
Abstract
Most osteosarcomas (OSs) develop from mesenchymal cells at the bone with abnormal growth in young patients. OS has an annual incidence of 3.4 per million people and a 60–70% 5-year surviving rate. About 20% of OS patients have metastasis at diagnosis, and only 27% of patients with metastatic OS survive longer than 5 years. Mutation of tumor suppressors RB1, TP53, REQL4 and INK4a and/or deregulation of PI3K/mTOR, TGFβ, RANKL/NF-κB and IGF pathways have been linked to OS development. However, the agents targeting these pathways have yielded disappointing clinical outcomes. Surgery and chemotherapy remain the main treatments of OS. Recurrent and metastatic OSs are commonly resistant to these therapies. Spontaneous canine models, carcinogen-induced rodent models, transgenic mouse models, human patient-derived xenograft models, and cell lines from animal and human OSs have been developed for studying the initiation, growth and progression of OS and testing candidate drugs of OS. The cell plasticity regulated by epithelial-to-mesenchymal transition transcription factors (EMT-TFs) such as TWIST1, SNAIL, SLUG, ZEB1 and ZEB2 plays an important role in maintenance of the mesenchymal status and promotion of cell invasion and metastasis of OS cells. Multiple microRNAs including miR-30/9/23b/29c/194/200, proteins including SYT-SSX1/2 fusion proteins and OVOL2, and other factors that inhibit AMF/PGI and LRP5 can suppress either the expression or activity of EMT-TFs to increase epithelial features and inhibit OS metastasis. Further understanding of the molecular mechanisms that regulate OS cell plasticity should provide potential targets and therapeutic strategies for improving OS treatment.
Similar content being viewed by others


Background
Osteosarcoma (OS) is the most common type of cancer that initiates at the bone, with a worldwide incidence of 3.4 per million people each year [1]. The 5-year survival rate for classic OS was only 20% during most of the twentieth century until the introduction of adjuvant chemotherapy in the 1970s [2]. The routine treatment of high-grade OS also shifted from amputation to chemotherapy and limb salvage by 1990, with a subsequent increase in overall survival rate to more than 65% [3, 4].
OS arises from primitively transformed cells with a mesenchymal origin [4]. The cancer cells in OS look like early forms of bone cells that normally help generate new bone tissues, but the bone tissues in OS are not as strong as that of normal bones [5]. Each year, about 800 to 900 new cases of OS are diagnosed in the United States [5]. Based on patient ages and causes of OS development, they can be classified into primary and secondary OSs. The primary OS typically develops in young patients as a result of abnormal bone development. The secondary OS occurs in patients over 65 years old, and is usually secondary to malignant conditions of Paget’s disease, post-irradiation exposure, severe bone infarct, osteochondroma and osteoblastoma [6]. Based on the location and appearance, OSs can be classified into intramedullary, juxtacortical, and extraskeletal OSs, and each of these OS types can be further divided into several subtypes [6]. Intramedullary OS is the most common and the fastest growing type, which accounts for nearly 80% of all OSs. Intramedullary OS develops in the medullary cavity of a long bone, such as femur and tibia. Furthermore, there are a number of subtypes of intramedullary OS according to the tumor cell types. The common subtypes include osteoblastic, chondroblastic, fibroblastic, small-cell, and epithelioid OSs. Juxtacortical OS is the second most common type, and they account for 10 to 15% of all OSs. This type of OS develops on the outer surface of the bone, or periosteum that is the dense layer of connective tissue covering the bone. Extraskeletal OS is rare and grows slowly, accounting for fewer than 5% of all OSs. Extraskeletal OS does not touch the bone and often arises from soft tissues that have experienced prior radiation therapy (Fig. 1a).

The types and subtypes (a) as well as the skeletal distribution (b) of osteosarcomas
Most OSs occur in children and young adults at ages from 10 to 30 years old, with the peak incidence in the second decade of life; however, people at any ages can develop OS [7]. OS is the third most common cancer in adolescence, accounting for approximately 3% of all adolescent cancers, with only lymphoma and brain tumor being more prevalent [1]. Adolescent OS usually develops in a region where the bone grows rapidly, such as the end region of the long leg or arm bones [8]. In some cases, OS occurs in the humerus, ulna, radius, fibula or pelvis (Fig. 1b) [9]. OS occurs slightly more common in boys versus girls, which may be related to the taller body height correlated with faster bone growth in boys on average. However, girls tend to develop OS slightly earlier, which may be related to their earlier growth spurt on average. The incidence of OS is higher in black population compared with white and other populations [10]. In the United States, more than 50% of all OS arising in patients over the age of 60 are the secondary OSs. Unlike OSs in children and young patients, OSs in elder patients more commonly develop at axial locations and in tissue areas that have previously received irradiation or have existing bone abnormalities [11]. OS patients older than 60 years are associated with a higher risk of metastatic disease [8]. Elder men are also associated with higher risks to develop OS than elder women do. In contrast to the OS disparity in black children, OS is more frequently observed in white elder adults compared with black and other elder adults [10]. The incidences of OS among 24-year-old and younger individuals are generally consistent in different countries in the world, with most cases diagnosed during puberty. However, the incidences of OS among elder men are higher in some countries including United Kingdom, Australia, and Canada compared with other countries [12].
Most OSs occur sporadically, and the exact causes for OS development are still not fully understood. Especially, the vast majority of OS cases in adolescents and young adults are sporadic with no known familial genetic or environmental causes [13,14,15]. However, it has been noticed that most OS cases occur in patients with certain rare inherited types of cancers or diseases such as retinoblastoma, Li–Fraumeni syndrome, and Rothmund–Thomson syndrome, which involve chromosomal abnormalities of the tumor-suppressors RB1 and TP53, as well as the DNA helicase REQL4 genes, respectively [6, 11, 16]. Individuals carrying germline RB1 mutations have approximately 1000-fold increased risk to develop OS [17]. Abnormalities in the CDKN2A gene, which codes for p16INK4a, a CDK4 inhibitor, and p14ARF, a p53 stabilizer, also increase the risk of OS development [6]. OS occurs more commonly at the regions of bone growth, which is presumably attributed to the genomic mutations acquired from the active cell proliferation. People receiving radiation therapies for treating other types of cancers may have a higher risk to develop OS later from the treated tissue sites. Indeed, people treated with radiotherapies at younger ages or received high irradiation doses have increased risks to develop OS [11]. In addition, people with certain non-cancerous bone diseases such as Paget’s disease and hereditary multiple osteochondromas also have increased risks to develop OS. Specifically, about 1% of Paget’s disease patients actually develop OS [11].
About 20% of OS have disseminated to the lung, brain or other organs at the time of diagnosis [18]. Chemotherapy and surgery are the most common treatments for most OS patients [1, 19]. Chemotherapies given before and/or after the surgery significantly reduce the risk of recurrence. The most commonly used chemotherapy drugs include methotrexate, doxorubicin and cisplatin, and two or more of these drugs are usually administrated in combination [19]. The 5-year survival rate of all-stage OSs is about 60%. However, if metastasis has happened at the time of diagnosis, the 5-year survival rate drops significantly to only 27%, which has not been improved significantly over the last four decades [16]. Recurrent or metastatic OSs are usually resistant to currently available standard treatments. Therefore, it is necessary to understand the detailed mechanisms responsible for OS development, progression and metastasis in order to identify novel therapeutic targets for treating OS.
Cellular and animal models for studying OS
Canine OS models
Spontaneous OSs are more common in large dogs compared with humans, making dog an attractive model to study this disease [20]. Canine OS is similar to that of human OS in terms of biological features and clinical symptoms [21, 22]. It is estimated that over 10,000 cases of canine OS occur annually in the United States. The major difference between canine OS and human OS is that canine OS is a disease of large breed elder dogs (6–12 years of age), which is considered as a limitation to use canine OS models to study human OS. The median disease-free intervals are 4 months after single surgery treatment, and 13 months after combined treatment of surgery and chemotherapy [23,24,25]. Many genomic alterations involved in human OS pathogenesis are also detected in canine OSs, such as the loss-of-function genetic alterations of the TP53 [26,27,28], RB [29] and PTEN [30] tumor suppressor genes in both human and canine OSs.
Radiation or chemical carcinogen induced OS models
Historically, rodent OS models began with the exposure of rats or mice to chemical and radioactive carcinogens [31, 32]. Murine OS models have been induced by exposing animals to radioactive substances such as radium, thorium and roentgen [33]. Despite their high incidences, these models probably represent secondary OSs developed in human patients who have received previous irradiation treatment, which might not share molecular mechanisms with primary OSs. Most of the chemically induced mouse OS models have been developed by injecting different chemical carcinogens directly into bones [33]. For example, rats treated with P32-orthophosphate have been shown to develop OS tumors that histologically resemble human OS [31, 34]. However, the carcinogen-induced murine model is more representative of a therapy-induced disease, while most human OSs are sporadic [35].
Animal OS-derived cell lines
Along with the rapid development of cancer immunotherapy, cancer cell lines isolated from spontaneously developed animal tumors have become important syngeneic models for studying immune suppression and activation, as well as interactions between immune and cancer cells in the same strain animals with identical genetic background (Table 1). The first batch of successfully established metastatic OS cell lines including K7, K8, K12, K14 and K37 were derived from a spontaneous OS in the distal femur of an 895-day-old female BALB/c mouse [34, 36,37,38,39,40]. These lines were metastatic in vivo and have been used for years to study the process of OS metastasis [41]. The Dunn cell line is another murine OS cell line derived from a spontaneous OS in the tail of a C3H/HeN mouse. This cell line is metastatic in vivo, and its xenograft tumors commonly metastasize to lung and liver in mice [42]. Multiple sublines derived from Dunn cell line have been used as OS angiogenesis and metastasis models in screening new compounds and testing candidate drugs [43,44,45,46]. The UMR 106-01 cell line was developed from a 32P-induced OS tumor in a Sprague-Dawley rat [32, 47]. This cell line has been well adopted to OS research due to its phenotypical similarities to human OS cells and rapid formation of pulmonary metastasis [48,49,50,51]. Besides murine, a number of cell lines have been derived from spontaneous canine OSs. Among these canine OS cell lines, the D-17 cell line was isolated from the lung metastasis of an 11-year-old female poodle. D-17 cells have been widely used in finding therapeutics for bone cancers in dogs [52, 53] (Table 1).
Human OS-derived cell lines
U2OS, the first human OS cell line, was established in the year of 1964, and has been used extensively in many in vitro studies. One limitation of this cell line is that it does not satisfy researchers’ needs for an in vivo metastatic model [54,55,56,57,58,59,60,61]. Unfortunately, this was also the case for other several subsequently established human OS cell lines such as HOS and SaOS2 cell lines [58, 62, 63]. In 2005, the HOS cells were treated with carcinogen or virus-mediated oncogene expression to induce genetic alterations, and the first metastatic human OS cell line 143B, along with several other derivative cell lines were established from these treated cells [58, 64,65,66,67,68,69,70,71]. Thereafter, many metastatic cell lines have been established in vitro, which have formed the basis for studying the cellular and molecular processes of OS (Table 1). A recent study characterized a set of 19 OS cell lines by profiling their gene expression and epigenetic patterns, and by comparing their differentiation, growth, invasion, and migration capacities in nude mice [72]. These valuable data should facilitate investigators to select appropriate OS cell lines for their researches.
Genetically engineered OS mouse models
Cell culture models may impose widespread genetic changes and loss of phenotypic heterogeneity that diverge from the characteristics of the original OSs. Taking SaOS2 as an example, OS cells maintained in culture demonstrate significant changes in phenotype over time [73]. In general, higher passage cells exhibit higher proliferation rates and lower alkaline phosphatase activity, while mineralization is more pronounced in late passage cells. Gene expression profiles may also change in culture over time. Genetically engineered mouse (GEM) models may help to provide spontaneously developed OS models with natural tumor environment for studying OS initiation, growth and metastasis.
The first GEM model of OS is the H2K-fos-tg mouse model, where c-fos is overexpressed in osteoblasts to induce OS development. The tumors developed in this model display similar histopathology to human osteoblastic OS, but these tumor cells do not produce distant metastasis that frequently occurs in human OSs [74]. Many murine OS models have been developed to recapitulate p53 and RB mutations in hereditary and sporadic human OSs [75]. Germ-line deletion of p53 results in an OS incidence of 4% in homozygous p53 null mice [76] and 25% in heterozygous p53 mice [77], indicating the importance of p53 loss in OS development. The higher OS incidence in heterozygous versus homozygous p53 knockout mice may be due to the development of other types of cancer that results in early death of the homozygous knockout animals. However, homozygous Rb knockout mice are lethal before birth, and heterozygous Rb knockout mice do not develop OS [78, 79].
The application of conditional gene manipulation and tissue specific Cre expression in mice have greatly enhanced the ability to induce OS from mesenchymal osteogenic cell lineages to model human OSs. Deletion of both Tp53 and Rb genes by Osterix-Cre leads to OS development with high penetrance [80]. Mice with Prx-1-Cre mediated deletion of both Tp53 and Rb genes induces OS development, and also generated poorly differentiated soft tissue sarcomas [81]. Mice with Osterix-Cre activated expression of a transgenic shRNA that targets p53 mRNA also develop osteoblastic OS at 100% penetrance. Although this model exhibits a longer latency to tumor onset, these OSs often develop in long bones and are highly metastatic to lung and liver. More importantly, this model does not develop any non-OS tumors [82].
In addition to c-fos and p53, other proteins such as TWIST1 [83], p14ARF [84], p16INK4a [85], PRKAR1A [86], and p21CIP [87] have been implicated in OS pathogenesis based on findings from human OS samples, and their involvements in OS development have also been demonstrated in GEM models. Their alterations appear to complement the defects in the p53 and Rb pathways. Although these models provide further insights into OS genetics and biology, the long latency combined with low penetrance makes these models less practical.
OS cell-derived xenograft models
Human or mouse OS cells are routinely inoculated into immunocompromised mice to grow xenografts and allografts as OS models. The injected cells usually develop solid tumors within days or weeks. The advantages of these models include quick onset, affordable cost, ease of handling and maintenance, and high reproducibility. Although OS cells are ectopically inoculated under the skin in some studies, cell grafts that grow orthotopically in or near bones are considered as more relevant preclinical OS models. These OS cell-derived xenograft models have been very useful in identification of factors that are involved in OS invasion and drug candidates that inhibit OS growth [36, 37, 41, 88]. An obvious limitation of these xenograft models is that they do not provide information about the initiation and etiology of OS since it uses fully immortalized OS cells.
Patient-derived xenograft (PDX) OS models
To establish a PDX line, small pieces of fresh tissue from either an incisional (open) biopsy or a percutaneous (needle) biopsy of an OS tumor in a patient are transplanted into multiple immune-defective mice to grow xenograft tumors [89]. In PDX models, the OS tumor cells are never cultured in vitro and always maintained in an OS-like tissue environment, and thus, PDX models are considered as a much more clinically relevant model to represent both the general features and heterogeneity of human OSs [90, 91]. The PDX models also allow the study of early-stage progression of OS metastasis in vivo. The PDX models have been broadly used for screening and testing drugs for developing new therapies [92,93,94]. However, like any other models, the PDX models are not perfect. Some initial generations of PDXs may take a long time to form tumors, suggesting that these OS cells still experience senescence, immortalization, and/or growth selection processes before a small population of tumor cells grows to form a tumor. Additionally, along with the tumor growth, most, if not all, of the stromal cells such as fibroblasts, immune cells, vascular cells and fat cells in the tumor environment are replaced with mouse cells, which means that the human OS tumor cells still grow in a mouse tissue and cellular environment.
The mesenchymal and epithelial plasticity in OS progression
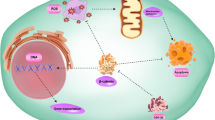
Epithelial-to-mesenchymal transition (EMT) and its reverse process, mesenchymal-to-epithelial transition (MET), are required for adapting cell plasticity in many physiological and pathophysiological processes, such as embryonic development, wound healing, fibrosis and cancer metastasis [95]. The cancer cells with an epithelial origin such as breast and prostate adenocarcinoma cells can undergo EMT to acquire mesenchymal gene markers and morphologies. Cancer cells at an EMT state are highly capable to escape from epithelial tumor cell clusters, invade into the stromal tissue, and disseminate to distant organs [96, 97]. Once getting in a distant organ, a MET process is thought to help the disseminated cancer cells to adapt the new tissue environment for proliferation and establishment of metastasis [96]. Cancer cells can have various degrees of EMT or MET statuses, ranging from full epithelial to full mesenchymal states (Fig. 2) [95]. Most sarcoma cells including OS cells exhibit an epithelial or a hybrid mesenchymal and epithelial phenotype. Since sarcoma cells are inherently locked in a mesenchymal state, they unlikely reprogram to a full epithelial state. However, depending on sarcoma’s histiotypes, they can reprogram their degrees of epithelial and mesenchymal states during their growth, progression and metastasis. Indeed, although the features of epithelial and mesenchymal plasticity are variable across OSs with different histiotypes, EMT and MET events are frequently observed and regulated by many molecular players [98].

The different cellular features associated with the different plasticity states of osteosarcoma (OS) cells. In this model, OS cells can maintain different states of hybrid epithelial/mesenchymal phenotypes characteristic of different expression levels of epithelial and mesenchymal markers. These OS cells can undergo either more epithelial or more mesenchymal transition states, which are correlated with different cellular features associated with the aggressiveness of OS. EMT, epithelial-to-mesenchymal transition; MET, mesenchymal-to-epithelial transition; V, vimentin; E, E-cadherin; N, nucleus. +++, strong or high; ++, moderate; +, weak; −, negative
EMT-promoting transcription factors (EMT-TFs) in OS and other sarcomas
EMT has been extensively studied in carcinomas in which EMT properties are associated with drug resistance, invasion and metastasis [96]. Sarcoma has a mesenchymal origin, and its mesenchymal phenotype is maintained by the functions of EMT-TFs, including TWIST1, SNAIL, SLUG, ZEB1 and ZEB2, and associated with more aggressive behaviors. Depending on different patient cohorts, variable percentages (32–56%) of human OSs have TWIST1 expression. The TWIST1-positve OS cells are associated with metastatic phase III OS tissues and also with poor clinical outcomes [99, 100]. SNAIL is widely expressed in OSs [101]. Knockdown of SNAIL in SaOS2 cells increases E-cadherin expression to promote MET, which is accompanied with decreased cell migratory and invasive properties. Conversely, overexpression of SNAIL in SaOS2 cells suppresses E-cadherin expression to promote EMT-like process, which promotes invasion and metastasis of these cells [102]. The expression of SLUG is associated with high-grade cranial bone OSs with high metastatic potentials [103]. Knockdown of SLUG in canine D-17 and human SaOS2 cells significantly decreases the migration and invasion capabilities of these OS cells by remodeling their actin cytoskeleton organization and disrupting their cellular protrusions. Knockdown of SLUG also inhibits the growth of these cell-derived xenografts in an in vivo chick chorioallantoic membrane (CAM) assay model. Ectopic expression of SLUG in these OS cells increases the expression level of WNT5A, decreases the expression level of the adhesion molecule osteoblast cadherin (OB-Cad), and increases cell motility by promoting the formation of actin-rich cellular protrusions [104]. The expression levels of ZEB1 protein in human sarcoma tissues are also positively correlated with lung metastasis, which is consistent with the finding showing that ZEB1 knockdown in MG-63 cells significantly inhibits cell invasive capability [105]. The mixed canine benign mammary tumors are composed of epithelial cells and cartilage or bone tissue, which is a species-specific type. The malignant canine mammary tumors include carcinomas, fibrosarcomas and OSs. In comparison with canine mammary epithelial cells and carcinomas, fibrosarcomas express high and OSs express even higher levels of a panel of homeobox genes including ZEB2. ZEB2 is an EMT-TF important for craniofacial bone formation [106]. Collectively, these studies suggest that the EMT-TFs play important roles in maintaining the mesenchymal status of sarcomas. Understanding the molecular mechanisms underlying these EMT-TFs regulated OS cell growth and metastasis may provide new opportunities to identify potential molecular targets for treating OSs.
MET in OS and other sarcomas
Since sarcomas have a mesenchymal cellular origin, more studies have been carried to understand the role of MET in sarcoma progression. Patients with more epithelial-like carcinomas tend to have better clinical outcomes compared with patients with more mesenchymal-like carcinomas, and a similar trend is also the case for patients with sarcomas. E-cadherin is the first epithelial marker detected in bone and soft tissue sarcomas [107]. A meta-analysis of 812 bone and soft tissue sarcoma tumors demonstrates that low E-cadherin expression is associated with poor five-year overall survival [108]. Ewing sarcoma/primitive neuroectodermal tumor (ES/PNET) cells frequently express epithelial markers such as cytokeratins, claudin-1 and ZO-1, and exhibit a partial epithelial differentiation state. This study showed evidence for expression of tight junction proteins such as claudin-1 and ZO-1 in over 50% of ES/PNET samples, suggesting partial epithelial differentiation in this kind of cancer [109]. It is also reported that patients with leiomyosarcomas that express high epithelial signature genes including E-cadherin also have a better prognosis [110, 111]. In OSs, E-cadherin expression levels have been found to be inversely correlated with metastasis potential but positively correlated with good prognosis [99]. However, it should be noted that the process of MET in sarcoma is characterized by increased expression of epithelial-like markers such as E-cadherin, whereas the typical mesenchymal markers including vimentin remain abundantly expressed in the sarcoma cells [111, 112].
The signaling pathways and EMT-TFs that regulate MET
Several signaling pathways can program a MET status of OS cells through regulating the transcriptional activities of EMT-TFs. These cross-talk regulatory networks are depicted in Fig. 3. Specifically, blocking Wnt/LDL receptor related protein 5 (LRP5) signaling by a soluble negative dominant form of LRP5 mutant in OS cells markedly upregulates the expression of E-cadherin, an epithelial marker, and downregulates the expression of N-cadherin, a mesenchymal marker. Inhibition of the Wnt/LRP5 signaling also downregulates the activity of EMT-TFs such as TWIST1 and SLUG [113]. Synovial sarcoma translocated-synovial sarcoma X1 and 2 (SYT-SSX1/2) interact with SNAIL and SLUG, respectively, to diminish their transcriptional repression activities on E-cadherin expression, resulting in an increase in E-cadherin expression and an acquisition of epithelial characteristics in synovial sarcoma cells [112]. In OS cells, the autocrine motility factor (AMF), also known as phosphoglucose isomerase (PGI), enhances SNAIL activity. Accordingly, silencing the expression of AMF/PGI can reduce SNAIL activity, which induces terminal differentiation of these OS cells into mature osteoblasts, resulting in suppression of the growth and pulmonary metastasis of these OS cell-derived xenografts in nude mice [114]. Ovo like zinc finger 2 (OVOL2) represses ZEB1 expression by binding to the ZEB1 promoter, so high OVOL2 expression is associated with low ZEB1 expression in human OS. In agreement with this finding, overexpression of OVOL2 in OS cells can promote MET and suppress cell migration and invasion [115]. In addition, miRNAs can directly regulate E-cadherin expression to induce MET or indirectly regulate E-cadherin expression through targeting EMT-TFs. For example, miR-30 and miR-9 can target TWIST1, SNAIL and ZEB1 mRNAs, and miR-23b, miR-29c, miR-194 and miR-200 can downregulate TWIST1 and ZEB1 mRNAs, resulting in upregulation of E-cadherin expression [116, 117]. It is interesting to notice that, expression of one mesenchymal factor is often sufficient to induce EMT in epithelial-derived carcinomas, while expression of two epithelial factors such as GRHL2 and miR-200 are required to drive MET in human rhabdomyosarcoma cells. This may suggest that the expression of epithelial genes in mesenchymal cells requires both transcriptional de-repression (e.g. via miR-200s) and transcriptional activation (e.g. via GRHL2) events to happen [118, 119].

The molecular regulatory mechanisms for MET in OS cells. The EMT-inducing transcription factors including SNAIL, SLUG, TWIST1 and ZEB1 are expressed in OS cells, which directly or indirectly repress the expression of epithelial genes such as E-cadherin to maintain mesenchymal cell features. MET is initiated by inhibiting EMT-TFs through activating upstream signaling pathways such as SYT-SSX1/2, OVOL2 and miRNAs or suppressing AMF/PGI or LRP5 in OS cells. Please refer to the text for related references. VDR vitamin D receptor, TIMP1 TIMP metallopeptidase inhibitor 1
Clinical implications and therapeutic opportunities of the EMT/MET plasticity of OS cells
The growing body of data on the regulation of sarcoma cell EMT/MET plasticity may offer clinical implications and therapeutic opportunities to patients. Specifically, the MET phenotype with epithelial marker expression in sarcomas may serve as prognostic markers. Withaferin-A (WFA), a naturally derived bioactive compound, is a vimentin inhibitor, which could be a promising drug against vimentin-expressing sarcoma cells. WFA treatment causes vimentin cleavage and induces sarcoma cell apoptosis. WFA also significantly inhibits growth, local recurrence and metastasis of the soft tissue sarcoma cell-derived xenografts in vivo [120]. This finding suggests that vimentin may be a good target for inhibiting soft tissue sarcomas.
EMT-TFs may also serve as potential therapeutic targets in sarcomas. It has been reported that TWIST1 is one of the 45 chemoresistant-signature genes that can predict OS patients’ response to neoadjuvant chemotherapy at the time of diagnosis. Knockdown of TWIST1 in multiple OS cell lines including HOS, SJSA-1 and 143B cells can largely overcome the chemoresistance of these cancer cells [121]. However, another study showed that TWIST1 can increase the chemosensitivity of SaOS2 and MG-63 OS cells to cisplatin treatment by downregulating endothelin-1 (ET-1) [122], because high ET-1 expression increases cell invasion and survival against cisplatin treatment in OS cells [123]. These different results from different studies might be due to the different functions of TWIST1-regulated target genes. Therefore, it may depend on specific OS cell context to determine whether TWIST1 can serve as a drug target. It has also demonstrated that OS cells that survived after cisplatin treatment at a sublethal dose exhibit a more mesenchymal phenotype and an elevated capacity to metastasize. Under these circumstances, inhibition of SNAIL can promote cisplatin sensitivity and prevent cisplatin treatment-induced EMT-like process, which results in diminished OS cell growth and survival [124]. Therefore, targeting these EMT-TFs in OS cells may help to induce MET and improve OS cell response to chemotherapy.
Conclusion remarks
Metastasis is the major cause of cancer-related death. When compared with numerous metastasis-related researches carried out for other cancer types such as breast and prostate cancers, much fewer studies of OS metastasis have been conducted so far. OS patients treated in recent years basically still receive essentially unchanged treatments applied in 1970s [16]. These may be due to the absence of consistent genetic mutations, and the relatively low incidence and high heterogeneity of the disease. Furthermore, our limited understanding of the biology about what drives OS cell dissemination from the primary bony sites and their subsequent proliferation at a second tissue environment such as the lung may have hindered our ability to develop new therapies for treating metastatic OSs. To impact the lives of patients suffering from metastatic OSs, it will be necessary to deepen our fundamental knowledge about OS metastasis and its specific vulnerabilities at cellular and molecular levels. The cell signaling pathways implicated in OS biology through genetic and other preclinical studies mainly include PI3K/mTOR [125], TGFβ [126], RANKL/NF-κB [127], and IGF [128]. Unfortunately, clinical studies evaluating the reagents that target these pathways have yielded disappointing results. Recently, although HER2 expression has been detected in certain OSs and tested as a therapeutic target in OS, targeting HER2 with trastuzumab, an FDA approved antibody drug for breast and gastric cancers with HER2 overexpression, is still not very effective to inhibit OS growth [129]. However, immunotherapy using HER2 chimeric antigen receptor (CAR) T-cells may be developed into a promising therapeutic approach for treating HER2-positive OSs [130]. Of note, the role of HER2 in the regulation of EMT/MET plasticity in OS cells is currently unknown.
It becomes obvious that new targets still need to be identified for developing new therapeutic strategies and drugs. Further understanding of the cell plasticity in OS progression could offer new opportunities to address these issues. EMT-TFs, especially TWIST1 and SLUG, play important roles in bone development and remodeling. These EMT-TFs regulate EMT and MET plasticity of OS cells, which is not identical to their regulations in solid tumors of epithelial origin [98]. Further investigation and deeper understanding of the EMT/MET-regulatory machineries in OS cells may help to identify druggable molecular targets. Furthermore, given the complexity of EMT/MET-like regulatory networks and the ability of cancer cells to adapt to stress conditions, targeting one protein or pathway may not be sufficient to completely impede EMT-related process or initiate MET. The future druggable targets that can be identified from the EMT/MET-like regulatory networks for treating metastasis of OS cells may be used in combination with the current surgery and chemotherapy treatments to achieve better clinical outcomes.
Availability of data and materials
Not applicable.
Abbreviations
- AMF:
-
Autocrine motility factor
- CAM:
-
Chick chorioallantoic membrane
- EMT:
-
Epithelial-to-mesenchymal transition
- EMT-TF:
-
Epithelial-to-mesenchymal transition transcription factor
- ES/PNET:
-
Ewing sarcoma/primitive neuroectodermal tumor
- ET-1:
-
Downregulating endothelin-1
- GEM:
-
Genetically engineered mouse
- LRP5:
-
LDL receptor related protein 5
- MET:
-
Mesenchymal-to-epithelial transition
- Meta:
-
Metastasis
- OB-cad:
-
Adhesion molecule osteoblast cadherin
- OS:
-
Osteosarcoma
- OVOL2:
-
Ovo like zinc finger 2
- PDX:
-
Patient-derived xenograft
- PGI:
-
Phosphoglucose isomerase
- SYT-SSX1/2:
-
Synovial sarcoma translocated-synovial sarcoma X 1/2
- TIMP1:
-
TIMP metallopeptidase inhibitor 1
- VDR:
-
Vitamin D receptor
- WFA:
-
Withaferin-A
References
Misaghi A, Goldin A, Awad M, Kulidjian AA. Osteosarcoma: a comprehensive review. SICOT J. 2018;4:12.
Eilber F, Giuliano A, Eckardt J, Patterson K, Moseley S, Goodnight J. Adjuvant chemotherapy for osteosarcoma: a randomized prospective trial. J Clin Oncol. 1987;5(1):21–6.
Simon MA, Aschliman MA, Thomas N, Mankin HJ. Limb-salvage treatment versus amputation for osteosarcoma of the distal end of the femur. J Bone Joint Surg Am. 1986;68(9):1331–7.
Gibbs CP Jr, Levings PP, Ghivizzani SC. Evidence for the osteosarcoma stem cell. Curr Orthop Pract. 2011;22(4):322–6.
Cokkinides V, Albano J, Samuels A, Ward M, Thum J. American cancer society: cancer facts and figures. Atlanta: American Cancer Society; 2005.
Kundu ZS. Classification, imaging, biopsy and staging of osteosarcoma. Indian J Orthop. 2014;48(3):238–46.
Ozaki T, Flege S, Liljenqvist U, Hillmann A, Delling G, Salzer-Kuntschik M, Jurgens H, Kotz R, Winkelmann W, Bielack SS. Osteosarcoma of the spine: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2002;94(4):1069–77.
Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, Kotz R, Salzer-Kuntschik M, Werner M, Winkelmann W, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002;20(3):776–90.
Tan JZ, Schlicht SM, Powell GJ, Thomas D, Slavin JL, Smith PJ, Choong PF. Multidisciplinary approach to diagnosis and management of osteosarcoma—a review of the St Vincent’s Hospital experience. Int Semin Surg Oncol. 2006;3:38.
Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer. 2009;115(7):1531–43.
Hayden JB, Hoang BH. Osteosarcoma: basic science and clinical implications. Orthop Clin N Am. 2006;37(1):1–7.
Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125(1):229–34.
Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, Schiffman JD. At-risk populations for osteosarcoma: the syndromes and beyond. Sarcoma. 2012;2012:152382.
Durfee RA, Mohammed M, Luu HH. Review of osteosarcoma and current management. Rheumatol Ther. 2016;3(2):221–43.
Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:548151.
Saraf AJ, Fenger JM, Roberts RD. Osteosarcoma: accelerating progress makes for a hopeful future. Front Oncol. 2018;8:4.
Rogers KM, Conran RM. Educational case: pediatric osteosarcoma. Acad Pathol. 2019;6:2374289519833902.
Miwa S, Shirai T, Yamamoto N, Hayashi K, Takeuchi A, Igarashi K, Tsuchiya H. Current and emerging targets in immunotherapy for osteosarcoma. J Oncol. 2019;2019:7035045.
Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop. 2006;30(6):445–51.
Misdorp W. Skeletal osteosarcoma. Animal model: canine osteosarcoma. Am J Pathol. 1980;98(1):285–8.
Mueller F, Fuchs B, Kaser-Hotz B. Comparative biology of human and canine osteosarcoma. Anticancer Res. 2007;27(1A):155–64.
Rankin KS, Starkey M, Lunec J, Gerrand CH, Murphy S, Biswas S. Of dogs and men: comparative biology as a tool for the discovery of novel biomarkers and drug development targets in osteosarcoma. Pediatr Blood Cancer. 2012;58(3):327–33.
Kirpensteijn J, Teske E, Kik M, Klenner T, Rutteman GR. Lobaplatin as an adjuvant chemotherapy to surgery in canine appendicular osteosarcoma: a phase II evaluation. Anticancer Res. 2002;22(5):2765–70.
Kirpensteijn J, Kik M, Rutteman GR, Teske E. Prognostic significance of a new histologic grading system for canine osteosarcoma. Vet Pathol. 2002;39(2):240–6.
Lascelles BD, Dernell WS, Correa MT, Lafferty M, Devitt CM, Kuntz CA, Straw RC, Withrow SJ. Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol. 2005;12(12):1073–83.
Kirpensteijn J, Kik M, Teske E, Rutteman GR. TP53 gene mutations in canine osteosarcoma. Vet Surg. 2008;37(5):454–60.
van Leeuwen IS, Cornelisse CJ, Misdorp W, Goedegebuure SA, Kirpensteijn J, Rutteman GR. P53 gene mutations in osteosarcomas in the dog. Cancer Lett. 1997;111(1–2):173–8.
Johnson AS, Couto CG, Weghorst CM. Mutation of the p53 tumor suppressor gene in spontaneously occurring osteosarcomas of the dog. Carcinogenesis. 1998;19(1):213–7.
Levine RA, Fleischli MA. Inactivation of p53 and retinoblastoma family pathways in canine osteosarcoma cell lines. Vet Pathol. 2000;37(1):54–61.
Levine RA, Forest T, Smith C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol. 2002;39(3):372–8.
Bensted JP, Blackett NM, Lamerton LF. Studies on the development of radiation-induced bone tumours. Acta Unio Int Contra Cancrum. 1959;15:559–60.
Martin TJ, Ingleton PM, Underwood JC, Michelangeli VP, Hunt NH, Melick RA. Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature. 1976;260(5550):436–8.
Jones KB. Osteosarcomagenesis: modeling cancer initiation in the mouse. Sarcoma. 2011;2011:694136.
Ek ET, Dass CR, Choong PF. Commonly used mouse models of osteosarcoma. Crit Rev Oncol Hematol. 2006;60(1):1–8.
Guijarro MV, Ghivizzani SC, Gibbs CP. Animal models in osteosarcoma. Front Oncol. 2014;4:189.
Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin Exp Metastasis. 2000;18(3):261–71.
Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, Trepel J, Meltzer P, Helman L. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res. 2001;61(9):3750–9.
Schmidt J, Strauss GP, Schon A, Luz A, Murray AB, Melchiori A, Aresu O, Erfle V. Establishment and characterization of osteogenic cell lines from a spontaneous murine osteosarcoma. Differentiation. 1988;39(3):151–60.
Buondonno I, Gazzano E, Jean SR, Audrito V, Kopecka J, Fanelli M, Salaroglio IC, Costamagna C, Roato I, Mungo E, et al. Mitochondria-targeted doxorubicin: a new therapeutic strategy against doxorubicin-resistant osteosarcoma. Mol Cancer Ther. 2016;15(11):2640–52.
Mortara L, Orecchia P, Castellani P, Borsi L, Carnemolla B, Balza E. Schedule-dependent therapeutic efficacy of L19mTNF-alpha and melphalan combined with gemcitabine. Cancer Med. 2013;2(4):478–87.
Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, Helman LJ. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10(2):182–6.
Dunn TB, Andervont HB. Histology of some neoplasms and non-neoplastic lesions found in wild mice maintained under laboratory conditions. J Natl Cancer Inst. 1963;31:873–901.
Okamoto T, Yamada N, Tsujimura T, Sugihara A, Nishizawa Y, Ueda H, Kashiwamura S, Tsutsui H, Futani H, Maruo S, et al. Inhibition by interleukin-18 of the growth of Dunn osteosarcoma cells. J Interferon Cytokine Res. 2004;24(3):161–7.
Obata H, Kuratsu S, Uchida A, Araki N, Myoui A, Ueda T, Yoshikawa H. Analysis of organ selectivity in the metastatic behavior of Dunn osteosarcoma. Clin Orthop Relat Res. 2002;398:212–22.
Asai T, Ueda T, Itoh K, Yoshioka K, Aoki Y, Mori S, Yoshikawa H. Establishment and characterization of a murine osteosarcoma cell line (LM8) with high metastatic potential to the lung. Int J Cancer. 1998;76(3):418–22.
Hara K, Kusuzaki K, Takeshita H, Kuzuhara A, Tsuji Y, Ashihara T, Hirasawa Y. Oral administration of 1 alpha hydroxyvitamin D3 inhibits tumor growth and metastasis of a murine osteosarcoma model. Anticancer Res. 2001;21(1A):321–4.
Ek ET, Dass CR, Contreras KG, Choong PF. Inhibition of orthotopic osteosarcoma growth and metastasis by multitargeted antitumor activities of pigment epithelium-derived factor. Clin Exp Metastasis. 2007;24(2):93–106.
Fisher JL, Mackie PS, Howard ML, Zhou H, Choong PF. The expression of the urokinase plasminogen activator system in metastatic murine osteosarcoma: an in vivo mouse model. Clin Cancer Res. 2001;7(6):1654–60.
Manolagas SC, Deftos LJ. Comparison of 1,25-, 25-, and 24,25-hydroxylated vitamin D3 binding in fetal rat calvariae and osteogenic sarcoma cells. Calcif Tissue Int. 1981;33(6):655–61.
Ng KW, Partridge NC, Niall M, Martin TJ. Epidermal growth factor receptors in clonal lines of a rat osteogenic sarcoma and in osteoblast-rich rat bone cells. Calcif Tissue Int. 1983;35(3):298–303.
Quan GM, Ojaimi J, Li Y, Kartsogiannis V, Zhou H, Choong PF. Localization of pigment epithelium-derived factor in growing mouse bone. Calcif Tissue Int. 2005;76(2):146–53.
Wolfesberger B, Tonar Z, Gerner W, Skalicky M, Heiduschka G, Egerbacher M, Thalhammer JG, Walter I. The tyrosine kinase inhibitor sorafenib decreases cell number and induces apoptosis in a canine osteosarcoma cell line. Res Vet Sci. 2010;88(1):94–100.
Nieves MA, Vahle J, Ackermann M, Howard M, Dietz AB, Carpenter SL, Cheville N. Production and characterization of canine osteosarcoma cell lines that induce transplantable tumors in nude mice. Am J Vet Res. 1998;59(3):359–62.
Eriksen MLI, Bunger EF. C: Bone morphogenetic protein-2 but not bone morphogenetic protein-4 and -6 stimulates chemotactic migration of human osteoblasts, human marrow osteoblasts, and U2-OS cells. Bone. 1996;18(1):53–7.
Agiostratidou G, Sgouros I, Galani E, Voulgari A, Chondrogianni N, Samantas E, Dimopoulos MA, Skarlos D, Gonos ES. Correlation of in vitro cytotoxicity and clinical response to chemotherapy in ovarian and breast cancer patients. Anticancer Res. 2001;21(1A):455–9.
Olkku A, Bodine PV, Linnala-Kankkunen A, Mahonen A. Glucocorticoids induce glutamine synthetase expression in human osteoblastic cells: a novel observation in bone. Bone. 2004;34(2):320–9.
Manara MC, Baldini N, Serra M, Lollini PL, De Giovanni C, Vaccari M, Argnani A, Benini S, Maurici D, Picci P, et al. Reversal of malignant phenotype in human osteosarcoma cells transduced with the alkaline phosphatase gene. Bone. 2000;26(3):215–20.
Rhim JS, Cho HY, Huebner RJ. Non-producer human cells induced by murine sarcoma virus. Int J Cancer. 1975;15(1):23–9.
Billiau A, Edy VG, Heremans H, Van Damme J, Desmyter J, Georgiades JA, De Somer P. Human interferon: mass production in a newly established cell line, MG-63. Antimicrob Agents Chemother. 1977;12(1):11–5.
Ponten J, Saksela E. Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer. 1967;2(5):434–47.
Mayr L, Pirker C, Lotsch D, Van Schoonhoven S, Windhager R, Englinger B, Berger W, Kubista B. CD44 drives aggressiveness and chemoresistance of a metastatic human osteosarcoma xenograft model. Oncotarget. 2017;8(69):114095–108.
Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;59(1):221–6.
Jia SF, Worth LL, Kleinerman ES. A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clin Exp Metastasis. 1999;17(6):501–6.
Luu HH, Kang Q, Park JK, Si W, Luo Q, Jiang W, Yin H, Montag AG, Simon MA, Peabody TD, et al. An orthotopic model of human osteosarcoma growth and spontaneous pulmonary metastasis. Clin Exp Metastasis. 2005;22(4):319–29.
McAllister RM, Nelson-Rees WA, Peer M, Laug WE, Isaacs H Jr, Gilden RV, Rongey RW, Gardner MB. Childhood sarcomas and lymphomas. Characterization of new cell lines and search for type-C virus. Cancer. 1975;36(5):1804–14.
Rhim JS, Putman DL, Arnstein P, Huebner RJ, McAllister RM. Characterization of human cells transformed in vitro by N-methyl-N’-nitro-N-nitrosoguanidine. Int J Cancer. 1977;19(4):505–10.
Rhim JS, Cho HY, Vernon ML, Arnstein P, Huebner RJ, Gilden RV. Characterization of non-producer human cells induced by Kirsten sarcoma virus. Int J Cancer. 1975;16(5):840–9.
Rhim JS, Kim CM, Arnstein P, Huebner RJ, Weisburger EK, Nelson Ress WA. Transformation of human osteosarcoma cells by a chemical carcinogen. J Natl Cancer Inst. 1975;55(6):1291–4.
Hensler PJ, Annab LA, Barrett JC, Pereira-Smith OM. A gene involved in control of human cellular senescence on human chromosome 1q. Mol Cell Biol. 1994;14(4):2291–7.
Rhim JS, Park DK, Arnstein P, Huebner RJ, Weisburger EK, Nelson-Rees WA. Transformation of human cells in culture by N-methyl-N’-nitro-N-nitrosoguanidine. Nature. 1975;256(5520):751–3.
Yuan J, Ossendorf C, Szatkowski JP, Bronk JT, Maran A, Yaszemski M, Bolander ME, Sarkar G, Fuchs B. Osteoblastic and osteolytic human osteosarcomas can be studied with a new xenograft mouse model producing spontaneous metastases. Cancer Invest. 2009;27(4):435–42.
Mohseny AB, Machado I, Cai Y, Schaefer KL, Serra M, Hogendoorn PC, Llombart-Bosch A, Cleton-Jansen AM. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab Invest. 2011;91(8):1195–205.
Hausser HJ, Brenner RE. Phenotypic instability of Saos-2 cells in long-term culture. Biochem Biophys Res Commun. 2005;333(1):216–22.
Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF. Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol. 1993;122(3):685–701.
Janeway KA, Walkley CR. Modeling human osteosarcoma in the mouse: from bedside to bench. Bone. 2010;47(5):859–65.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–21.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7.
Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7(4):480–4.
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359(6393):288–94.
Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008;22(12):1662–76.
Lin PP, Pandey MK, Jin F, Raymond AK, Akiyama H, Lozano G. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis. 2009;30(10):1789–95.
Mutsaers AJ, Ng AJ, Baker EK, Russell MR, Chalk AM, Wall M, Liddicoat BJ, Ho PW, Slavin JL, Goradia A, et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA. Bone. 2013;55(1):166–78.
Entz-Werle N, Choquet P, Neuville A, Kuchler-Bopp S, Clauss F, Danse JM, Simo-Noumbissie P, Guerin E, Gaub MP, Freund JN, et al. Targeted apc;twist double-mutant mice: a new model of spontaneous osteosarcoma that mimics the human disease. Transl Oncol. 2010;3(6):344–53.
Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448(7156):943–6.
Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413(6851):86–91.
Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J, Kashyap M, Hu P, Maiuri T, Narala SR, et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest. 2010;120(9):3310–25.
Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61(16):6234–8.
Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26(3):513–23.
Rainusso N, Cleveland H, Hernandez JA, Quintanilla NM, Hicks J, Vasudevan S, Marco RAW, Allen-Rhoades W, Wang LL, Yustein JT. Generation of patient-derived tumor xenografts from percutaneous tumor biopsies in children with bone sarcomas. Pediatr Blood Cancer. 2019;66(4):e27579.
Crnalic S, Hakansson I, Boquist L, Lofvenberg R, Brostrom LA. A novel spontaneous metastasis model of human osteosarcoma developed using orthotopic transplantation of intact tumor tissue into tibia of nude mice. Clin Exp Metastasis. 1997;15(2):164–72.
Kresse SH, Meza-Zepeda LA, Machado I, Llombart-Bosch A, Myklebost O. Preclinical xenograft models of human sarcoma show nonrandom loss of aberrations. Cancer. 2012;118(2):558–70.
Loh AHP, Stewart E, Bradley CL, Chen X, Daryani V, Stewart CF, Calabrese C, Funk A, Miller G, Karlstrom A, et al. Combinatorial screening using orthotopic patient derived xenograft-expanded early phase cultures of osteosarcoma identify novel therapeutic drug combinations. Cancer Lett. 2019;442:262–70.
Nomura M, Rainusso N, Lee YC, Dawson B, Coarfa C, Han R, Larson JL, Shuck R, Kurenbekova L, Yustein JT. Tegavivint and the beta-Catenin/ALDH Axis in chemotherapy-resistant and metastatic osteosarcoma. J Natl Cancer Inst. 2019;111(11):1216–27.
Lowery CD, Dowless M, Renschler M, Blosser W, VanWye AB, Stephens JR, Iversen PW, Lin AB, Beckmann RP, Krytska K, et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin Cancer Res. 2019;25(7):2278–89.
Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. 2019;49(3):361–74.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–36.
Sannino G, Marchetto A, Kirchner T, Grunewald TGP. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transition in mesenchymal tumors: a paradox in sarcomas? Cancer Res. 2017;77(17):4556–61.
Yin K, Liao Q, He H, Zhong D. Prognostic value of Twist and E-cadherin in patients with osteosarcoma. Med Oncol. 2012;29(5):3449–55.
Lei P, Ding D, Xie J, Wang L, Liao Q, Hu Y. Expression profile of Twist, vascular endothelial growth factor and CD34 in patients with different phases of osteosarcoma. Oncol Lett. 2015;10(1):417–21.
Yang H, Zhang Y, Zhou Z, Jiang X, Shen A. Snail-1 regulates VDR signaling and inhibits 1,25(OH)-D(3) action in osteosarcoma. Eur J Pharmacol. 2011;670(2–3):341–6.
Yang H, Zhang Y, Zhou Z, Jiang X, Shen A. Transcription factor Snai1-1 induces osteosarcoma invasion and metastasis by inhibiting E-cadherin expression. Oncol Lett. 2014;8(1):193–7.
Sharili AS, Allen S, Smith K, Hargreaves J, Price J, McGonnell I. Expression of Snail2 in long bone osteosarcomas correlates with tumour malignancy. Tumour Biol. 2011;32(3):515–26.
Sharili AS, Allen S, Smith K, Price J, McGonnell IM. Snail2 promotes osteosarcoma cell motility through remodelling of the actin cytoskeleton and regulates tumor development. Cancer Lett. 2013;333(2):170–9.
Shen A, Zhang Y, Yang H, Xu R, Huang G. Overexpression of ZEB1 relates to metastasis and invasion in osteosarcoma. J Surg Oncol. 2012;105(8):830–4.
Wensman H, Goransson H, Leuchowius KJ, Stromberg S, Ponten F, Isaksson A, Rutteman GR, Heldin NE, Pejler G, Hellmen E. Extensive expression of craniofacial related homeobox genes in canine mammary sarcomas. Breast Cancer Res Treat. 2009;118(2):333–43.
Sato H, Hasegawa T, Abe Y, Sakai H, Hirohashi S. Expression of E-cadherin in bone and soft tissue sarcomas: a possible role in epithelial differentiation. Hum Pathol. 1999;30(11):1344–9.
Wang N, He YL, Pang LJ, Zou H, Liu CX, Zhao J, Hu JM, Zhang WJ, Qi Y, Li F. Down-regulated E-cadherin expression is associated with poor five-year overall survival in bone and soft tissue sarcoma: results of a meta-analysis. PLoS ONE. 2015;10(3):e0121448.
Schuetz AN, Rubin BP, Goldblum JR, Shehata B, Weiss SW, Liu W, Wick MR, Folpe AL. Intercellular junctions in Ewing sarcoma/primitive neuroectodermal tumor: additional evidence of epithelial differentiation. Mod Pathol. 2005;18(11):1403–10.
Tian W, Wang G, Yang J, Pan Y, Ma Y. Prognostic role of E-cadherin and Vimentin expression in various subtypes of soft tissue leiomyosarcomas. Med Oncol. 2013;30(1):401.
Yang J, Eddy JA, Pan Y, Hategan A, Tabus I, Wang Y, Cogdell D, Price ND, Pollock RE, Lazar AJ, et al. Integrated proteomics and genomics analysis reveals a novel mesenchymal to epithelial reverting transition in leiomyosarcoma through regulation of slug. Mol Cell Proteomics. 2010;9(11):2405–13.
Saito T, Nagai M, Ladanyi M. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: a potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res. 2006;66(14):6919–27.
Guo Y, Zi X, Koontz Z, Kim A, Xie J, Gorlick R, Holcombe RF, Hoang BH. Blocking Wnt/LRP5 signaling by a soluble receptor modulates the epithelial to mesenchymal transition and suppresses met and metalloproteinases in osteosarcoma Saos-2 cells. J Orthop Res. 2007;25(7):964–71.
Niinaka Y, Harada K, Fujimuro M, Oda M, Haga A, Hosoki M, Uzawa N, Arai N, Yamaguchi S, Yamashiro M, et al. Silencing of autocrine motility factor induces mesenchymal-to-epithelial transition and suppression of osteosarcoma pulmonary metastasis. Cancer Res. 2010;70(22):9483–93.
Liu J, Wu Q, Wang Y, Wei Y, Wu H, Duan L, Zhang Q, Wu Y. Ovol2 induces mesenchymal-epithelial transition via targeting ZEB1 in osteosarcoma. Onco Targets Ther. 2018;11:2963–73.
Yang J, Du X, Wang G, Sun Y, Chen K, Zhu X, Lazar AJ, Hunt KK, Pollock RE, Zhang W. Mesenchymal to epithelial transition in sarcomas. Eur J Cancer. 2014;50(3):593–601.
Berlanga P, Munoz L, Piqueras M, Sirerol JA, Sanchez-Izquierdo MD, Hervas D, Hernandez M, Llavador M, Machado I, Llombart-Bosch A, et al. miR-200c and phospho-AKT as prognostic factors and mediators of osteosarcoma progression and lung metastasis. Mol Oncol. 2016;10(7):1043–53.
Ware KE, Gilja S, Xu S, Shetler S, Jolly MK, Wang X, Bartholf Dewitt S, Hish AJ, Jordan S, Eward W, et al. Induction of mesenchymal-epithelial transitions in sarcoma cells. J Vis Exp. 2017;122:e55520.
Somarelli JA, Shetler S, Jolly MK, Wang X, Bartholf Dewitt S, Hish AJ, Gilja S, Eward WC, Ware KE, Levine H, et al. Mesenchymal-epithelial transition in sarcomas is controlled by the combinatorial expression of microRNA 200s and GRHL2. Mol Cell Biol. 2016;36(19):2503–13.
Lahat G, Zhu QS, Huang KL, Wang S, Bolshakov S, Liu J, Torres K, Langley RR, Lazar AJ, Hung MC, et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS ONE. 2010;5(4):e10105.
Man TK, Chintagumpala M, Visvanathan J, Shen J, Perlaky L, Hicks J, Johnson M, Davino N, Murray J, Helman L, et al. Expression profiles of osteosarcoma that can predict response to chemotherapy. Cancer Res. 2005;65(18):8142–50.
Zhou Y, Zang X, Huang Z, Zhang C. TWIST interacts with endothelin-1/endothelin A receptor signaling in osteosarcoma cell survival against cisplatin. Oncol Lett. 2013;5(3):857–61.
Zhao Y, Liao Q, Zhu Y, Long H. Endothelin-1 promotes osteosarcoma cell invasion and survival against cisplatin-induced apoptosis. Clin Orthop Relat Res. 2011;469(11):3190–9.
Fang S, Yu L, Mei H, Yang J, Gao T, Cheng A, Guo W, Xia K, Liu G. Cisplatin promotes mesenchymal-like characteristics in osteosarcoma through Snail. Oncol Lett. 2016;12(6):5007–14.
Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA. 2014;111(51):E5564-73.
Rodriguez Calleja L, Jacques C, Lamoureux F, Baud’huin M, Tellez Gabriel M, Quillard T, Sahay D, Perrot P, Amiaud J, Charrier C, et al. DeltaNp63alpha silences a miRNA program to aberrantly initiate a wound-healing program that promotes TGFbeta-induced metastasis. Cancer Res. 2016;76(11):3236–51.
Chen Y, Di Grappa MA, Molyneux SD, McKee TD, Waterhouse P, Penninger JM, Khokha R. RANKL blockade prevents and treats aggressive osteosarcomas. Sci Transl Med. 2015;7(317):317ra197.
Kolb EA, Kamara D, Zhang W, Lin J, Hingorani P, Baker L, Houghton P, Gorlick R. R1507, a fully human monoclonal antibody targeting IGF-1R, is effective alone and in combination with rapamycin in inhibiting growth of osteosarcoma xenografts. Pediatr Blood Cancer. 2010;55(1):67–75.
Ebb D, Meyers P, Grier H, Bernstein M, Gorlick R, Lipshultz SE, Krailo M, Devidas M, Barkauskas DA, Siegal GP, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol. 2012;30(20):2545–51.
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–96.
Acknowledgements
Not applicable.
Funding
This work is partially supported by the Gordon Cain Endowed Professorship in Cell Biology at Baylor College of Medicine and the NIH Grant R01 CA193455 to J. Xu.
Author information
Authors and Affiliations
Contributions
JX and XY performed and conceived the review. XY wrote the manuscript and drew all the figures. JX and JY revised the manuscript and figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors do not have any conflict of interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, X., Yustein, J.T. & Xu, J. Research models and mesenchymal/epithelial plasticity of osteosarcoma. Cell Biosci 11, 94 (2021). https://doi.org/10.1186/s13578-021-00600-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00600-w