
Abstract
Background
The ability to image vascular endothelial growth factor (VEGF) could enable prospective, non-invasive monitoring of patients receiving anti-angiogenic therapy. This study investigates the specificity and pharmacokinetics of 111In-bevacizumab binding to VEGF and its use for assessing response to anti-angiogenic therapy with rapamycin.
Specificity of 111In-bevacizumab binding to VEGF was tested in vitro with unmodified radiolabelled bevacizumab in competitive inhibition assays. Uptake of 111In-bevacizumab in BALB/c nude mice bearing tumours with different amounts of VEGF expression was compared to that of isotype-matched control antibody (111In-IgG1κ) with an excess of unlabelled bevacizumab. Intratumoural VEGF was evaluated using ELISA and Western blot analysis. The effect of anti-angiogenesis therapy was tested by measuring tumour uptake of 111In-bevacizumab in comparison to 111In-IgG1κ following administration of rapamycin to mice bearing FaDu xenografts. Uptake was measured using gamma counting of ex vivo tumours and effect on vasculature by using anti-CD31 microscopy.
Results
Specific uptake of 111In-bevacizumab in VEGF-expressing tumours was observed. Rapamycin led to tumour growth delay associated with increased relative vessel size (8.5 to 10.3, P = 0.045) and decreased mean relative vessel density (0.27 to 0.22, P = 0.0015). Rapamycin treatment increased tumour uptake of 111In-bevacizumab (68%) but not 111In-IgGκ and corresponded with increased intratumoural VEGF165.
Conclusions
111In-bevacizumab accumulates specifically in VEGF-expressing tumours, and changes after rapamycin therapy reflect changes in VEGF expression. Antagonism of mTOR may increase VEGF in vivo, and this new finding provides the basis to consider combination studies blocking both pathways and a way to monitor effects.
Similar content being viewed by others


Background
Angiogenesis is central to tumour growth and invasion [1, 2]. As with all cancer therapies, anti-angiogenic therapy is only effective in a subgroup of patients and hence there is a need to develop new methods to predict and monitor response to treatment. In contrast to histological studies, radionuclide imaging has the advantage of offering non-invasive, prospective longitudinal assessment of angiogenesis, of the whole tumour and metastases, by targeting pathways involved in the process [3]. This is also important because of their toxicity and expense.
The vascular endothelial growth factor (VEGF) pathway is one of the key effectors of angiogenesis and primarily mediated by the interaction of VEGF with the receptor VEGF receptor 2 (VEGFR2) [4, 5]. High VEGF concentration in the blood or in tumour tissue has been associated with an elevated risk of recurrence, metastasis and poor survival [6, 7]. Imaging VEGF provides a tool for non-invasively assessing the levels of VEGF within tumour deposits. One method of imaging of VEGF involves radiolabelling bevacizumab (Avastin), a humanised [8] monoclonal immunoglobulin (IgG1κ) that binds all isoforms of human VEGF [9, 10]. Bevacizumab has been previously labelled with 89Zr [11, 12], 64Cu [13], 86Y [14], 124I [15], 125I [16] and 111In [12] for imaging. Of these, 111In- and 89Zr-labelled bevacizumab have been tested in clinical trials and have shown decreased tracer uptake in patients treated with anti-angiogenic agents [17, 18].
The inhibitors of the mammalian target of rapamycin (mTOR) have potent anti-angiogenic effect [19]. However, the effect of mTOR inhibitors on tumour vasculature in patients with renal and breast cancer remains unexplored. Also, despite several previous studies, the optimal time of imaging after administration of bevacizumab is unclear. In this context, we report here the results of in vitro and in vivo investigation to test the utility of 111In-bevacizumab in the detection of the response to the anti-angiogenic mTOR inhibitor rapamycin, analogues of which (everolimus) are used to treat renal and breast cancer.
Methods
Conjugation and 111In labelling of bevacizumab
Bevacizumab (25 mg/mL, Roche, USA) buffered with sodium bicarbonate (0.1 M, pH 8.2, Sigma-Aldrich) was reacted with 7-fold molar excess of 2-(4-isothiocyanatobenzyl) diethylenetriamine pentaacetic acid (DTPA, Macrocyclics, Dallas, USA) dissolved in anhydrous dimethyl sulfoxide (DMSO, Sigma-Aldrich) for 45 min at room temperature. Unreacted DTPA was removed by gel-filtration chromatography using a G-50 Sephadex (Sigma-Aldrich) column. Purified DTPA-bevacizumab was buffer-exchanged into sodium citrate (0.1 M, pH 5.0, Sigma-Aldrich) and incubated with 111In chloride (1–2 MBq/μg) (PerkinElmer) for 1 h at room temperature. Radiolabelling yield was measured using instant thin layer chromatography (ITLC) in sodium citrate (0.1 M, pH 5.0) and was always >95%.
Cell lines and xenografts
MDA-MB-231 (human breast adenocarcinoma, triple receptor negative) cell lines were a gift from Dr. Helen Sheldon (Weatherall Institute of Molecular Imaging, University of Oxford, UK). The cell lines were selected from clones infected with a retrovirus to express VEGF: 2F11 (244.7 ± 10.0 pg/mg protein, as determined by ELISA) and IE3 (3.6 ± 0.3 pg/mg protein). LS174T (human colorectal adenocarcinoma), 786-O (human renal adenocarcinoma), and FaDu (laryngeal carcinoma) cell lines were obtained from the American Type Culture Collection (ATCC). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% foetal bovine serum (FBS) (Gibco), L-glutamine (20 mM), penicillin G (100 u/mL) and streptomycin (100 mg/mL) (Sigma-Aldrich) 37 °C in 5% CO2 humidified atmosphere.
In vitro evaluation
The immunoreactivity of 111In-bevacizumab was assessed by competitive binding to VEGF expressed on 2F11 cells. Unlabelled bevacizumab (0.01 to 1000 nM) and 1 nM of 111In-bevacizumab were added to wells seeded with cells (2 × 105) and incubated at 4 °C for 2 h. Following removal of supernatant and washing, the cells were lysed using sodium hydroxide (250 μL, 0.1 M) and the radioactivity of the lysate was measured in an automated gamma-counter (Wizard, PerkinElmer).
In vivo biodistribution, kinetics and specificity
All animal procedures were performed in accordance with the Animals Scientific Procedures Act (ASPA) of 1986 (UK). Female athymic BALB/c nu/nu (nude) mice (Charles Rivers) were kept in a pathogen-controlled environment with access to food and water ad libitum. Xenografts were established by subcutaneous injection into the flank of mice with 1 × 106 (FaDu) or 4 × 106 cells (MDA-MB231 IE3, LS 174T and 786-O). Experiments were initiated once tumours reached around 7 mm in diameter. Kinetics of uptake were investigated in LS174T xenograft-bearing mice. The mice were euthanised 1, 3, 5 or 7 days after administration of 111In-bevacizumab (5 MBq/3 μg), and the organs and tumours were harvested, weighed and radioactivity measured.
Specificity of uptake was determined by administering either 111In-bevacizumab (5 MBq, 3 μg); an isotype-matched, 111In-labelled, non-specific antibody, 111In-IgG1κ (Southern Biotech) conjugated and labelled using the same method as for 111In-bevacizumab (5 MBq, 3 μg); or 111In-bevacizumab (5 MBq, 3 μg) plus a hundred fold (300 μg) excess of unlabelled bevacizumab. The animals were euthanised on day 5 when organs and tumour were excised to determine uptake of tracer and for protein and histological analyses.
The effect of rapamycin on 111In-bevacizumab uptake
An initial dose-escalation study was performed to determine the optimal dose of rapamycin in FaDu xenograft-bearing BALB/c mice. FaDu xenografts were chosen as they have moderate VEGF production and relatively consistent tumour growth rates. The mice received 1, 5 or 20 mg/kg of rapamycin or vehicle via intraperitoneal injection daily for 10 days. The mice were assessed for systemic side effects of therapy, and tumour sizes were measured daily. Daily 20 mg/kg rapamycin appeared to have greater growth inhibition than the other doses and did not lead to significant systemic effects in the mice.
Subsequently, FaDu xenograft-bearing mice (n = 6–7/group) received rapamycin (20 mg/kg; LC laboratories) or vehicle (DMSO, 5% TWEEN-80 (Sigma-Aldrich), 5% polyethylene glycol 300 (PEG300) (Sigma-Aldrich) and 0.9% NaCl (Braun)) by daily intraperitoneal injection for 10 days. On day 5 of rapamycin therapy, the mice received 111In-bevacizumab (5 MBq, 3 μg) or 111In-IgG1κ (5 MBq, 3 μg) intravenously. After treatment, the mice were euthanised. Tumour size was measured with callipers, and volume was calculated using the formula: V = (a × b 2)/2, where a and b are the largest and the smallest perpendicular diameters, respectively.
Autoradiography and immunohistochemistry
After resection, 8-μm frozen tumour sections were cut, applied to slides and then imaged in a storage phosphor-imager (Cyclone Plus, PerkinElmer). The distribution of VEGF and bound bevacizumab in frozen-embedded tumour sections were determined by immunohistochemistry using anti-VEGF (ab46154, Abcam) and anti-IgG (709-176-149, Jackson Laboratories) antibodies, respectively. Images were acquired using a confocal immunofluorescence microscope using ×100 magnification (LSM710, Zeiss).
VEGF quantification by ELISA
Homogenising buffer (ethylenediaminetetraacetic acid (EDTA, 1.5 mM) (Sigma-Aldrich), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 20 mM) (Sigma-Aldrich) and one protease inhibitor tablet (Complete Mini, Roche) per 10 mL, pH 7.5) was added to frozen tumour samples at 0.02 mL/mg. Following homogenisation, the suspension was centrifuged at 4 °C (3000g for 10 min). The supernatant was removed and ultracentrifuged at 4 °C (225,000g for 40 min). The supernatant was used for protein analysis. Protein concentrations were determined using the Bio-Rad colorimetric DC protein assay. Tumour VEGF concentration was determined using the VEGF Quantikine kit (R&D systems). VEGF levels were normalised for protein concentration.
VEGF isoform analysis by Western blotting
Homogenised samples from individual tumours were diluted with PBS to give protein concentrations of 0.25–0.4 mg/mL and run on a 4–12% Bis (2-hydroxyethyl) imino-tris (hydroxymethyl) methane-HCl (Bis-Tris) precast gel (Invitrogen). The following standard proteins were used: VEGF121 (4644-VS-010, R&D Systems), VEGF165 (293-VE-010, R&D Systems) and VEGF189 (ab106307, Abcam). Primary antibodies, rabbit anti-VEGF (sc152, Santa Cruz) and rabbit anti-beta actin (ab8227, Abcam), were added to the blot followed by a secondary antibody, goat, anti-rabbit horseradish peroxidase (HRP) conjugate (656120, Invitrogen). Analysis of blots was performed using ImageJ (National Institutes for Health).
Vessel analysis
Phycoerythrin (PE)-conjugated anti-mouse CD31 antibody (100 μL; Biolegend) was administered intravenously 10 min before the mice were euthanised. Immediately after resection, tumour specimens were examined using confocal microscopy (Leica Microsystems Ltd) as previously described [20]. For each image (Fig. 1a), a mask of the tumour region was created and contrast of fainter vessels was improved using histogram equalisation. Vessels were detected by applying a line operator [21], non-maximal suppression [22] and binary thinning to the images. The line operator was applied with an angular resolution of 4° and over a scale range of 5–20 pixels. This resulted in three output images: a binary image showing the central line of vessels (Fig. 1b), an image showing vessel orientation (Fig. 1c) and an image showing vessel width (Fig. 1d). Vessel density was then calculated as the sum of the vessel width image divided by the area of the image mask. Mean vessel size was calculated as the sum of the vessel width image divided by the sum of the binary line image using an in-house Matlab (Mathworks) programme.

An example of vessel analysis from CD31 immunohistochemistry images. a Original image. b The binary line image. c Orientation image indicating the angulation of vessels (scale represents degrees from 0 to 180). d Scale image representing the size of individual vessels (scale represents pixels)
SPECT-CT imaging
SPECT-CT imaging was performed using a nanoSPECT/CT system (Bioscan) equipped with parallel pinhole collimators. The mice (n = 6) were anaesthetised with isofluorane, and air mixture and imaging was conducted as reported before [23].
Statistics
All statistical analyses were performed using Prism version 5.04 (GraphPad Software) using either two-tailed t tests or one-way analysis of variances (ANOVAs) as appropriate. Pearson coefficients were used to determine correlations. For the competitive binding assay, a one site-fit logIC50 curve was used. P values of ≤0.05 were considered statistically significant. Data are plotted as means ± standard error of the mean (SEM), based on three independent experiments.
Results
Radiopharmaceutical synthesis
The synthesis of radiolabelled bevacizumab was performed successfully with yields of up to 20% and very high radiochemical purity (>95%). Binding of 111In-bevacizumab to VEGF was unaltered compared to unmodified antibody in several (>6) competitive binding assays (Additional file 1).
In vivo kinetics and biodistribution
111In-bevacizumab uptake in tumours peaked at day 5 post-injection (p.i.) (26.5 ± 3.2%ID/g, Additional file 2). In contrast, normal tissue uptake was maximal at day 1. For subsequent experiments and biodistribution imaging was performed at 5 days p.i.
Uptake of 111In-bevacizumab at 5 days p.i. was 14.3 ± 1.7, 22.3 ± 1.5 and 17.5 ± 0.9%ID/g in FaDu, LS 174T and 786-O xenografts, respectively (Fig. 2). Apart from the blood, the uptake in all normal organs was less than 10%ID/g, yielding good visualisation of the tumour. With respect to the FaDu xenograft, there was a wide range of uptakes demonstrated with a standard deviation of 8.1, which is reflected in the variation of mean uptakes between experiments.

Distribution of 111In-bevacizumab after 5 days in mice bearing different tumour xenografts. Black = FaDu, hatched = LS 174T, white = 786-O. Data are presented as mean ± SEM, n = minimum of 4 mice/group
Specificity
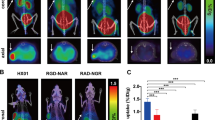
Uptake within tumours was not influenced by tumour mass (R = −0.06, P = 0.67) (Additional file 3). Tumour uptake of 111In-bevacizumab was greater than that of the non-specific control probe, 111In-IgG1κ (FaDu, 20.3 ± 1.4 versus 3.1 ± 0.5%ID/g; LS 174T, 19.6 ± 1.5 versus 2.8 ± 0.1%ID/g; 786-O, 17.5 ± 0.9 versus 4.4 ± 0.4%ID/g, P < 0.0001). When administered in combination with a 100-fold excess of non-radiolabelled bevacizumab, tumour uptake of 111In-bevacizumab was reduced compared to 111In-bevacizumab alone (FaDu, 20.3 ± 1.4 versus 7.2 ± 0.4%ID/g, P < 0.001; LS 174T, 19.6 ± 1.5 versus 8.8 ± 1.1%ID/g, P < 0.001; 786-O, 17.5 ± 0.9 versus 13.1 ± 0.4%ID/g, P < 0.05). Uptake of 111In-bevacizumab was low in xenografts derived from MDA-MB231 IE3 (2.1 ± 0.3%ID/g), which expresses low levels of VEGF (Fig. 3a). Taken together, these data indicate specific accumulation of 111In-bevacizumab in VEGF-expressing tumours. This is supported by selective SPECT-CT imaging of the mice (n = 6) bearing FaDu xenografts, which displayed intense uptake of tracer within the tumours in the mice injected with 111In-bevacizumab compared to the mice injected with 111In-IgG1κ (Fig. 3b).

a Specificity of uptake of 111In-bevacizumab. Uptake of 111In-bevacizumab (black); 111In-bevacizumab + excess of unlabelled bevacizumab (hatched) and 111In-IgG1κ (white) in different tumour models. Data are presented as mean ± SEM, *P < 0.05; ****P < 0.0001, n = minimum of 4 mice/group. b SPECT-CT transaxial image of FaDu xenograft-bearing mice injected with either 111In-bevacizumab or 111In-IgG1κ (tumours in circles)
Autoradiography of tumours removed from the animals following administration of 111In-bevacizumab was performed to qualitatively evaluate the intratumoural distribution of 111In (Fig. 4). Areas of relatively high radionuclide accumulation in tumours (indicated by dark areas on the autoradiographs) showed a similar distribution to bevacizumab. This suggests that intratumoural radioactivity results from accumulation of intact radiolabelled antibody. Further evidence that the antibody has not been substantially degraded is provided by the low uptake in the spleen and liver, which would have been expected to be higher if the tracer had dissociated in vivo and released free 111In into the blood (Fig. 2). There was no significant correlation between uptake of 111In-bevacizumab and intratumoural VEGF level (Additional file 4) (r = 0.20, P = 0.19). The similar distribution of 111In on autoradiography with immunostaining for VEGF (Fig. 4) suggests that the tracer binds specifically to tumour-associated VEGF.

Comparison of the distribution of VEGF, bevacizumab, and 111In in FaDu xenograft sections. The rows are examples of two sections from different FaDu xenografts injected with 111In-bevacizumab and an excess of unlabelled bevacizumab at 5 days. The left panels (green) are immunofluorescence images of VEGF staining, the centre panels (red) are immunofluorescence images of bevacizumab staining, and the right panels (grayscale) are autoradiographs of 111In distribution
Response to rapamycin therapy
FaDu tumour xenograft-bearing mice were treated with either rapamycin (20 mg/kg daily) or vehicle for 10 days (Fig. 5). Rapamycin led to tumour growth inhibition (140 ± 22 mm3 versus 547 ± 101 mm3, P = 0.0004). Representative images of tumour vasculature from the two groups are shown in Fig. 5b. We found a significant decrease in relative vessel density (0.27 ± 0.01 to 0.22 ± 0.02, P = 0.0015) and an increase in relative mean vessel size (8.5 ± 0.2 to 10.3 ± 0.4, P = 0.045) compared to vehicle-treated mice. These data are consistent with vascular normalisation [24], although the extensive vessel regression observed in our model would be expected to be associated with decreased rather than increased perfusion.

The effect of rapamycin on FaDu xenografts. a Changes in tumour volume with rapamycin therapy. On day 10, tumour volumes are significantly different (P = 0.0004). b Select images of CD31 confocal microscopy of tumours from mice treated with rapamycin. c Changes in relative mean vessel size with rapamycin therapy. d Changes of relative mean vessel density of rapamycin. e Uptake changes of 111In-bevacizumab and 111In-IgGκ in FaDu xenografts treated with rapamycin (20 mg/kg). The control groups were treated with vehicle injections. Data are presented as mean ± SEM, **P < 0.01, ***P < 0.001, ****P < 0.0001, n = 6–7/group
Tumour uptake of 111In-bevacizumab significantly increased (68%, P < 0.01) with rapamycin therapy (Fig. 5e). In contrast, 111In-IgG1κ exhibited very low uptake, which did not significantly change (P > 0.05) after rapamycin therapy, indicating that the increase in 111In-bevacizumab uptake is specific and not related to vascular changes.
Comparison with overall VEGF levels (measured by ELISA) within the tumours suggested that the increased uptake of 111In-bevacizumab was associated with an increase in VEGF (Fig. 6a) (from 283 ± 56 to 419 ± 47 pg/g of protein) measured by ELISA; however, this did not reach statistical significance (P = 0.07). This may be explained by analysis of Western blots (Fig. 6b), which demonstrated a significant increase in VEGF165 (P < 0.01) but no significant change in the VEGF121 and VEGF189 isoforms (Fig. 6c).

The effect of rapamycin therapy on VEGF levels in FaDu xenografts. a Changes in VEGF levels as measured by ELISA. Differences in levels were not statistically significant. b Changes in VEGF isoforms assessed by Western blot in FaDu xenografts treated with either rapamycin (20 mg/kg/day) or vehicle control for 10 days. c Changes in VEGF isoforms were measured using densitometry in the corresponding blots. Data are presented as mean ± SEM, **P < 0.01, n = 11/group
Discussion
Great progress has been made in the development of anti-angiogenic therapies targeting VEGF or its upstream targets, such as phosphoinositide 3-kinase (PI3K), mTOR and AKT [19, 24, 25]. However, these agents are only effective in a small and, at present, unpredictable subset of patients. This fact, coupled with high drug costs and adverse side effects has highlighted the need for accurate biomarkers for assessing angiogenesis and its response to therapy. Hence, non-invasive monitoring of angiogenesis by imaging is urgently needed to better assess and modify anti-angiogenic therapies. Successful implementation of imaging and treatment monitoring using radiotracers requires detailed knowledge of their pharmacokinetics and specificity. In this context, we investigated the role of 111In-bevacizumab in detecting response to anti-angiogenic therapy targeting mTOR.
Preclinical studies have provided evidence for the specific binding of 111In-bevacizumab to VEGF [16, 26]. However, in clinical trials involving patients with renal cell carcinoma (14 patients) [18] and metastatic colorectal cancer (12 patients) [27], no correlation was observed between uptake of tracer and intratumoural VEGF expression level, measured using either enzyme-linked immunosorbent assays (ELISAs), in situ hybridisation (ISH) or immunohistochemistry (IHC). In contrast, in a study of 111In-bevacizumab used in patients with melanoma [17], correlation between uptake of the tracer in the tumours and IHC of VEGF in tumour specimens was observed (nine patients).
Pharmacokinetic analyses in our study demonstrated that the greatest uptake and greatest tumour to background ratio occurs at around 5 days after injection. This differs from the data reported by Stollman et al. [16] who showed in the same murine tumour model that uptake was greatest on day 3 and the tumour to background ratios increased over the week with day 7 post-injection having the highest values. However, their work did not measure uptake values on day 5 and so may have missed the peak signal to background ratio. The overall prediction of our pharmacokinetics is that in patients, the optimal time for imaging would be 3–5 days post-injection. Our study corroborates a recent clinical study that demonstrated best visualisation and quantification of 111In-bevacizumab is 4 days post-injection in melanoma patients [17].
We showed that 111In-bevacizumab accumulated specifically in tumours. This was demonstrated in three VEGF-expressing xenograft models by suppression of uptake through competitive inhibition by an excess of unlabelled bevacizumab and by comparison with an 111In-labelled IgG isotype-matched control antibody. A direct relationship to VEGF, however, was not supported by comparing uptake with VEGF ELISAs of the tumour tissue. VEGF has many isoforms, and there are differences in their concentration, localisation and affinity for bevacizumab. Thus, a single ELISA or indeed other methods may not correlate with localisation of the antibody. Indeed, it may be that antibody uptake is the most important clinical variable to study for response, rather than VEGF and all its isoforms. We hypothesised that different isoforms of VEGF detected by 111In-bevacizumab would be predominantly matrix and cell membrane-associated VEGF (e.g., VEGF165 and VEGF189), whereas ELISA measures all isoforms, including diffusible forms such as VEGF121 [26]. This is supported by the spatial distribution of VEGF observed in tumour histological sections that co-localised with the tracer on autoradiography. In addition, uptake of 111In-bevacizumab has been shown to be only specific in blocking studies with tumours expressing VEGF165 and VEGF189 in comparison to those expressing VEGF121 [26]. Our results also are in agreement with the reports of recent clinical trials with111In-bevacizumab in patients with colorectal cancer liver metastases and renal cell carcinoma (RCC) that failed to identify a correlation between uptake of 111In-bevacizumab and VEGF levels measured by ELISA and ISH [18, 27]. Altogether, these data build on previous evidence supporting that circulating VEGF measurements have been of little value in monitoring or predicting response to bevacizumab in previous studies [28].
For the assessment of response of 111In-bevacizumab uptake to anti-angiogenic therapy, we used the mTOR inhibitor rapamycin at a dose previously demonstrated to have strong anti-angiogenic activity [29, 30]. Rapamycin led to significant tumour growth delay associated with profound vascular regression as reported before [31]. Therefore, the increased uptake of 111In-bevacizumab after treatment with rapamycin therapy is unlikely to be due to improved perfusion due to the strong anti-angiogenic effect. This contrasts with a prior clinical study where treatment with sorafenib, a multikinase inhibitor targeting mainly VEGF receptors, in RCC led to decreased 111In-bevacizumab uptake and did not correlate with VEGF levels [18]. This was hypothesised to be due to destruction of the vasculature, preventing tracer delivery to the tumour. This discrepancy is difficult to interpret, though may relate to the relative small size of tumours in mouse models compared to patients, where diffusion of the radiotracer may potentially overcome poor delivery by destroyed vasculature.
In our work, quantification of VEGF levels demonstrated a trend towards increased VEGF levels post-rapamycin, associated with an increase in uptake of 111In-bevacizumab. The lack of statistical significance in the increase of overall VEGF levels could be attributed to relative differences in VEGF isoforms. Indeed, the changes in VEGF levels were predominantly due to an increase in VEGF165, which may reflect the importance of VEGF165 over other isoforms in tumour angiogenesis [32]. VEGF121 and VEGF189 levels did not appear to change with rapamycin therapy. These results are surprising as they conflict with the mechanistic hypothesis and experimental evidence that mTOR inhibition decreases VEGF levels [30, 33, 34]. In particular, a similar study by van der Bilt et al. [35] used 89Zr-bevacizumab to assess response to everolimus in ovarian cancer xenografts. They showed that VEGF-A and 89Zr-bevacizumab uptake decreased in the xenografts after treatment with 10 mg/kg of everolimus. A potential explanation for this contrast is the dose of rapamycin used in our study could have led to increase in hypoxia by vascular regression and hence VEGF induction via activation of HIF-1α. Alternatively, it is well recognised that inhibition of mammalian target of rapamycin complex 1 (mTORC1) by rapamycin can result in a feedback upregulation of AKT and PI3K pathways [36, 37], and both pathways are known to increase VEGF expression [19, 38]. So it will be interesting in future studies to use 111In-bevacizumab to compare drugs that block both mTOR and this feedback activation.
Conclusions
Collectively, these data show that 111In-bevacizumab is a specific radiotracer to visualise VEGF within tumours and monitor response to anti-angiogenic therapy mediated by mTOR inhibition. 111In-bevacizumab was specific for matrix and cell membrane-associated forms of VEGF. This work provides important insight and support for further exploring 111In-bevacizumab for non-invasive imaging of VEGF in the clinical setting.
Abbreviations
- AKT:
-
Protein kinase B
- ANOVA:
-
Analysis of variance
- Bis-Tris:
-
[Bis (2-hydroxyethyl) imino-tris (hydroxymethyl) methane-HCl]
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- DTPA:
-
Diethylenetriamine pentaacetic acid
- ECM:
-
Extracellular matrix
- EDTA:
-
Ethylenediaminetetraacetic acid
- ELISA:
-
Enzyme-linked immunosorbent assay
- FBS:
-
Foetal bovine serum
- HEPES:
-
4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid
- HRP:
-
Horseradish peroxidase
- IC50 :
-
Half maximal inhibitory concentration
- IgG:
-
Immunoglobulin G
- IHC:
-
Immunohistochemistry
- ISH:
-
In situ hybridisation
- ITLC:
-
Instant thin layer chromatography
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
Mammalian target of rapamycin complex 1
- PEG:
-
Polyethylene glycol
- PI3K:
-
Phosphoinositide 3-kinase
- RCC:
-
Renal cell cancer
- SEM:
-
Standard error of the mean
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
References
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49.
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–30.
Dijkgraaf I, Boerman OC. Radionuclide imaging of tumor angiogenesis. Cancer Biother Radiopharm. 2009;24(6):637–47.
Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2(10):795–803.
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–27.
Hyodo I, Doi T, Endo H, Hosokawa Y, Nishikawa Y, Tanimizu M, et al. Clinical significance of plasma vascular endothelial growth factor in gastrointestinal cancer. Eur J Cancer. 1998;34(13):2041–5.
Lee JC, Chow NH, Wang ST, Huang SM. Prognostic value of vascular endothelial growth factor expression in colorectal cancer patients. Eur J Cancer. 2000;36(6):748–53.
Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57(20):4593–9.
Varey AH, Rennel ES, Qiu Y, Bevan HS, Perrin RM, Raffy S, et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer. 2008;98(8):1366–79.
Liang W-C, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J, et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281(2):951–61.
Nagengast WB, de Korte MA, Oude Munnink TH, Timmer-Bosscha H, den Dunnen WF, Hollema H, et al. 89Zr-Bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51(5):761–7.
Nagengast WB, de Vries EG, Hospers GA, Mulder NH, de Jong JR, Hollema H, et al. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med. 2007;48(8):1313–9.
Paudyal B, Paudyal P, Oriuchi N, Hanaoka H, Tominaga H, Endo K. Positron emission tomography imaging and biodistribution of vascular endothelial growth factor with 64Cu-labeled bevacizumab in colorectal cancer xenografts. Cancer Sci. 2011;102(1):117–21.
Nayak TK, Garmestani K, Baidoo KE, Milenic DE, Brechbiel MW. PET imaging of tumor angiogenesis in mice with VEGF-A targeted (86)Y-CHX-A″-DTPA-bevacizumab. Int J Cancer. 2011;128(4):920–6.
Christoforidis JB, Carlton MM, Knopp MV, Hinkle GH. PET/CT imaging of I-124–radiolabeled bevacizumab and ranibizumab after intravitreal injection in a rabbit model. Invest Ophthalmol Vis Sci. 2011;52(8):5899–903.
Stollman TH, Scheer MGW, Leenders WPJ, Verrijp KCN, Soede AC, Oyen WJG, et al. Specific imaging of VEGF-A expression with radiolabeled anti-VEGF monoclonal antibody. Int J Cancer. 2008;122(10):2310–4.
Nagengast WB, Hooge MNL-d, van Straten EME, Kruijff S, Brouwers AH, den Dunnen WFA, et al. VEGF-SPECT with 111In-bevacizumab in stage III/IV melanoma patients. Eur J Cancer. 2011;47(10):1595–602.
Desar IME, Stillebroer AB, Oosterwijk E, Leenders WPJ, van Herpen CML, van der Graaf WTA, et al. 111In-Bevacizumab imaging of renal cell cancer and evaluation of neoadjuvant treatment with the vascular endothelial growth factor receptor inhibitor sorafenib. J Nucl Med. 2010;51(11):1707–15.
Seeliger H, Guba M, Kleespies A, Jauch KW, Bruns CJ. Role of mTOR in solid tumor systems: a therapeutical target against primary tumor growth, metastases, and angiogenesis. Cancer Metastasis Rev. 2007;26(3-4):611–21.
Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res. 2012;72(1):239–48.
Zwiggelaar R, Parr TC, Taylor CJ. Finding orientated line patterns in digital mammographic images. Proc of 7th British Machine Vision Conference. 1996;715–24. http://www.bmva.org/bmvc/1996/zwiggelaar_1.html.
Sonka M, Hlavac V, Boyle R. Image processing, analysis and machine vision. London: Chapman and Hall; 1993.
Cornelissen B, Able S, Kersemans V, Waghorn PA, Myhra S, Jurkshat K, et al. Nanographene oxide-based radioimmunoconstructs for in vivo targeting and SPECT imaging of HER2-positive tumors. Biomaterials. 2013;34(4):1146–54.
Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–27.
Fokas E, Yoshimura M, Prevo R, Higgins G, Hackl W, Maira SM, et al. NVP-BEZ235 and NVP-BGT226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat Oncol. 2012;7:48.
Stollman TH, Scheer MGW, Franssen GM, Verrijp KN, Oyen WJG, Ruers TJM, et al. Tumor accumulation of radiolabeled bevacizumab due to targeting of cell- and matrix-associated VEGF-A isoforms. Cancer Biother Radiopharm. 2009;24(2):195–200.
Scheer MGW, Stollman TH, Boerman OC, Verrijp K, Sweep FCGJ, Leenders WPJ, et al. Imaging liver metastases of colorectal cancer patients with radiolabelled bevacizumab: lack of correlation with VEGF-A expression. Eur J Cancer. 2008;44(13):1835–40.
Garcia-Donas J, Rodriguez-Antona C, Jonasch E. Molecular markers to predict response to therapy. Semin Oncol. 2013;40(4):444–58.
Huynh H, Chow PK, Palanisamy N, Salto-Tellez M, Goh BC, Lee CK, et al. Bevacizumab and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J Hepatol. 2008;49(1):52–60.
Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, et al. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71(5):1573–83.
Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8(2):128–35.
Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18(1):4–25. doi:10.1210/er.18.1.4.
Medici D, Olsen BR. Rapamycin inhibits proliferation of hemangioma endothelial cells by reducing HIF-1-dependent expression of VEGF. PLoS ONE. 2012;7(8):e42913.
van Asselt SJ, Oosting SF, Brouwers AH, Bongaerts AHH, de Jong JR, Lub-de Hooge MN, et al. Everolimus reduces 89Zr-bevacizumab tumor uptake in patients with neuroendocrine tumors. J Nucl Med. 2014;55(7):1087–92.
van der Bilt ARM, van Scheltinga AGTT, Timmer-Bosscha H, Schröder CP, Pot L, Kosterink JGW, et al. Measurement of tumor VEGF-A levels with 89Zr-bevacizumab PET as an early biomarker for the antiangiogenic effect of everolimus treatment in an ovarian cancer xenograft model. Clin Cancer Res. 2012;18(22):6306–14.
Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 2014;13(11):2477–88.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–74.
Castellino RC, Durden DL. Mechanisms of disease: the PI3K-Akt-PTEN signaling node—an intercept point for the control of angiogenesis in brain tumors. Nat Clin Pract Neuro. 2007;3(12):682–93.
Funding
This work was funded through the EPSRC/CRUK Oxford Cancer Imaging Centre.
Authors’ contributions
NP, KV and AH contributed to the study conception and design. KV, AH and FG contributed to the project supervision. NP, EF, DA and SA contributed to the acquisition of the data. NP, DA, KV, AH and BC contributed to the analysis and interpretation of the data. NP, DA and SA contributed to the drafting of the manuscript. EF, KV, AH and BC the contributed to critical revision. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
This article does not contain any studies with human participants performed by any of the authors.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Competitive binding of 111In-bevacizumab to VEGF. The IC50 value for unlabeled bevacizumab was 0.84 ± 0.45 nM when competed with 1 nM of 111In-bevacizumab for binding to cells expressing VEGF. Data are presented as mean ± SEM, n = 12.
Additional file 2:
Kinetics of uptake of 111In-bevacizumab in LS174T xenografts and organs in female BALB/c nude mice. ANOVA demonstrate a statistically significant difference between the means of each day (P = 0.03). Error bars are SEM, n = 4/group.
Additional file 3:
Comparison of tumour mass with uptake of 111In-bevacizumab. There is no correlation.
Additional file 4:
Comparison of VEGF in tumours, as measured by ELISA, with uptake of 111In-bevacizumab. There is no correlation.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Patel, N., Able, S., Allen, D. et al. Monitoring response to anti-angiogenic mTOR inhibitor therapy in vivo using 111In-bevacizumab. EJNMMI Res 7, 49 (2017). https://doi.org/10.1186/s13550-017-0297-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13550-017-0297-9