
Abstract
Background
While ageing is associated with increased insulin resistance (IR), the molecular mechanisms underlying increased IR in the muscle, the primary organ for glucose clearance, have yet to be elucidated in older individuals. As epigenetic processes are suggested to contribute to the development of ageing-associated diseases, we investigated whether differential DNA methylation was associated with IR in human primary muscle stem cells (myoblasts) from community-dwelling older individuals.
Methods
We measured DNA methylation (Infinium HumanMethylationEPIC BeadChip) in myoblast cultures from vastus lateralis biopsies (119 males/females, mean age 78.24 years) from the Hertfordshire Sarcopenia Study extension (HSSe) and examined differentially methylated cytosine phosphate guanine (CpG) sites (dmCpG), regions (DMRs) and gene pathways associated with HOMA2-IR, an index for the assessment of insulin resistance, and levels of glycated hemoglobin HbA1c.
Results
Thirty-eight dmCpGs (false discovery rate (FDR) < 0.05) were associated with HOMA2-IR, with dmCpGs enriched in genes linked with JNK, AMPK and insulin signaling. The methylation signal associated with HOMA2-IR was attenuated after the addition of either BMI (6 dmCpGs), appendicular lean mass index (ALMi) (7 dmCpGs), grip strength (15 dmCpGs) or gait speed (23 dmCpGs) as covariates in the model. There were 8 DMRs (Stouffer < 0.05) associated with HOMA2-IR, including DMRs within T-box transcription factor (TBX1) and nuclear receptor subfamily-2 group F member-2 (NR2F2); the DMRs within TBX1 and NR2F2 remained associated with HOMA2-IR after adjustment for BMI, ALMi, grip strength or gait speed. Forty-nine dmCpGs and 21 DMRs were associated with HbA1c, with cg13451048, located within exoribonuclease family member 3 (ERI3) associated with both HOMA2-IR and HbA1c. HOMA2-IR and HbA1c were not associated with accelerated epigenetic ageing.
Conclusions
These findings suggest that insulin resistance is associated with differential DNA methylation in human primary myoblasts with both muscle mass and body composition making a significant contribution to the methylation changes associated with IR.
Similar content being viewed by others

Background
Insulin resistance (IR) and impaired insulin secretion are hallmarks of type 2 diabetes mellitus (T2D) [1] and are common in older adults with multiple long-term conditions. Concomitant with an increasingly ageing population, the prevalence of IR and T2D are rising exponentially, with the incidence of T2D amongst older adults currently more than twice that of middle-aged adults [2]. The aetiology of IR and T2D is multifactorial, involving a complex integration of both genetics and environmental exposures [3, 4]. Genetic factors identified by GWAS that influence the pathogenesis of T2D currently explain less than 20% of the heritability [5, 6], with environmental factors that include ageing [7], sedentary behaviour [8, 9], poor nutrition/high-calorie intake [10], obesity [11] and intrauterine environment [12] all causally implicated. Such environmental factors have been suggested to act at least in part through epigenetic processes to modulate IR/T2D risk, with genome-wide DNA methylation analyses showing altered methylation of key regulators of glucose metabolism in both pancreatic islet cells and muscle tissue of individuals with prediabetes and T2D [13, 14].
The skeletal muscle is the primary organ implicated in glucose clearance and is responsible for 80% of postprandial glucose uptake from the circulation [15]. Skeletal muscle IR, which has been reported to be present decades before the onset of β cell failure and symptomatic T2D [15, 16], is caused by desensitization of the muscle to insulin leading to elevated blood glucose levels [16]. During ageing, there are significant changes in body composition with the loss of muscle mass and function (termed sarcopenia) and/or increased adiposity [17]. Both sarcopenia and adiposity lead to structural and functional modifications in the skeletal muscle including impaired mitochondrial bioenergetics, oxidative stress, increased senescence, intramyocellular lipid accumulation, modified activity of insulin sensitivity regulatory enzymes, decreased autophagy and a decline in satellite cell function [18]. Such changes have been suggested to impair the skeletal muscle insulin sensitivity and increase the risk of IR and T2D, although specific underlying mechanisms for increased incidence of T2D in older people have yet to be elucidated.
The skeletal muscle is maintained by muscle stem cells termed satellite cells, which lie beneath the basal lamina, and which become activated (termed myoblasts) and proliferate upon stress or injury; these cells then either differentiate to regenerate or repair muscle fibers or return to a quiescence state to repopulate the satellite cell niche [19]. During ageing, the number and self-renewal capacity of satellite cells have been shown to decline, with alterations in the epigenetic control of quiescence pathways impairing the replenishment of the muscle stem cell pool and contributing to a decreased regenerative response in older age [20,21,22,23]. In rodent models of IR/T2D, impaired muscle regeneration has also been observed and this was associated with impaired satellite cell proliferation and myoblast accumulation [24]. Myoblasts from individuals with T2D have also been reported to retain many of the diabetic phenotypes observed in muscle tissue, such as impaired glucose uptake and lipid oxidation [25]. Moreover, differences in DNA methylation have been reported using the 450 K HumanMethylation array in myoblasts from individuals with T2D [26], suggesting epigenetic changes related to T2D are also present in stem cells within the muscle, which may contribute to both impaired myogenesis and regeneration as well as the diabetic phenotype. IR often precedes the onset of T2D, but the relationship between myoblast DNA methylation and IR or the age-related decline in glycaemic control is not known. To gain insight into the underlying gene regulatory mechanisms associated with IR, we used the Infinium Human MethylationEPIC BeadChip (interrogating over 850,000 CpGs sites) to characterise the associations between DNA methylation in human primary myoblasts from community-dwelling older individuals with Homeostatic Model Assessment of Insulin Resistance (HOMA2-IR, an index for the assessment of insulin resistance) and levels of glycated hemoglobin HbA1c, together with the pathways enriched amongst the differentially methylated genes. In addition, we investigated the influence of sarcopenia and adiposity on the HOMA2-IR- and HbA1c-associated methylation signatures to further our understanding of how changes in body composition may contribute to the development of insulin resistance and dysglycaemia in older age.
Methods
Study participants
Participants were recruited from the UK Hertfordshire Sarcopenia Study Extension (HSSe), investigating life course influences on muscle function in community-dwelling older people [27]. This study received ethical approval from the Hertfordshire Research Ethics Committee (number 07/Q0204/68) and was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments. Of the 168 HSSe participants, 138 had sufficient DNA from the myoblast cultures for methylation analysis. After quality control of the array data set (outlined below), 119 samples were used for subsequent analyses (Table 1).
Procedures
Fasting blood samples (overnight) were taken from the anterior cubital fossa, and serum, plasma and fluoride oxalate aliquots were prepared in 1 ml tubes and frozen at − 80℃ until further analyses. Fasting insulin (serum), glucose (plasma) and HbA1c (whole blood) concentrations were measured using Beckman Coulter Access Ultrasensitive Insulin assay (paramagnetic particle, chemiluminescent immunoassay), Beckman Coulter enzymatic UV test (hexokinase method) and Sebia CAPILLARYS HbA1c (separation and quantification of the HbA1c glycated fraction of haemoglobin by capillary electrophoresis), respectively. HOMA2-IR was calculated using the HOMA2 Calculator (v.2.23, University of Oxford) available online at https://www.dtu.ox.ac.uk/homacalculator/. After an overnight fast, percutaneous vastus lateralis muscle biopsies (Weil-Blakesley conchotome) containing between 10 and 60 mg of the tissue were conducted under local anesthetic, using a standardised protocol [28], and muscle cell cultures were isolated and frozen down.
Skeletal muscle stem cell isolation and myoblast culture
Details of muscle stem cell isolation and myoblast culture are described in [23]. Briefly, muscle biopsies were minced and digested in 0.5 mg/mL collagenase (Sigma, UK), before pre-plating to remove fibroblasts. Cells were frozen down at passage 2. Myoblast cultures were then thawed and sorted using CD56 MicroBeads. CD56-positive cells were expanded and plated out for all experiments at passage 4 (P4). Passage 4 cells showed that they were ≥ 96% positive for CD56. Comparison of DNA methylation patterns between HSSe-cultured myoblasts (P4) and muscle biopsies from which the cells were isolated showed a correlation between myoblast and muscle DNA methylation for the 353 Horvath epigenetic clock CpG loci used to determine biological age (r = 0.914, p ≤ 2.2 × 10−16) and for probes within the myogenesis gene set (r = 0.960, p = 2.0 × 10−16) (Supplementary Fig. 1). For this comparison 40 paired HSSe myoblast/muscle samples were assessed, as these were the only muscle biopsy samples where there was sufficient DNA available.
Infinium Human MethylationEPIC BeadChip array
Genomic DNA (gDNA) was extracted from actively proliferating myoblasts using the GenElute™ Mammalian Genomic DNA miniprep Kit (Merck) following the manufacturer’s instructions. Quality and quantity were determined by Nanodrop (Thermo Scientific) and Qubit™ (Invitrogen). 750 ng of genomic DNA was treated with sodium bisulfite using the Zymo EZ DNA Methylation kit (ZymoResearch, Irvine, California, USA) and hybridised to the Infinium Human MethylationEPIC BeadChip array (Illumina, Inc. CA, USA). This work was carried out at the Centre for Molecular Medicine and Therapeutics (CMMT) (http://www.cmmt.ubc.ca).
Infinium HumanMethylationEPIC BeadChip array data processing
Myoblast Infinium Human MethylationEPIC data was processed using the Bioconductor package minfi (v1.32.0) [29] in R (v3.6.2). We applied beta-mixture quantile (BMIQ) normalization using the ChAMP package (v.2.16.2) to remove array biases and correct probe design. Methylation profiling was carried out in 142 DNA samples, which included 4 technical replicates. CpGs is known to cross-hybridise to other locations in the genome (n = 3027) [30], coinciding with SNPs (n = 95,193), with a detection p value > 0.01 (n = 13,564), and a beadcount < 3 (n = 10,368) were removed from the dataset. 16,338 probes aligning to the sex chromosomes and 2815 non-CpG probes were also removed from the dataset. The 4 technical replicate samples were included, for which the Euclidean distance was calculated, and hierarchical clustering, using complete linkage clustered the pairs together. Data were further assessed by visualization of methylation density plots and calculation of median absolute deviation (MAD) scores [31]. The correlation difference for each sample was calculated which is the difference between the average of all the pairwise correlations that involve the sample and the average of all the pairwise correlations that do not. A MAD score (robust Z score) is then calculated from the correlation difference by subtracting the medians and dividing it by median absolute deviations (MAD). To determine outliers a standard cutoff (− 5) was set [32]. Technical replicates were removed after normalization but before inference (the duplicate with the lowest MAD score was removed). Twelve samples showed aberrant methylation densities and MAD scores lower than − 5 were removed from subsequent analysis along with 7 samples which had a predicted sex differing to their actual sex. This resulted in 119 samples which were taken forward for further analysis. The methylation data sets analysed generated in this study can be accessed on the gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/), under accession number GSE221540.
Infinium Human MethylationEPIC BeadChip array data analysis and statistical analysis
Robust regression models were run using limma (v3.38.3) [33]. Models were adjusted for age, sex, position on array, batch and two surrogate variables (SVs) to account for unknown sources of variation in the data. Surrogate variables (SVs) were calculated using surrogate variable analysis (SVA) [34,35,36] in bioconductor using all probes. SVs were checked for correlations against phenotypic data to ensure that they were not removing biological data. Both “position on array” and “batch” were used as categorical variables in the linear regression model. Inflation of p values was assessed (genomic inflation factor lambda), quantile–quantile (Q-Q) plots generated and bacon (v1.10.1) [37] used to control for genomic inflation of test statistics where lambda > 1.2. Since obesity is a major risk factor for T2D, a sensitivity analysis was carried out with additional adjustment for BMI. Analyses took account of multiple tests with the Benjamini–Hochberg adjustment for false discovery rate (FDR), using an FDR < 0.05. Log fold change (logFC) was calculated as part of the linear regression model where the slope is the logFC. Analysis of differentially methylated regions (DMRs) was conducted using DMRcate (v.2.8.5) [38]. DMR significance was ranked by Stouffer score. All statistical analyses were carried out in R (v3.6.2).
Gene pathway and transcription factor enrichment analysis
Protein–protein interaction (PPI) networks were examined for the top 100 differentially methylated CpG (dmCpG)-associated genes using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) and visualized in Cytoscape using a maximum of 2 additional molecules. The top 100 differentially methylated genes were chosen as an arbitrary cutoff to provide sufficient input into the PPI analysis to generate exploratory dmCpG-associated pathways and networks. Pathways were considered if they generated a significant PPI enrichment score (P < 0.05) and contained 3 or more genes present within the EWAS top 100. Gene ontology (GO, Biological Process) and KEGG pathway enrichment were determined from the largest network cluster. To investigate upstream regulators and effector networks, ingenuity pathway analysis (IPA) (Qiagen, UK) version 68752261 was used.
Muscle epigenetic age estimation
Epigenetic age acceleration was calculated as the residuals of regressing the epigenetic age estimated by the Muscle Epigenetic Age Test (MEAT) [39] over chronological age.
Results
Participant characteristics
Table 1 shows characteristics of the 119 participants of which 89 were female and 30 male. The mean (SD) age was 78.24 (2.59), body mass index (BMI) (kg/m2) 27.25 (4.06), HOMA2-IR 1.03 (0.58), fasting insulin (mU/L) 7.73 (4.42), fasting glucose (mmol/l) 5.43 (0.65) and HbA1c 37.79 (5.33). Of the 119 participants with DNA methylation data, 115 had phenotypic data for HOMA2-IR and 114 for HbA1c. Of the HOMA2-IR characterised participants, 115 had phenotypic data for grip strength, 111 for appendicular lean mass (ALMi) and 115 for gait speed. For HbA1c-characterised participants, 114 had phenotypic data for grip strength, 110 for ALMi and 114 for gait speed.
Identification of differentially methylated CpGs in myoblasts associated with HOMA2-IR
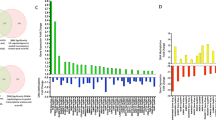
There were 38 differentially methylated CpGs (dmCpGs) significantly (FDR < 0.05) associated with HOMA2-IR (Fig. 1 A, B, C, D, E, Table 2, Supplementary Table 1). The three dmCpGs with the strongest associations were cg08944484, located within the 3′ untranslated region (UTR) of the saccharine dehydrogenase (SCCPDH) gene on chromosome 1 (FDR = 3.27 × 10−03), cg01351409 located in an intergenic region of chromosome 7 (FDR = 3.37 × 10−03) and cg20625334 found within the gene body of signal transducing adapter molecule 2 (STAM2) (FDR = 3.37 × 10−03).

A Volcano plot showing the dmCpGs with respect to HOMA2-IR (FDR < 0.05 = red, top 10 FDR < 0.05 labelled). B Manhattan plot showing the dmCpGs with respect to HOMA2-IR (FDR < 0.05 = red, top 10 FDR < 0.05 labelled). C Locations of dmCpGs with respect to HOMA2-IR (FDR < 0.05). D Overlap of dmCpGs (FDR < 0.05) with respect to HOMA2-IR and HbA1c. E Overlap of top 100 dmCpG-associated genes with respect to HOMA2-IRand HbA1c
To gain an understanding of the functional significance of the methylation changes associated with HOMA2-IR, the top 100 dmCpG-associated genes were inputted into STRING to generate a PPI network. The PPI enrichment P value for the overall network for HOMA2-IR was 4.3 × 10−02 indicating biological connection between the proteins. Selection of the largest gene cluster identified within the network was found to be enriched for tissue homeostasis (FDR 6.60 × 10−04) anatomical structure homeostasis (FDR 7.50 × 10−04) and regulation of JNK cascade (FDR 1.90 × 10−03) within gene ontology (GO) pathways for biological process, and AMPK signalling (FDR 1.41 × 10−02) and Insulin signalling (FDR 1.41 × 10−02) within the KEGG ontology (Table 3, Supplementary Table 2). To identify potential causal networks and upstream regulators that may mediate changes in IR, the HOMA2-IR dmCpG-associated genes were analysed using IPA. The top networks were cellular movement, connective tissue development and function, and skeletal and muscular system development and function (Fig. 2, Supplementary Table 3), with the top upstream regulators identified as T cell factor (TCF) (p 3.80E − 04), Clock Circadian Regulator (CLOCK) (p 1.58 × 10−03) and MAPK (p 3.76 × 10−03) (Supplementary Table 4).

Network diagram showing the top IPA gene network of cellular movement, connective tissue development and function and skeletal and muscular system development and function and associated molecules. Grey nodes indicate molecules within the input dataset, while white nodes are generated by IPA core analysis and represent molecules not in the input dataset added to the network from knowledge base to generate the interaction network
Influence of BMI on the HOMA2-IR associated DNA methylation signature
To assess the influence of BMI on the HOMA2-IR associated methylation signature in myoblasts, BMI was added as a covariate to the regression model; after adjustment for BMI, there were 6 dmCpGs associated (FDR ≤ 0.05) with HOMA2-IR. The strongest associations were with cg13451048 located within the gene body of exoribonuclease family member 3 (ERI3) on chromosome 1 (FDR 3.60 × 10−03), and cg01351409 and cg22007860 located in intergenic regions of chromosome 7 (FDR 4.20 × 10−03) and chromosome 3 (FDR 1.40 × 10−02), respectively (Supplementary Table 5). Of the 6 dmCpGs associated with HOMA2-IR after adjustment for BMI, 3 overlapped with the dmCpGs identified without adjustment for BMI, these being cg13451048 within ERI3, and the intergenic CpGs cg01351409 and cg10416784.
Pathway analysis of the top 100 dmCpG-associated genes after adjustment for BMI showed enrichment of regulation of epidermal growth factor receptor (FDR 1.01 × 10−02) and regulation of protein kinase B signalling (FDR 1.89 × 10−02) and anatomical structure morphogenesis (FDR 4.06 × 10−02) within the GO pathways for biological process, and relaxin signalling (FDR 5.98 × 10−05) and endocrine resistance (FDR 2.00 × 10−04) amongst the KEGG ontology (FDR 4.00 × 10−04) (Supplementary Table 6).
Influence of sarcopenia on HOMA2-IR-associated DNA methylation signature
As sarcopenia has been implicated in the development of insulin resistance, we adjusted the regression model for each of the 3 definitional components of sarcopenia, namely ALMi, grip strength and gait speed to determine how these measures of muscle mass, strength and function may influence the HOMA2-IR-associated DNA methylation signal in myoblasts (Supplementary Tables 7, 8 and 9). After addition of ALMi as a covariate in the model, the number of dmCpGs associated with HOMA2-IR was reduced from 38 to 7. The top dmCpGs were cg01351409, an intergenic CpG located on chromosome 7, cg18292904 located within the macrophage-stimulating protein receptor gene (MSTR1, also known as RON kinase) and cg14878421 an intergenic CpG located on chromosome 4 (Supplementary Table 7). Only 5 of the 38 dmCpGs identified in the unadjusted analysis remained significantly associated with HOMA2-IR; these were cg20625334 (STAM2), cg13451048 (ERI3) and the intergenic CpGs: cg01351409, cg10703001 and cg00623421, which all showed the same direction of association, effect size and significance. Adjustment for grip strength in the model also attenuated the methylation signal associated with HOMA2-IR with 15 dmCpGs associated with HOMA2-IR after adjustment; 12 of which overlapped with the dmCpGs from the unadjusted analysis (Supplementary Table 8), while the addition of gait speed to the model resulted in the association of 23 dmCpGs with HOMA2-IR, of which 18 of the dmCpGs overlapped with those identified in the unadjusted analyses (Supplementary Table 9).
PPI analysis revealed that the top pathways enriched amongst biological process GO category were regulation of protein kinase B signalling (FDR 2.20 × 10−05) and regulation of protein phosphorylation (FDR 4.78 × 10−05) after adjustment for ALMi (Supplementary Table 10), regulation of protein kinase B signalling (FDR 2.00 × 10−03) and positive regulation of protein kinase B signalling (FDR 4.00 × 10−03) after adjustment for grip strength (FDR 2.00 × 10−03) (Supplementary Table 11), and generation of neurons (FDR 1.60 × 10−03) and positive regulation of kinase activity (FDR 1.90 × 10−03) after adjustment for gait speed (Supplementary Table 12). The top KEGG pathways were MAPK (FDR 8.30 × 10−05) and insulin signalling (FDR 8.30 × 10−05) after adjustment for ALMi, Prolactin Signalling (FDR 2.90 × 10−03) and longevity regulation (FDR 1.34 × 10−02) after adjustment for grip strength, and insulin signalling (FDR4.70 × 10−03) and bacterial invasion of epithelial cells (FDR 4.70 × 10−03) after adjustment for gait speed.
HbA1c was associated with differential DNA methylation of skeletal muscle myoblasts
To identify methylation changes associated with longer term glycemia, we examined associations between DNA methylation and HbA1c levels. Forty-nine dmCpGs were associated with HbA1c (FDR < 0.05) (Supplementary Table 13), with the top 3 CpGs being cg19477361 located in a CpG island and 5′UTR region of guanine nucleotide-binding protein subunit gamma-7 (GNG7) (FDR 2.42 × 10−03), cg13451048 located in the gene body of exoribonuclease family member 3 (ERI3) on chromosome 1 (FDR 3.06 × 10−03), and cg22337620 located in the gene body of the DLGAP1 gene which encodes for disk large-associated protein 1 (DAP-1), also known as guanylate kinase-associated protein (GKAP) (FDR 3.81 × 10−03). Only one dmCpG associated with both HOMA2-IR and HbA1c, which was cg13451048 within ERI3 (Fig. 1D, Supplementary Table 14). After addition of BMI in the regression model, there were 48 dmCpGs associated with HbA1c (Supplementary Table 15), with 36 of the dmCpGs overlapping with the dmCpGs in the unadjusted model. The addition of ALMi as a covariate resulted in 44 dmCpGs associated with Hb1Ac, with 18 dmCpGs in common with those from the unadjusted analysis (Supplementary Table 16), while the addition of grip strength resulted in 70 dmCpGs associated with Hb1Ac levels with 37 in common with the unadjusted analyses (Supplementary Table 17); adjustment for gait speed led to 64 dmCpGs associated with HbA1c, with 38 overlapping with those identified in the unadjusted analysis (Supplementary Table 18).
Network analysis of HbA1c dmCpG-associated genes showed a PPI enrichment P value for the overall network of 1.38 × 10−02. Within the largest cluster the top GO biological process pathways were nervous system development (FDR 1.27 × 10−05), multicellular organism development (FDR 4.23 × 10−05) and cell differentiation (FDR 4.23 × 10−05), whilst in the KEGG ontology top pathways were Hippo signalling (FDR 1.60 × 10−03) and pathways in cancer (FDR 1.60 × 10−03) (Supplementary Table 19). The top pathways enriched within the biological process category, after the addition of BMI as a covariate, were cell communication (FDR 3.79 × 10−06), Signalling (FDR 3.79 × 10−06) and developmental process (FDR 3.79 × 10−06) (Supplementary Table 20). Top pathways enriched were neuron migration (FDR 8.17 × 10−05), cell communication (FDR 8.17 × 10−05) and signal transduction FDR 8.17 × 10−05) after addition of ALMi in the model (Supplementary Table 21); enzyme-linked receptor protein signalling pathways (FDR 4.90 × 10−03), cell differentiation (FDR 4.90 × 10−03) and signaling (FDR = 5.50 × 10−03) after adjustment for grip strength (Supplementary Table 22), and cell differentiation (FDR 8.98 × 10−06), generation of neurons (FDR 8.98 × 10−06) and cell communication (FDR 1.10 × 10−04) after adjustment for gait speed (Supplementary Table 23). Similar KEGG pathways were enriched in both the unadjusted and adjusted analyses.
HOMA2-IR and HbA1c are associated with multiple differentially methylated regions
Regional analysis identified DMRs associated with HOMA2-IR and HbA1c (Table 4, Supplementary Tables 24 and 25). 8 DMRs were associated with HOMA2-IR (Stouffer < 0.05), with the top 3 DMRs located within the Homobox A3 (HOXA3) gene, consisting of 8 CpGs (Stouffer 9.70 × 10−03), the T-box transcription factor (TBX1) gene consisting of 4 CpGs (Stouffer 1.0 × 10−03) and the nuclear receptor subfamily 2 group F member 2 (NR2F2) consisting of 4 CpGs (Stouffer 1.3 × 10−02), respectively (Table 4). After adjustment for BMI, 12 DMRs were associated with HOMA2-IR, with the top 3 located in CTD-2562J15.6, TBX1 and N2RF2, which were all associated with HOMA2-IR in the unadjusted analysis (Supplementary Table 26) Adjustment for each of the definitional components of sarcopenia resulted in 4 DMRs associated with HOMA2-IR; with DMRs being located within TBX1, RNF126P1, HOXA3 and NR2F2 after adjustment for either grip strength or gait speed (Supplementary Tables 27 and 28), and DMRs within NR2F2, RNF126P and TBX1 together with an intergenic region of Chr 20 after adjustment for ALMi (Supplementary Table 29). HbA1c was associated with 21 DMRs, with the top DMR located in the HLA Complex Group 9 (HGC9) gene (Stouffer 1.9 × 10−04) (Supplementary Table 25). There were no DMRs that overlapped between HOMA2-IR and HbA1c. There were 32 DMRs associated with HbA1c after adjustment for BMI (Supplementary Table 30), and 9 after adjustment for either grip strength or gait speed (Supplementary Tables 31 and 32), the top DMR in these analyses being located within HCG9 which is also the top DMR in the unadjusted analysis. Twenty-four DMRs were associated with HbA1c after adjustment for ALMi, the top DMR being located within FERM domain containing 4A (FRMD4A) (Supplementary Table 33).
DmCpGs associated with HOMA2-IR or HbA1c were not related to accelerated epigenetic ageing in skeletal muscle myoblasts
To determine whether the methylation changes associated with HOMA2-IR or HbA1c represented accelerated epigenetic ageing within skeletal muscle myoblasts, epigenetic age was calculated [39]. Epigenetic age as determined by the muscle epigenetic age estimator MEAT was strongly correlated with chronological age (P = 0.002), but there were no associations between accelerated epigenetic age and HOMA2-IR (P = 0.556) or HbA1c (P = 0.552) unadjusted, or after adjustment for BMI, ALMi, grip strength or gait speed (Supplementary Table 34).
Discussion
Our analyses have shown differential methylation of CpGs in human primary myoblasts from older individuals with respect to HOMA2-IR and HbA1c. The HOMA2-IR-associated dmCpGs were enriched in genes associated with insulin and AMPK signalling pathways, as well as muscle development and tissue homeostasis. The IR-associated methylation signal was attenuated after adjustment for ALMi or BMI, suggesting that muscle mass and body composition make a significant contribution to the methylation changes associated with insulin resistance in myoblasts.
There were changes in DNA methylation in the myoblasts at both the single CpG and regional level associated with HOMA2-IR. Of the top three HOMA2-IR-associated dmCpGs located within a gene, cg08944484 was located within the 3′ untranslated region of the SCCPDH gene, which encodes for saccharopine dehydrogenase, a protein which localises to the mitochondrion and is predicted to enable oxidoreductase activity and glycolipid biosynthetic processes [40]; cg20625334 located in the gene body of STAM2 which encodes a protein that acts downstream of Janus Kinase (JAK) and regulates receptor signalling and trafficking in mammalian cells [41], and cg19423170 located within the APP gene which encodes for the amyloid precursor protein. Currently neither SCCPDH or STAM2 have been directly linked with IR/T2D although STAM2 has been shown to be modulated by maternal diabetes in the developing embryo in an animal experimental model [42] suggesting a role for STAM2 in T1D and as a target for perturbations induced by maternal diabetes and transgenerational inheritance of phenotype. The APP protein has been linked with somatic metabolic disorders related to T2D; for example, animal experimental models using App−/− mice have shown higher levels of insulin degrading enzyme (IDE) mRNA, protein and activity in the skeletal muscle as well as lower fasting levels of blood insulin and a larger increase in response to glucose compared to controls [43]. Mice overexpressing APP protein have further been shown to exhibit mitochondrial dysfunction within the skeletal muscle and a substantial reduction in tricarboxylic acid (TCA) cycle activity indicating a switch from aerobic to anaerobic glucose metabolism [44]. Changes to the methylation status of the APP gene associated with IR may indicate a switch in glucose metabolism linked to changes in components of the insulin signalling pathways through modulation of muscle mitochondrial function in older people.
As both sarcopenia and obesity have been implicated in the development of IR, we performed sensitivity analyses additionally adjusting the regression model for the 3 definitional components of sarcopenia and BMI. Adjusting for ALMi attenuated the methylation signature associated with HOMA2-IR suggesting that ALMi makes a significant contribution to the methylation changes associated with insulin resistance in myoblasts. Low muscle mass will impair glucose clearance, and reduce physical inactivity, increasing the risk of IR. Although it has been suggested that there is a bidirectional relationship between IR and sarcopenia, as increased insulin resistance which is characterized by a proinflammatory phenotype and oxidative stress, can impair muscle cell function, leading to losses in the skeletal muscle mass and strength and potentially leading to the development of sarcopenia [45]. A reduction in the HOMA2-IR-associated DNA methylation signal was also observed after addition of grip strength and gait speed as covariates in the analysis. Grip strength is correlated with ALMi and gait speed in this subset of individuals from the HSSe, so the attenuation in the methylation signature after adjustment for grip strength may reflect the lower muscle mass of the individuals.
Increased adiposity is a major risk factor in the development of IR. We found that after adjustment for BMI, 6 dmCpGs were significantly associated with HOMA2-IR, compared to 38 dmCpGs in the unadjusted model, suggesting that body composition contributes to the methylation patterns associated with HOMA2-IR and/or body composition may induce changes in DNA methylation at specific loci, which may in turn influence IR risk. In contrast, dmCpGs not confounded by BMI may be associated with IR via mechanisms independent of body composition. While BMI is only a crude measure of adiposity there was a marked difference in the dmCpGs associated with HOMA2-IR after adjustment for BMI and ALMi with only one dmCpG in common (cg13451048 located within ERI3), in accordance with BMI and ALMI reflecting different aspects of body composition.
Pathway analysis revealed that the HOMA2-IR-associated dmCpGs were enriched in genes involved in tissue homeostasis, regulation of JNK, AMPK and insulin signalling pathways. The JNK signalling pathway is involved in stress activation and has increasingly been recognised as an important mediator of IR. For example, animal experimental models have shown that activation of JNK induces IR and ß-cell dysfunction in obesity [46]. While the AMPK signalling pathway also affects insulin action and glucose metabolism; AMPK is an energy sensor which upon activation promotes glucose transport, mitochondrial function, mTOR signalling, fatty acid oxidation and sirtuin expression, while inhibiting inflammation and JNK signalling, oxidative stress and chemokine expression [47]. In model systems and human studies, sustained decreases in AMPK activity induced by obesity or overnutrition accompany IR, whereas AMPK activation is associated with increased insulin sensitivity [48,49,50,51]. Differential methylation of genes within the JNK, Insulin and AMPK pathways suggests such changes may contribute to the dysfunction of these pathways leading to IR. Similar pathways were found to be enriched amongst differentially expressed transcripts in muscle tissue from elderly individuals with insulin resistance [52], suggesting epigenetic processes may consolidate or mediate such changes in gene expression resulting in aberrant insulin and downstream signalling perpetuating the insulin-resistant phenotype. After addition of either ALMi, grip strength and gait speed to the model there was enrichment for PKB/AKT signalling pathways, known to play a pivotal role in JNK, AMPK and insulin signalling pathways; this suggests that the epigenetic dysregulation of these pathways in insulin resistant individuals may in part be independent of muscle mass and strength. Interestingly, after adjustment for BMI, although there was still enrichment amongst insulin and JNK signalling pathways, there was also enrichment amongst epidermal growth factor (EGFR) and relaxin signalling pathways suggesting potential effects of IR independent of body composition on muscle stem cell activation, differentiation and survival [53,54,55].
The top DMRs associated with HOMA2-IR were located within HOXA3, TBX1 and NR2F2. HOXA3 is a DNA binding factor which has been previously reported to be hypermethylation in aged muscle tissue. TBX1 is a gene that encodes for a member of the T box gene family of DNA binding transcription factors and has been linked to the regulation of insulin signalling and glucose homeostasis within adipose tissue [56]. Within murine muscle Tbx1 has been shown to modulate muscle fiber type and oxidative metabolism in myotubes as well as stimulate myoblast differentiation [57]. Here, 4 dmCpGs were located within the identified DMR in the gene body or 3′ UTR region of three TBX1 transcripts. A DMR was also identified within NR2F2, also known as COUP transcription factor 2 (COUP-TFII) associated with HOMA2-IR, in both the unadjusted and adjusted analyses. COUP-TFII through its interaction with the glucocorticoid receptor (GR) is known to control gluconeogenesis, through direct binding of COUP-TFII/GR complexes to the promoters of gluconeogenic enzyme genes [58, 59]. In the skeletal muscle of mice, COUP-TFII regulates Glut4 expression, and in humans, single-nucleotide polymorphisms within the COUP-TFII promoter are associated with insulin sensitivity [59]. Recently in an animal experimental model, it has also been shown that COUP-TFII plays an important role in myogenesis and myoblast fusion, whereby COUP-TFII represses the transcription of Nephronectin (Npnt), integrin subunit beta 1 (Itgb1D) and Caveolin 3 (Cav3), genes important for cell–cell fusion [58]. DMRs within TBX1 and NR2F2 remained associated with HOMA2-IR after the addition of BMI, ALMi, grip strength or gait speed as covariates, suggesting these differentially methylated regions are associated with HOMA2-IR independent of body composition, muscle mass or strength. In the BMI-adjusted model, there was also a second DMR containing 10 dmCpG sites identified within NR2F2, 8.6 kb downstream of the first DMR; this second DMR overlaps with the NR2F2-AS1 gene, suggesting that DNA methylation of this region may affect the antisense RNA produced from this locus and the regulation of NR2F2 through a post-transcriptional mechanism.
DNA methylation was also associated with HbA1c levels; 73% of the dmCpGs associated with HbA1c in the unadjusted analysis overlapped with those in the BMI adjusted analysis, 45% after adjustment for ALMi, 84% after adjustment for grip strength and 78% for gait speed. The dmCpGs associated with HbA1c that were robust to adjustments for BMI, ALMi, grip strength and gait speed included G-protein subunit gamma 7 (GNG7), a tumour suppressor gene known to induce autophagy and cell death via the mTOR pathways and inhibit cell division [60] and ERI3, a gene involved in RNA processing. The top pathways enriched amongst the HbA1c associated dmCpGs were the nervous system development, cell differentiation, Hippo signalling and pathways in cancer driven by the differential methylation of genes such as BMP7, APC regulator of WNT signalling pathway (APC) and transcription factor 7 like 1 (TCF7L1). BMP7 has been reported to increase glucose uptake in muscle by stimulating Glut4 translocation to the plasma membrane through activation of pyruvate dehydrogenase kinase 1 (PDK1) and AKT, suggesting that differential methylation of BMP7 may affect glucose uptake in myoblasts and potentially blood glucose levels. As insulin resistance is strongly associated with ageing, we investigated whether the dmCpGs associated with HOMA2-IR or HbA1c were associated with accelerated ageing using the MEAT epigenetic clock. We found that although chronological age was strongly related to epigenetic age, the dmCpGs associated with HOMA2-IR or HbA1c were not associated with accelerated ageing, suggesting that although there is an increasing incidence of IR with age, the epigenetic changes associated with IR are distinct from cellular ageing.
Previously there have been limited studies on DNA methylation changes in myoblasts associated with IR. Davegardhl et al. studied differences in DNA methylation in differentiating myoblasts from middle-aged individuals with T2D. They found that pathways involved in steroid biosynthesis, PPAR signalling and biosynthesis of unsaturated fatty acids (USFAs) were upregulated, and the pentose phosphate pathway downregulated upon myoblast differentiation in individuals with T2D compared to normal glucose tolerance (NGT) individuals [26]. In this study, where we compared DNA methylation in actively dividing myoblasts from elderly individuals; we found a significant change in DNA methylation in relation to insulin resistance suggesting DNA methylation changes are present in myoblasts prior to their differentiation into myotubes implying such changes may perpetuate the insulin resistant phenotype.
Strengths of this study are, that to our knowledge, this is the first to examine the association between measures of IR and dysglycemia in aged individuals with DNA methylation in human primary myoblasts. Moreover, we examined the association between DNA methylation and measures of IR across a relatively large number of human primary myoblast cultures isolated from 119 community dwelling older individuals. We were also able to assess the contribution that BMI and measures of muscle mass and strength made to the IR-associated methylation signature. Limitations to the study firstly include the use of cultured human primary myoblasts, where the process of cell culturing may in itself induce epigenetic changes within the cells. However, in the present study, all myoblasts were used at the same early passage number and previous research has demonstrated that cultured primary human myoblasts derived from individuals with T2D retain many of the diabetic phenotypes observed in muscle tissue, such as impaired glucose uptake and lipid oxidation [25]. Secondly, although we carried out the analyses with and without adjustment for BMI, studies have suggested that the visceral fat depot is a major risk factor in the development of IR [61]; further studies investigating the contribution of different fat depots to the HOMA2-IR DNA methylation signal would provide an additional level of understanding of the role of obesity on the methylation changes associated with IR. Thirdly, due to the limited number of early passage cells, we were unable to measure whether the isolated and cultured myoblasts displayed insulin resistance or whether the methylation changes reported here were associated with a corresponding changes in RNA expression or functional changes in the myoblasts themselves. We therefore cannot ascertain whether the changes in CpG methylation play a causal role in the development of IR or are a result of the downstream effects of IR on DNA methylation; however, the demonstration of DNA methylation changes in genes involved in key insulin regulating pathways suggest that they may contribute at least in part to the mechanism by which IR develops and/or is maintained in older skeletal muscle.
Conclusions
These findings show widespread changes in the methylome of human myoblasts that are associated with HOMA2-IR and HbA1c. Furthermore, we show that HOMA2-IR associated dmCpGs were enriched in genes implicated in insulin signalling and skeletal and muscular system development and function, supporting the premise that epigenetic changes may contribute to the age-related decline in glycaemic control and suggesting that it may be possible to ameliorate the impairments in glycaemic control and improve muscle health in old age by developing intervention strategies which reset the epigenetic landscape of myoblasts.
Availability of data and materials
The methylation data sets generated in this study are available on the gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/), under accession number GSE221540.
Abbreviations
- IR:
-
Insulin resistance
- HSSe:
-
Hertfordshire Sarcopenia Study Extension
- dmCpG:
-
Differentially methylated cytosine phosphate guanine
- DMRs:
-
Differentially methylated regions
- HOMA2-IR:
-
Homeostatic model assessment of insulin resistance
- HbA1c:
-
Glycosylated hemoglobin, type A1C
- FDR:
-
False discovery rate
- JNK:
-
C-Jun N-terminal kinase
- AMPK:
-
5′ AMP-activated protein kinase
- BMI:
-
Body mass index
- ALMi:
-
Appendicular Lean Mass Index
- TBX1 :
-
T-box transcription factor
- NR2F2 :
-
Nuclear receptor subfamily 2 group F member 2
- ERI3 :
-
Exoribonuclease family member 3
- T2D:
-
Type 2 diabetes mellitus
- GWAS:
-
Genome-wide association studies
- gDNA:
-
Genomic DNA
- BMIQ:
-
Beta-mixture quantile
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- MAD:
-
Median absolute deviation
- SVs:
-
Surrogate variables
- Q-Q:
-
Quantile-quantile
- PPI:
-
Protein–protein interaction
- STRING:
-
Search Tool for the Retrieval of Interacting Genes/Proteins
- GO:
-
Gene Ontology
- IPA:
-
Ingenuity pathway analysis
- MEAT:
-
Muscle epigenetic age test
- UTR:
-
3′ Untranslated region
- SCCPDH :
-
Saccharine dehydrogenase
- STAM2 :
-
Signal transducing adapter molecule 2
- TCF:
-
T cell factor
- CLOCK:
-
Clock circadian regulator
- MAPK:
-
Mitogen-activated protein kinase
- MSTR1 :
-
Macrophage-stimulating protein receptor
- GNG7 :
-
Guanine nucleotide-binding protein subunit gamma-7
- DAP-1:
-
Disks large-associated protein 1
- GKAP:
-
Guanylate kinase-associated protein
- HOXA3 :
-
Homobox A3
- RNF126P1 :
-
Ring finger protein 126 pseudogene 1
- HGC9 :
-
HLA complex group 9
- FRMD4A :
-
FERM domain containing 4A
- JAK:
-
Janus kinase
- APP :
-
Amyloid beta precursor protein
- T1D:
-
Type 1 diabetes
- IDE:
-
Insulin degrading enzyme
- TCA:
-
Tricarboxylic acid
- PKB/AKT:
-
Protein kinase B
- EGFR:
-
Epidermal growth factor
- COUP-TF11 :
-
Nuclear receptor subfamily 2 group F member 2
- GR:
-
Glucocorticoid receptor
- Npnt :
-
Nephronectin
- Itgb1D :
-
Integrin subunit beta 1
- Cav3 :
-
Caveolin 3
- GNG7:
-
G-protein subunit gamma 7
- APC:
-
APC regulator of WNT signalling pathway
- BMP7 :
-
Bone morphogenetic protein 7
- APC :
-
APC regulator of WNT signalling pathway
- TCF7L1 :
-
Transcription factor 7 like 1
- Glut4:
-
Glucose transporter type 4
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- USFAs:
-
Unsaturated fatty acids
- NGT:
-
Normal glucose tolerance
References
Li M, et al. Trends in insulin resistance: insights into mechanisms and therapeutic strategy. Signal Transduct Target Ther. 2022;7(1):216.
Kalyani RR, Egan JM. Diabetes and altered glucose metabolism with aging. Endocrinol Metab Clin North Am. 2013;42(2):333–47.
Galicia-Garcia U, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275.
Redondo MJ, et al. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia. 2020;63(10):2040–8.
DeForest N, Majithia AR. Genetics of type 2 diabetes: implications from large-scale studies. Curr Diab Rep. 2022;22(5):227–35.
Vujkovic M, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52(7):680–91.
Fazeli PK, Lee H, Steinhauser ML. Aging is a powerful risk factor for type 2 diabetes mellitus independent of body mass index. Gerontology. 2020;66(2):209–10.
Li D-D, et al. Sedentary lifestyle and body composition in type 2 diabetes. Diabetol Metab Syndr. 2022;14(1):8.
Hamilton MT, Hamilton DG, Zderic TW. Sedentary behavior as a mediator of type 2 diabetes. Med Sports Sci. 2014;60:11–26.
Sarkar C, Webster C, Gallacher J. Are exposures to ready-to-eat food environments associated with type 2 diabetes? A cross-sectional study of 347 551 UK Biobank adult participants. Lancet Planetary Health. 2018;2(10):e438–50.
Al-Goblan AS, Al-Alfi MA, Khan MZ. Mechanism linking diabetes mellitus and obesity. Diabetes Metab Syndr Obes. 2014;7:587–91.
Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35(7):595–601.
Zhou Z, et al. DNA methylation landscapes in the pathogenesis of type 2 diabetes mellitus. Nutr Metab. 2018;15:47–47.
Dayeh T, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160.
DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32 Suppl 2(Suppl 2):S157–63.
Merz KE, Thurmond DC. Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol. 2020;10(3):785–809.
St-Onge MP, Gallagher D. Body composition changes with aging: the cause or the result of alterations in metabolic rate and macronutrient oxidation? Nutrition. 2010;26(2):152–5.
Shou J, Chen PJ, Xiao WH. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr. 2020;12(1):14.
Feige P, et al. Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell. 2018;23(5):653–64.
Sousa-Victor P, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–21.
Conboy IM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302(5650):1575–7.
Roth SM, et al. Skeletal muscle satellite cell populations in healthy young and older men and women. Anat Rec. 2000;260(4):351–8.
Bigot A, et al. Age-associated methylation suppresses SPRY1, leading to a failure of re-quiescence and loss of the reserve stem cell pool in elderly muscle. Cell Rep. 2015;13(6):1172–82.
Nguyen M-H, Cheng M, Koh TJ. Impaired muscle regeneration in Ob/ob and Db/db mice. ScientificWorldJOURNAL. 2011;11:827190.
Gaster M, et al. The diabetic phenotype is conserved in myotubes established from diabetic subjects: evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes. 2002;51(4):921–7.
Davegardh C, et al. VPS39-deficiency observed in type 2 diabetes impairs muscle stem cell differentiation via altered autophagy and epigenetics. Nat Commun. 2021;12(1):2431.
Westbury LD, et al. Associations between objectively measured physical activity, body composition and sarcopenia: findings from the Hertfordshire sarcopenia study (HSS). Calcif Tissue Int. 2018;103(3):237–45.
Baczynska AM, et al. Human vastus lateralis skeletal muscle biopsy using the Weil-Blakesley conchotome. J Vis Exp. 2016;109:e53075.
Aryee MJ, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Pidsley R, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):208.
Antoun E, et al. Epigenome-wide association study of sarcopenia: findings from the Hertfordshire Sarcopenia Study (HSS). J Cachexia Sarcopenia Muscle. 2021;13:240–53.
Zhao S, et al. QuickRNASeq lifts large-scale RNA-seq data analyses to the next level of automation and interactive visualization. BMC Genomics. 2016;17:39.
Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–35.
Saffari A, et al. Effect of maternal preconceptional and pregnancy micronutrient interventions on children’s DNA methylation: findings from the EMPHASIS study. Am J Clin Nutr. 2020;112(4):1099–113.
Perrier F, et al. Identifying and correcting epigenetics measurements for systematic sources of variation. Clin Epigenetics. 2018;10:38.
van Iterson M, et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18(1):19.
Peters TJ, et al. Calling differentially methylated regions from whole genome bisulphite sequencing with DMRcate. Nucleic Acids Res. 2021;49(19):e109.
Voisin S, et al. An epigenetic clock for human skeletal muscle. J Cachexia Sarcopenia Muscle. 2020;11(4):887–98.
Zhao X, et al. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol Cell Proteomics. 2011;10(1):M110.000299-M110.000299.
Pandey A, et al. Identification of a novel immunoreceptor tyrosine-based activation motif-containing molecule, STAM2, by mass spectrometry and its involvement in growth factor and cytokine receptor signaling pathways*. J Biol Chem. 2000;275(49):38633–9.
Pavlinkova G, Salbaum JM, Kappen C. Maternal diabetes alters transcriptional programs in the developing embryo. BMC Genomics. 2009;10(1):274.
Kulas JA, et al. Ablation of amyloid precursor protein increases insulin-degrading enzyme levels and activity in brain and peripheral tissues. Am J Physiol Endocrinol Metab. 2019;316(1):E106–20.
Boncompagni S, et al. Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J Biol Chem. 2012;287(24):20534–44.
Mesinovic J, et al. Sarcopenia and type 2 diabetes mellitus: a bidirectional relationship. Diabetes Metab Syndr Obes. 2019;12:1057–72.
Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–6.
Entezari M, et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: a pre-clinical and clinical investigation. Biomed Pharmacother. 2022;146:112563.
Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl). 2011;89(7):667–76.
Ruderman NB, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4):E751–60.
Xu XJ, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53(4):792–801.
Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–78.
Shou J, Chen PJ, Xiao WH. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr. 2020;12:14.
Leroy MC, et al. Epidermal growth factor receptor down-regulation triggers human myoblast differentiation. PLoS ONE. 2013;8(8):e71770.
Plesneva SA, et al. Study of structural-functional arrangement of the adenylyl cyclase signaling mechanism of action of insulin-like growth factor 1 revealed in muscle tissue of representatives of vertebrates and invertebrates. J Evol Biochem Physiol. 2008;44(5):542–51.
Kuznetsova LA, et al. Disturbance of regulation of NO synthase activity by peptides of insulin family in rat skeletal muscles in streptozotocin model of neonatal type 2 diabetes mellitus. Dokl Biochem Biophys. 2010;432(1):123–5.
Markan KR, et al. Adipose TBX1 regulates β-adrenergic sensitivity in subcutaneous adipose tissue and thermogenic capacity in vivo. Mol Metab. 2020;36:100965.
Motohashi N, et al. Tbx1 regulates inherited metabolic and myogenic abilities of progenitor cells derived from slow- and fast-type muscle. Cell Death Differ. 2019;26(6):1024–36.
Lee H-J, et al. Dysregulation of nuclear receptor COUP-TFII impairs skeletal muscle development. Sci Rep. 2017;7(1):3136.
Boutant M, et al. Glucose-dependent regulation of NR2F2 promoter and influence of SNP-rs3743462 on whole body insulin sensitivity. PLoS ONE. 2012;7(5):e35810.
Jiang J, et al. Circular RNA CDR1as inhibits the metastasis of gastric cancer through targeting miR-876-5p/GNG7 axis. Gastroenterol Res Pract. 2021;2021:5583029.
Preis SR, et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity (Silver Spring). 2010;18(11):2191–8.
Acknowledgements
The authors would like to thank Oscar Urtatiz for assistance in microarray processing. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal Skeletal Muscle.
Funding
This work was supported by grant funding from the Medical Research Council (MC_U47585827, MC_ST_U2055, MC_PC_21003; MC_PC_21001), Benevolent AI Limited, Arthritis Research UK, Royal Osteoporosis Society, International Osteoporosis Foundation, Cohen Trust, NIHR Southampton Biomedical Research Centre, University of Southampton and University Hospital Southampton NHS Foundation Trust, NIHR Musculoskeletal Biomedical Research Unit, and University of Oxford. K.M.G. is supported by the UK Medical Research Council (MC_UU_20/4), the US National Institute On Aging of the National Institutes of Health (award number U24AG047867), the UK Economic and Social Research Council and the Biotechnology and Biological Sciences Research Council (award number ES/M0099X/), the National Institute for Health Research (as an NIHR Senior Investigator (NF-SI-055–0042) and through the NIHR Southampton Biomedical Research Centre, and the European Union’s Erasmus + Capacity-Building ImpENSA Project. H.P.P. is supported by the National Institute for Health Research through the NIHR Southampton Biomedical Research Centre. This report is independent research, and the views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. The grant funders had no role in the design, collection, analysis and interpretation of the data, writing of the paper or decision to submit for publication. For the purpose of Open Access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
K A. Lillycrop, M A. Burton and E S. Garratt conceived and designed the research; M A Burton, E S. Garratt and E Antoun performed the research and acquired the data. M A. Burton, M O. Hewitt and H Y. Sharkh analysed the data and M A. Burton, and K A. Lillycrop interpreted the data. M A Burton and K A. Lillycrop drafted the manuscript. All authors were involved in revising the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study received ethical approval from the Hertfordshire Research Ethics Committee (number 07/Q0204/68) and was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments.
Consent for publication
Not applicable.
Competing interests
K.M. Godfrey and H.P. Patel have received reimbursement for speaking at conferences sponsored by companies selling nutritional products. N.C. Harvey reports personal fees, consultancy, lecture fees and honoraria from Alliance for Better Bone Health, AMGEN, MSD, Eli Lilly, UCB, Kyowa Kirin, Servier, Shire, Consilient Healthcare and Internis Pharma, outside the submitted work. C. Cooper has received consultancy fees and honoraria from Amgen, Danone, Eli Lilly, GlaxoSmithKline, Medtronic, Merck, Nestlé, Novartis, Pfizer, Roche, Servier, Shire, Takeda, and UCB. M.A. Burton, E.S. Garratt, E. Antoun, K.M. Godfrey, and K.A. Lillycrop are part of academic research programmes that have received research funding from BenevolentAI Bio Ltd., Nestec, and Danone. The other authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
(A) Correlation of the methylation beta values of the Horvath Pan Tissue CpGs and (B) bland altman plot of these CpGs between the muscle tissue and myoblasts. (C) Correlation of the methylation beta values of the CpGs associated with the genes in the myogenesis geneset (https://www.gsea-msigdb.org/gsea/msigdb/cards/HALLMARK_MYOGENESIS) and (D) bland altman plot of these CpGs between the muscle tissue and myoblasts.
Additional file 2: Supplementary Table 1.
List of HOMA2-IR dmCpGs. Supplementary Table 2. Top 20 GO and KEGG terms associated with the HOMA2-IR networks. Supplementary Table 3. IPA Networks from top 100 dmCpG associated genes with HOMA2-IR. Supplementary Table 4. IPA upstream regulators from top 100 dmCpG associated genes with HOMA2-IR. Supplementary Table 5. List of HOMA2-IR dmCpGs after adjustment for BMI. Supplementary Table 6. Top 20 GO and KEGG terms associated with the HOMA2-IR networks after adjustment for BMI. Supplementary Table 7. List of HOMA2-IR associated dmCpGs after adjustment for ALMi. Supplementary Table 8. List of HOMA2-IR associated dmCpGs after adjustment for grip strength. Supplementary Table 9. List of HOMA2-IR associated dmCpGs after adjustment for gait speed. Supplementary Table 10. Top 20 GO and KEGG terms associated with the HOMA2-IR networks after adjustment for ALMi. Supplementary Table 11. Top 20 GO and KEGG terms associated with the HOMA2-IR networks after adjustment for grip strength. Supplementary Table 12. Top 20 GO and KEGG terms associated with the HOMA2-IR networks after adjustment for gait speed. Supplementary Table 13. List of HbA1C associated dmCpGs. Supplementary Table 14. Common Overlapping dmCpGs. Supplementary Table 15. List of fasting HbA1c associated dmCpGs after adjustment for BMI. Supplementary Table 16. List of hbA1c associated dmCpGs after adjustment for ALMi. Supplementary Table 17. List of hbA1c associated dmCpGs after adjustment for grip strength. Supplementary Table 18. List of hbA1c associated dmCpGs after adjustment for gait speed. Supplementary Table 19. Top 20 GO and KEGG terms associated with the HbA1c networks. Supplementary Table 20. Top 20 GO and KEGG terms associated with the HbA1c networks after adjustment for BMI. Supplementary Table 21. Top 20 GO and KEGG terms associated with the hbA1c networks after adjustment for ALMi. Supplementary Table 22. Top 20 GO and KEGG terms associated with the hbA1c networks after adjustment for grip strength. Supplementary Table 23. Top 20 GO and KEGG terms associated with the hbA1c networks after adjustment for gait speed. Supplementary Table 24. HOMA2-IR asscoiated differentially methylated regions. Supplementary Table 25. Hba1c asscoiated differentially methylated regions. Supplementary Table 26. HOMA2-IR asscoiated differentially methylated regions adjusted for BMI. Supplementary Table 27. HOMA2-IR asscoiated differentially methylated regions after adjustment for grip strength. Supplementary Table 28. HOMA2-IR asscoiated differentially methylated regions after adjustment for gait speed. Supplementary Table 29. HOMA2-IR asscoiated differentially methylated regions after adjustment for ALMi. Supplementary Table 30. HbA1c asscoiated differentially methylated regions after adjustment for BMI. Supplementary Table 31. HbA1c asscoiated differentially methylated regions after adjustment for grip strength. Supplementary Table 32. HbA1c asscoiated differentially methylated regions after adjustment for gait speed. Supplementary Table 33. HbA1c asscoiated differentially methylated regions after adjustment for ALMi. Supplementary Table 34. MEAT methylation clock for HOMA2-IR and HbA1c unadjusted and adjusted for BMI.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Burton, M.A., Garratt, E.S., Hewitt, M.O. et al. DNA methylation of insulin signaling pathways is associated with HOMA2-IR in primary myoblasts from older adults. Skeletal Muscle 13, 17 (2023). https://doi.org/10.1186/s13395-023-00326-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13395-023-00326-y