Actinobacteria, FamilyXIII, Peptococcaceae, and Gammaproteobacteria were associated with a notably decreased risk of general MG. A total of nine SMs were significantly associated with general MG. Three of these factors (kynurenine, glycylvaline, and vanillin) were linked with an increased general MG risk, whereas six factors (prasterone sulfate, oxidized bilirubin, decanoylcarnitine, androsterone sulfate, methionine, and leucylleucine) were substantially associated with reduced risk of general MG (Fig. 1). Furthermore, it was found that metabolites significantly related to general MG were mainly enriched in methionine and cysteine metabolism (p = 0.08) and chlorophyll and porphyrin metabolism (p = 0.07) (Supplementary File 1). The estimated value of associations obtained from MR-PRESSO and MR-Egger regression analysis were consistent with the data of the IVW method (Supplementary Table 1). The MR-Egger intercept test, Cochrane’s Q test, and LOO analysis revealed no strong evidence of heterogeneity and directional horizontal pleiotropy (Fig. 1, Supplementary Fig. 1). Steiger’s directionality analysis revealed that all the data were consistent with the correct result orientation exposure.

Forest plot for the causal effect of cytokines, gut microbiotas, and serum metabolites on the risk of MG derived from inverse variance weighted (IVW); MG: myasthenia gravis; OR, odds ratio; CI, confidence interval
Associations with early-onset and late-onset MG risk
As Figs. 4 and 5 showed, fifteen and eighteen significant exposure factors were identified associated with EOMG and LOMG respectively, with SNP numbers ranging from 6 to 42 and PVE (%) values ranging from 3.5 to 23.0. In the analysis of CKs, EOMG was significantly correlated with VEGF, while LOMG was significantly correlated with IL-2 RA and MCP 1 MCAF, indicating protective effects. Furthermore, the results of LOMG were more consistent with general MG while compare with EOMG. The GM analysis revealed that EOMG and LOMG were markedly correlated with seven and five species, respectively. In EOMG, the OR value calculation results showed that Gammaproteobacteria, Intestinibacter, Defluviitaleaceae, Defluviitaleaceae, and Blautia were protective factors, while Coprobacter and Faecalibacterium were risk factors. Meanwhile, in LOMG analysis, Actinobacteria and FamilyXIII showed protective effects, whereas Rhodospirillaceae, Butyricicoccus, and Clostridia were pathogenic factors. Moreover, Gammaproteobacteria was indicated to serve as a protective factor in EOMG and general MG, while Faecalibacterium was a significant pathogenic factor. For LOMG and general MG, Actinobacteria and FamilyXIII were identified as protective factors. In the exploration of SM, seven and eleven metabolites were found in EOMG and LOMG, respectively, which were significantly correlated with outcome. Additionally, it was found that choline, docosapentaenoic acid, and 1-methylxanthine were risk factors when EOMG was the outcome event, and oxidized bilirubin, 5-dodecenoate, prasterone sulfate, biliverdin, and oleoylcarnitine were protective factors. The LOMG analysis indicated four metabolites as risk factors, including succinylcarnitine, glycylvaline, kynurenine, and O-methylascorbate, and seven were protective factors, including methionine, glucose, androsterone sulfate, decanoylcarnitine, xanthine, prasterone sulfate,and stearamide. Compared with the results of serum metabolite analysis of general MG showed that oxidized bilirubin, prasterone sulfate, and oleoylcarnitine were common significant causal factors for EOMG, whereas common significant associations factors for LOMG included glycylvaline, methionine, prasterone sulfate, kynurenine, androsterone sulfate, and decanoylcarnitine. Notably, prasterone sulfate was significantly associated with all three outcomes. MR-PRESSO results were consistent with the IVW method (Supplementary Tables 2–3). The test of pleiotropy and heterogeneity did not show statistical significance. Metabolic pathway analysis found that metabolites significantly associated with EOMG were enriched in the porphyrin and chlorophyll metabolism (p = 0.003) and caffeine metabolism (p = 0.03) pathways, and those associated with LOMG were enriched in the glycolysis /gluconeogenesis (p = 0.08) pathway (Supplementary files 2 and 3). LOO analysis demonstrated that the MR results were robust and reliable (Supplementary Figs. 2 and 3).

Forest plot for the causal effect of cytokines, gut microbiotas, and serum metabolites on the risk of EOMG derived from inverse variance weighted (IVW); EOMG: early-onset myasthenia gravis; OR, odds ratio; CI, confidence interval

Forest plot for the causal effect of cytokines, gut microbiotas, and serum metabolites on the risk of EOMG derived from inverse variance weighted (IVW); LOMG: late-onset myasthenia gravis; OR, odds ratio; CI, confidence interval
Overall, the results of MR, general MG, and EOMG analyses provided more significant causal factor coverage in the GM, and LOMG and general MG had more significant associations in CKs and SM. This suggests that different factors may be highly correlated with MG at different onset stages.
Results of genetic correlation analysis
The LDSC regression revealed that Peptococcaceae (rg = 0.653, se = 0.260, p = 0.012), oxidized biliverdin (rg = 0.296, se = 0.142, p = 0.037), and kynurenine (rg = 0.228, se = 0.105, p = 0.030) have a significant positive correlation with general MG. Whereas EOMG was positively correlated with Intestinibacter (rg = 1.240, se = 0.402, p = 0.002) and kynurenine (rg = 0.245, se = 0.111, p = 0.016) and glucose (rg = 0.341, se = 0.144, p = 0.017) demonstrated a positive genetic correlation with LOMG (Supplementary File 1–3).
Discussion
Myasthenia gravis belongs to the largest group of neuromuscular junction disorders. It is a B cell-mediated autoimmune disease caused by pathogenic autoantibodies against the postsynaptic muscle endplate and manifests with muscle fatigue and weakness [5, 33]. Although the prognosis for MG patients is good, and the treatments include immunosuppressive, symptomatic, and supportive treatment, 10‒15% of patients show forms of uncontrollable disease and almost all patients require long-term medication [34]. MG patients are categorized into subgroups based on prognosis, optimum therapy, and diagnosis. MG is a reversible disorder that can be treated with intensity and optimism. Therefore, identifying high-risk factors according to subgroups will help further understand their pathogenesis and the development of better prevention and treatment.
It has been suggested that the incidence of MG is increasing because of the alterations in external causative factors, including infection and diet [35]. Previous reports have found that MG may be associated with CKs, GMs, and SMs. Furthermore, we have summarized the relevant methods and conclusions of the MR studies related to MG in the past five years (Table 2). This research is the first to elucidate the genetic correlations and causalities between CKs, GMs, and SMs and the risk of general MG, EOMG, and LOMG using the GWAS dataset. First, the genetic instrumental variables, which were substantially associated with different outcomes, were selected through MR and sensitivity analyses. Furthermore, an enrichment analysis of metabolite pathways that were significantly associated was conducted, and LDSC was employed to analyze the genetic correlation between these significantly associated exposure factors and outcomes.
The results identified 20 significant exposure factors in general MG, including two CKs, six intestinal flora, and nine SMs. In the analysis of MG with different onset times, slightly different results were found from general MG, with fifteen and eighteen remarkably associated exposure factors found in EOMG and LOMG, respectively. Further analysis demonstrated that EOMG and general MG have a significant similarity in IM-related factors, while LOMG has a high degree of overlap in CKs and SMs, which indicates that there are differences in exposure factors related to the high risk of MG in different onset periods.
Myasthenia gravis and cytokines
The literature suggests that in MG, various CKs and lymphocytes induce pathogenic inflammation and autoantibodies at the neuromuscular junction. Therefore, treatment targeting CKs could benefit patients with refractory MG [43,44,45,46,47]. . Furthermore, it has been indicated that serum IL-21, a follicular Th cell-related CK [41], is linked with elevated QMG score in MG patients, which decreases after steroid treatment [48]. Moreover, the rate of IL-10 is increased after the treatment of MG [49]. IL-6, Th 22, and TNF-α have been reported to take part in the pathogenesis and treatment of MG [50, 51]. This research significantly associated IL-2 RA and MCP 1 MCAF with general MG and LOMG risk. However, VEGF was more significantly associated with EOMG. It has been found in many studies that IL-2 is elevated in the serum of MG patients, and the receptor of IL-2 has great potential in the current field of immunotherapy research [52, 53], which validates the results of this investigation. However, MCP1, MCAF, and VEGF have not been reported in MG studies, which may be the direction for further experimental verification.
Myasthenia gravis and intestinal microbiota
Recently, it has been suggested that alterations in the gut microbiome are associated with the incidence and development of MG, and some changes in the abundance of gut microbes may weaken or promote an immune response, suggesting potential diagnostic biomarkers for MG prevention, diagnosis, and treatment [54, 55]. Qiu et al. indicated that in comparison with the healthy cohort, the gut microbiota abundance of bacterial taxa was altered in the MG group, with a sharp decrease in microbial abundance, especially in Clostridium [56]. In addition, in MG patients, microbial dysbiosis was associated with serum levels of inflammatory biomarkers. Moris et al. found that the relative proportions of Verrucomicrobiaceae and Bifidobacteriaceae were lower in MG patients, while Bacteroidetes and Desulfovibrionaceae were increased [57]. However, the causal relationships between intestinal flora and MG have not been studied. Here, it was found that EOMG and general MG have more similar GM biomarkers, indicating that intestinal flora might be crucially involved in the early development of MG, which is conducive to the early diagnosis of MG. Many studies have indicated that Faecalibacterium, Actinobacteria, and Clostridia are related to the occurrence and development of MG, which is verified in this research [57,58,59].
Myasthenia gravis and serum metabolites
In recent years, metabolomic analysis has significantly increased the understanding of MG [60]. Derrick Blackmore et al. identified 5 library-matched unique and 7 putative metabolites, including 2-methylbutyrylglycine, 3-hydroxybenzoic acid, and 3-methoxytyramine [61]. Yonghai Lu used LC-MS combined with multivariate statistical analysis to identify and classify SM associated with MG. Different metabolic profiles were observed at EOMG and LOMG, and 9 biomarkers, including gamma-aminobutyric acid and 1-phosphosphingosine, were identified [62]. This MR analysis identified 486 SM; of these, 10 to 12 metabolites were substantially associated with general MG, EOMG, and LOMG, respectively, in addition to the as-yet-undefined metabolites. Furthermore, kynurenine showed a significant genetic correlation with general MG and LOMG in LDSC analysis, consistent with previous studies [63].
Limitations
This investigation has several limitations. (1) Although subgroup analyses of EOMG and LOMG were performed separately, other MG subgroups, including thymoma- and MUSK-associated MG, antibody-negative generalized MG, LRP4-linked with MG subtypes, and ocular MG, were not analyzed. However, the incidence of these subtypes is very low (2–4%), and they have no available GWAS data. Further analysis of subgroup risk factors can be conducted in the future. (2) Although the selected SNPs indicated a strong association, shared SNPs cannot be considered exact proxies for exposure as they do not adequately clarify the overall variance in complicated traits. (3) Research on other ethnicities should be performed to verify the study conclusions since all the GWAS data was on patients of European ancestry.
Conclusion
In summary, this comprehensive two-sample MR study employed recent instrument variables to support potential CKs, GMs, and SMs markers associated with MG risk, stratified by MG subtypes. Furthermore, this work furnishes evidence on various risk factors associated with MG risk, varying with the timing of MG onset, highlighting the heterogeneity of the disease, which may help identify biomarkers for MG screening and improve understanding of MG occurrence.
Data availability
The original contributions presented in the study are included in the article or Supplementary Material. Further inquiries can be directed to the corresponding authors.
References
Gilhus NE, Skeie GO, Romi F, et al. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259–68. https://doi.org/10.1038/nrneurol.2016.44
Lascano AM, Lalive PH. Update in immunosuppressive therapy of myasthenia gravis. Autoimmun Rev. 2021;20(1):102712. https://doi.org/10.1016/j.autrev.2020.102712
Hansen JS, Danielsen DH, Somnier FE, et al. Mortality in myasthenia gravis: a nationwide population-based follow-up study in Denmark. Muscle Nerve. 2016;53(1):73–7. https://doi.org/10.1002/mus.24697
Carr AS, Cardwell CR, McCarron PO, et al. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:46. https://doi.org/10.1186/1471-2377-10-46
Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023–36. https://doi.org/10.1016/S1474-4422(15)00145-3
Andersen JB, Engeland A, Owe JF, et al. Myasthenia gravis requiring pyridostigmine treatment in a national population cohort. Eur J Neurol. 2010;17(12):1445–50. https://doi.org/10.1111/j.1468-1331.2010.03089.x
Guptill JT, Soni M, Meriggioli MN, Current Treatment. Emerging translational therapies, and new therapeutic targets for Autoimmune Myasthenia Gravis. Neurotherapeutics. 2016;13(1):118–31. https://doi.org/10.1007/s13311-015-0398-y
Floege J, Lüscher B, Müller-Newen G. Cytokines and inflammation. Eur J Cell Biol. 2012;91(6–7):427. https://doi.org/10.1016/j.ejcb.2012.01.003
Borish LC, Steinke JW. 2. Cytokines and chemokines. J Allergy Clin Immunol. 2003;111(2 Suppl):460–75. https://doi.org/10.1067/mai.2003.108
Wastyk HC, Fragiadakis GK, Perelman D, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. 2021;184(16):4137–53.e14. https://doi.org/10.1016/j.cell.2021.06.019
Zhou CB, Zhou YL, Fang JY. Gut microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer. 2021;7(7):647–60. https://doi.org/10.1016/j.trecan.2021.01.010
Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–43. https://doi.org/10.1016/j.immuni.2013.04.005
Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. https://doi.org/10.1126/science.aaf1098
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. https://doi.org/10.1093/hmg/ddu328
Smith GD, Ebrahim S. Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. https://doi.org/10.1093/ije/dyg070
Zhong H, Jiao K, Huan X, et al. Herpesvirus entry mediator on T cells as a protective factor for myasthenia gravis: a mendelian randomization study. Front Immunol. 2022;13:931821. https://doi.org/10.3389/fimmu.2022.931821
Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. https://doi.org/10.7554/eLife.34408
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25. https://doi.org/10.1093/ije/dyv080
Ahola-Olli AV, Würtz P, Havulinna AS, et al. Genome-wide Association Study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. 2017;100(1):40–50. https://doi.org/10.1016/j.ajhg.2016.11.007
Wang J, Kurilshikov A, Radjabzadeh D, et al. Meta-analysis of human genome-microbiome association studies: the MiBioGen consortium initiative. Microbiome. 2018;6(1):101. https://doi.org/10.1186/s40168-018-0479-3
Shin SY, Fauman EB, Petersen AK, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543–50. https://doi.org/10.1038/ng.2982
Chia R, Saez-Atienzar S, Murphy N, et al. Identification of genetic risk loci and prioritization of genes and pathways for myasthenia gravis: a genome-wide association study. Proc Natl Acad Sci U S A. 2022;119(5). https://doi.org/10.1073/pnas.2108672119
Glanville KP, Coleman JRI, O’Reilly PF, et al. Investigating Pleiotropy between Depression and Autoimmune diseases using the UK Biobank. Biol Psychiatry Glob Open Sci. 2021;1(1):48–58. https://doi.org/10.1016/j.bpsgos.2021.03.002
Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. https://doi.org/10.1038/nature15393
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–52. https://doi.org/10.1093/ije/dyq151
Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in mendelian randomization with some Invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14. https://doi.org/10.1002/gepi.21965
Verbanck M, Chen CY, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–98. https://doi.org/10.1038/s41588-018-0099-7
Burgess S. Sample size and power calculations in mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol. 2014;43(3):922–9. https://doi.org/10.1093/ije/dyu005
Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–41. https://doi.org/10.1038/ng.3406
Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11):e1007081. https://doi.org/10.1371/journal.pgen.1007081
Chong J, Xia J. Using MetaboAnalyst 4.0 for Metabolomics Data Analysis, Interpretation, and Integration with Other Omics Data. Methods Mol Biol 2020;2104:337-60. https://doi.org/10.1007/978-1-0716-0239-3_17
Wickham H, Averick M, Bryan J, et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4(43). https://doi.org/10.21105/joss.01686
Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8(5):475–90. https://doi.org/10.1016/s1474-4422(09)70063-8
Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. 2013;86(2):255–60.
Pedersen EG, Hallas J, Hansen K, et al. Late-onset myasthenia not on the increase: a nationwide register study in Denmark, 1996–2009. Eur J Neurol. 2013;20(2):309–14. https://doi.org/10.1111/j.1468-1331.2012.03850
Fan Y, Huang H, Chen X, et al. Causal effect of vitamin D on myasthenia gravis: a two-sample mendelian randomization study. Front Nutr. 2023;10:1171830. https://doi.org/10.3389/fnut.2023.1171830
Sun D, Tu L, Wang X, et al. Association between COVID-19 and myasthenia gravis (MG): a genetic correlation and mendelian randomization study. Brain Behav. 2023;13(11):e3239. https://doi.org/10.1002/brb3.3239
Liu C, Mao C, Li S, et al. Myasthenia gravis and ischemic stroke: a bidirectional mendelian randomization study. Curr Neurovasc Res. 2023;20(2):270–79. https://doi.org/10.2174/1567202620666230703122140
Xie W, Li J, Du H, et al. Causal relationship between PCSK9 inhibitor and autoimmune diseases: a drug target mendelian randomization study. Arthritis Res Ther. 2023;25(1):148. https://doi.org/10.1186/s13075-023-03122-7
Li J, Wang F, Zhang C, et al. Genetically predicted effects of physical activity and sedentary behavior on myasthenia gravis: evidence from mendelian randomization study. BMC Neurol. 2023;23(1):299. https://doi.org/10.1186/s12883-023-03343-y
Li K, Ouyang Y, Yang H. Myasthenia gravis and five autoimmune diseases: a bidirectional mendelian randomization study. Neurol Sci. 2023. https://doi.org/10.1007/s10072-023-07163-3
Su T, Yin X, Ren J, et al. Causal relationship between gut microbiota and myasthenia gravis: a bidirectional mendelian randomization study. Cell Biosci. 2023;13(1):204. https://doi.org/10.1186/s13578-023-01163-8
Uzawa A, Kawaguchi N, Kanai T, et al. Serum high mobility group box 1 is upregulated in myasthenia gravis. J Neurol Neurosurg Psychiatry. 2015;86(6):695–7. https://doi.org/10.1136/jnnp-2014-309232
Uzawa A, Kawaguchi N, Kanai T, et al. Increased serum peroxiredoxin 5 levels in myasthenia gravis. J Neuroimmunol. 2015;287:16–8. https://doi.org/10.1016/j.jneuroim.2015.07.013
Xie Y, Li HF, Jiang B, et al. Elevated plasma interleukin-17A in a subgroup of Myasthenia Gravis patients. Cytokine. 2016;78:44–6. https://doi.org/10.1016/j.cyto.2015.06.011
Kang SY, Kang CH, Lee KH. B-cell-activating factor is elevated in serum of patients with myasthenia gravis. Muscle Nerve. 2016;54(6):1030–33. https://doi.org/10.1002/mus.25162
Uzawa A, Kanai T, Kawaguchi N, et al. Changes in inflammatory cytokine networks in myasthenia gravis. Sci Rep. 2016;6:25886. https://doi.org/10.1038/srep25886
Li Y, Rauniyar VK, Yin WF, et al. Serum IL-21 levels decrease with glucocorticoid treatment in myasthenia gravis. Neurol Sci. 2014;35(1):29–34. https://doi.org/10.1007/s10072-013-1460-3
Yilmaz V, Oflazer P, Aysal F, et al. B cells produce less IL-10, IL-6 and TNF-α in myasthenia gravis. Autoimmunity. 2015;48(4):201–7. https://doi.org/10.3109/08916934.2014.992517
Aricha R, Mizrachi K, Fuchs S, et al. Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis. J Autoimmun. 2011;36(2):135–41. https://doi.org/10.1016/j.jaut.2010.12.001
Robat-Jazi B, Hosseini M, Shaygannejad V, et al. High frequency of Tc22 and Th22 cells in Myasthenia Gravis patients and their significant reduction after Thymectomy. Neuroimmunomodulation. 2018;25(2):80–8. https://doi.org/10.1159/000490855
Huan X, Zhao R, Song J, et al. Increased serum IL-2, IL-4, IL-5 and IL-12p70 levels in AChR subtype generalized myasthenia gravis. BMC Immunol. 2022;23(1):26. https://doi.org/10.1186/s12865-022-00501-8
Shumei Y, Yi L, Huanyu M, et al. IL-2 gene polymorphisms affect tacrolimus response in myasthenia gravis. Eur J Clin Pharmacol. 2019;75(6):795–800. https://doi.org/10.1007/s00228-019-02642-z
Kang Y, Li L, Kang X, et al. Gut microbiota and metabolites in myasthenia gravis: early diagnostic biomarkers and therapeutic strategies. Clin Immunol. 2022;245:109173. https://doi.org/10.1016/j.clim.2022.109173
Kapoor B, Gulati M, Gupta R, et al. Microbiota dysbiosis and myasthenia gravis: do all roads lead to Rome? Autoimmun Rev. 2023;22(5):103313. https://doi.org/10.1016/j.autrev.2023.103313
Qiu D, Xia Z, Jiao X, et al. Altered gut microbiota in Myasthenia Gravis. Front Microbiol. 2018;9:2627. https://doi.org/10.3389/fmicb.2018.02627
Moris G, Arboleya S, Mancabelli L, et al. Fecal microbiota profile in a group of myasthenia gravis patients. Sci Rep. 2018;8(1):14384. https://doi.org/10.1038/s41598-018-32700-y
Totzeck A, Ramakrishnan E, Schlag M, et al. Gut bacterial microbiota in patients with myasthenia gravis: results from the MYBIOM study. Ther Adv Neurol Disord. 2021;14:17562864211035657. https://doi.org/10.1177/17562864211035657
Zheng P, Li Y, Wu J, et al. Perturbed Microbial Ecology in Myasthenia gravis: evidence from the gut microbiome and fecal metabolome. Adv Sci (Weinh). 2019;6(18):1901441. https://doi.org/10.1002/advs.201901441
de Candia P, De Rosa V, Gigantino V, et al. Immunometabolism of human autoimmune diseases: from metabolites to extracellular vesicles. FEBS Lett. 2017;591(19):3119–34. https://doi.org/10.1002/1873-3468.12733
Blackmore D, Li L, Wang N, et al. Metabolomic profile overlap in prototypical autoimmune humoral disease: a comparison of myasthenia gravis and rheumatoid arthritis. Metabolomics. 2020;16(1):10. https://doi.org/10.1007/s11306-019-1625-z
Sengupta M, Cheema A, Kaminski HJ, et al. Serum metabolomic response of myasthenia gravis patients to chronic prednisone treatment. PLoS ONE. 2014;9(7):e102635. https://doi.org/10.1371/journal.pone.0102635
Adamczyk-Sowa M, Bieszczad-Bedrejczuk E, Galiniak S, et al. Oxidative modifications of blood serum proteins in myasthenia gravis. J Neuroimmunol. 2017;305:145–53. https://doi.org/10.1016/j.jneuroim.2017.01.019
Acknowledgements
The authors thank all investigators of the three GWAS summary datasets used in this study, for sharing them publicly for research, and would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work has been supported by grants from China’s National Natural Science Foundation (Nos. 82373680, 82273728 and 82273729). National Natural Science Foundation of China,China.
Author information
Authors and Affiliations
Contributions
W, LH, WLX-JL F-JL W, H-ML Z-XH, W H-ML F-JL Z-XH WW-W, LW, CL and JL-X designed the study. WW-W, WG and JL-F analyzed the data and wrote the main manuscript text. ML-H and XH-Z collected data, searched literature and prepared figures. LW and CL co-supervised the study. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the ethical committees of the original GWAS studies analyzed in this study. All original studies have obtained ethical approval and informed consent from the participants. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Because the current study is based on published research and public databases, no additional ethical approval or consent to participate is required.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Consent for publication
Not applicable.
Publisher’s note
All claims expressed in this article are those of the author and do not necessarily represent the views of their affiliated organizations, nor those of publishers, editors and reviewers. Any product that may be evaluated in this article, or claimed, may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ling Wang is the main corresponding author and Chen Li is co-corresponding author.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, W., Ge, W., Feng, J. et al. Mendelian randomization analyses of known and suspected risk factors and biomarkers for myasthenia gravis overall and by subtypes. BMC Neurol 24, 33 (2024). https://doi.org/10.1186/s12883-024-03529-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-024-03529-y