
Abstract
As age increases, the risk of developing type 2 diabetes increases, which is associated with senile skeletal muscle dysfunction. During skeletal muscle aging, mitochondrial dysfunction, intramyocellular lipid accumulation, increased inflammation, oxidative stress, modified activity of insulin sensitivity regulatory enzymes, endoplasmic reticulum stress, decreased autophagy, sarcopenia and over-activated renin-angiotensin system may occur. These changes can impair skeletal muscle insulin sensitivity and increase the risk of insulin resistance and type 2 diabetes during skeletal muscle aging. This review of the mechanism of the increased risk of insulin resistance during skeletal muscle aging will provide a more comprehensive explanation for the increased incidence of type 2 diabetes in elderly individuals, and will also provide a more comprehensive perspective for the prevention and treatment of type 2 diabetes in elderly populations.

Similar content being viewed by others


Background
Globally, the incidence of diabetes and the number of patients with diabetes are rising sharply, which has resulted in a worldwide public health problem that seriously threatens human health. According to the international diabetes federation (IDF), there are nearly 500 million people with diabetes in the world, and approximately 80% of people with diabetes live in low- income and middle-income countries. In 2017, there were approximately 425 million people with diabetes in the world between the ages of 20 and 79 years, and this number will grow to 629 million by 2045 [51]. The incidence of type 2 diabetes is significantly higher in the elderly than in young people [22].
Skeletal muscle is an important tissue involved in the body’s glucose metabolism (after a mixed meal, the skeletal muscle has important role in glucose disposal) and has an important effect on insulin sensitivity. Skeletal muscle dysfunction is involved in the development of type 2 diabetes. As age increases, many changes and dysfunctions occur in skeletal muscle. Therefore, the increased incidence of type 2 diabetes in the elderly may be closely related to skeletal muscle aging, but the underlying mechanism has not yet been elucidated. This review of the mechanism underlying the increased risk of insulin resistance during the skeletal muscle aging process will provide a more comprehensive explanation for the increased incidence of type 2 diabetes in elderly individuals and will also provide a more comprehensive perspective for the prevention and treatment of type 2 diabetes in elderly populations.
Aging skeletal muscle is an independent risk factor for insulin resistance
The risk of type 2 diabetes increases with age. Studies have shown that the prevalence of diabetes, impaired fasting glucose or impaired glucose tolerance in 20 to 39-year-olds in the United States was 20.9%, whereas the prevalence in individuals from 40 to 59 years old was 46.9%, the prevalence in individuals from 40 to 59 years old was 67.4%, and the prevalence in individuals ≥ 75 years old was 75.6% [22]. In addition, with increasing age, insulin sensitivity gradually decreased, the body’s glucose regulation ability decreased, and muscle atrophied [47]. Studies have shown that, compared with younger males (30 years old), older males (65–70 years) have reduced glucose metabolism and decreased expression of skeletal muscle glucose transporter 4 (GLUT4) [70]. Older people also exhibit lower skeletal muscle insulin-stimulated Akt activity [59], impaired insulin signaling, and decreased skeletal muscle insulin sensitivity. In addition, skeletal muscle insulin sensitivity in aged mice is reduced, and insulin resistance occurs [24, 84]. These data indicate that the glucose metabolism capacity of the skeletal muscle decreased and the risk of insulin resistance increased. The underlying mechanism is related to a series of changes in skeletal muscle during the aging process.
The pathophysiological changes in aging skeletal muscle
Skeletal muscle aging refers to the inevitable deterioration of skeletal muscle cell structure and biological function with age increased [23]. Major features such as decreased mitochondrial function, increased intramyocellular lipid, increased inflammatory levels, increased oxidative and endoplasmic reticulum stress, weakened enzyme activities, decreased autophagic capacity, decreased muscle mass and over-activated renin-angiotensin system will appear. These factors can lead to an increased risk of insulin resistance and will be discussed in details.
Mitochondrial dysfunction
Mitochondrial dysfunction during skeletal muscle aging
Changes in skeletal muscle mitochondrial structure and function are important features of skeletal muscle aging. In terms of the morphological structure, there is an imbalance in mitochondrial fusion and fission during skeletal muscle aging; mitochondria are larger and rounder, mitochondrial density is reduced, the number of mitochondria is decreased, and the mitochondria are characterized by matrix vacuolation and shorter cristae. Functionally, the ability to synthesize mitochondria and the oxidative capacity are significantly reduced [63], the production of mitochondrial reactive oxygen species (ROS) is increased, the antioxidant capacity is decreased, and the autophagy ability is correspondingly reduced in aging skeletal muscle. Studies have shown that the maximum oxygen uptake of skeletal muscle [85] and the steady-state oxygen uptake [80] decrease with age. Additionally, the maximum ATP production rate and the steady-state ATP production rate of aging skeletal muscle are also reduced [86], and this reduction is more notable in the slow muscle fibers [9]. In addition, the mitochondrial protein synthesis rate in aging skeletal muscle decreased by 40% [82], and mitochondrial protein homeostasis was impaired. These data suggest that a series of changes during skeletal muscle aging cause mitochondrial dysfunction.
Mitochondrial dysfunction can increase the risk of insulin resistance during skeletal muscle aging
Mitochondria play an important role in skeletal muscle insulin signaling [18], and the normal structure and function of mitochondria are also closely related to skeletal muscle insulin sensitivity. Studies have shown that there is a positive correlation between the number of skeletal muscle mitochondria and insulin sensitivity in elderly individuals [21]. Mitochondria are the major site of ROS production [25], and mitochondrial dysfunction will cause an increase in ROS production, and high levels of ROS impair insulin signaling pathways and induce skeletal muscle insulin resistance [18]. Moreover, due to the decreased oxidative phosphorylation and β-oxidation abilities of aging skeletal muscle, increased intramyocellular lipid accumulation also increases the risk of skeletal muscle insulin resistance. In addition, insulin is critical in maintaining normal mitochondrial function. Insulin inhibits FOXO1 activity, thereby maintaining the integrity of the mitochondrial electron transport chain (METC) and the ratio of NAD +/NADH [18] and protecting mitochondrial function. Therefore, insulin and mitochondria are interdependent; mitochondria require insulin for normal function, mitochondria are required for insulin signaling [99], and skeletal structure and function disorders in aging skeletal muscle increase the risk of insulin resistance.
Intramyocellular lipid accumulation
Intramyocellular lipid accumulation increases in aging skeletal muscle
With increasing age, the intramyocellular lipid (IMCL) content also increases gradually [27]. Studies have shown that older men have higher IMCL levels than younger individuals [89]. Moreover, skeletal muscle in older mice also contains higher levels of ceramide (CER) and diacylglycerol (DAG). Compared with young mice, the CER content of skeletal muscle in elderly mice increased two fold, and the DAG content also increased significantly [81]. These data indicate that IMCL accumulation increases in aging skeletal muscle. There are many mechanisms that cause IMCL accumulation. An important mechanism is the decrease in the oxidative phosphorylation and β-oxidation abilities of senile skeletal muscle mitochondria, which leads to a reduction in the oxidative decomposition of lipids in the skeletal muscle.
Intramyocellular lipid accumulation can increase the risk of insulin resistance in aging skeletal muscle
The accumulation of IMCL can impair skeletal muscle insulin signaling and promote skeletal muscle insulin resistance. Studies have shown that lipid infusion can lead to reduced insulin-induced skeletal muscle protein synthesis and decreased insulin sensitivity in healthy humans [33]. Reducing IMCL levels can improve skeletal muscle insulin sensitivity in obese mice [45]. However, the accumulation of simple triglycerides alone does not lead to insulin resistance [95]. As lipids accumulate, the levels of the CER and DAG increase, which impairs the insulin signaling pathway and promotes insulin resistance [96]. Studies have shown that increased CER levels in skeletal muscle in older individuals can promote insulin resistance [14], while reducing CER and DAG synthesis can improve skeletal muscle insulin sensitivity [61].
In addition, the accumulation of IMCL can also promote the inflammatory pathway [44], thereby promoting skeletal muscle insulin resistance. Studies have shown that skeletal muscle IMCL accumulation can increase the levels of inflammatory factors such as tumor necrosis factor-α (TNF-α), Toll-like receptor 2 (TLR2) and interleukin-1β (IL-1β) [71], thereby promoting the inflammatory pathway. Moreover, the expression of TLR2, TNF-α and IL-1β in old mice is also increased [81], thereby inhibiting Akt and mTOR activity and promoting insulin resistance [62]. Therefore, IMCL accumulation increases the risk of insulin resistance in aging skeletal muscle by impairing the insulin signaling pathway or promoting the inflammatory pathway.
Inflammation
Inflammation increases in aging skeletal muscle
The level of inflammation increases during skeletal muscle aging [41]. Studies have shown that, compared with young mice, the expression levels of the skeletal muscle inflammatory markers TLR2, TNF-α and IL-1β are elevated and the level of inflammation is increased in older mice [81]. There are many mechanisms by which the level of inflammation increases in aging skeletal muscle. For example, mitochondrial dysfunction, increased ROS production, IMCL accumulation, and ER stress caused by skeletal muscle aging can promote skeletal muscle inflammation.
Inflammation increases the risk of insulin resistance in aging skeletal muscle
Inflammation is closely related to skeletal muscle insulin resistance, and increased inflammation promotes skeletal muscle insulin resistance [38]. Studies have shown that TNF-α, monocyte chemoattractant protein-1 (MCP-1), C-reactive protein (CRP), ILs and other inflammatory factors can promote skeletal muscle insulin resistance [78]. After knocking out animal inflammatory factor-related receptors such as TNF-α receptors, skeletal muscle insulin resistance was improved [97]. These data suggest that inflammation promotes skeletal muscle insulin resistance.
Inflammation primarily impairs insulin signaling by activating the IKKβ/NF-κB and JNK pathways, thereby promoting skeletal muscle insulin resistance [26]. Studies have shown that IL-1 can activate the skeletal muscle IKKβ/NF-κB pathway, reduce IRS-1 activity, and promote skeletal muscle insulin resistance [93]. Skeletal muscle insulin resistance improves after the IKKβ/NF-κB pathway is inhibited [48]. Moreover, IL-6 also promotes the expression of cytokine signaling inhibitor 1 (SOCS1) and SOCS3, thereby degrading IRS-1 [93] and promoting insulin resistance. The JNK pathway and TNF-α impair insulin signaling by inducing IRS-1 serine phosphorylation [26]. In addition, inflammation can also promote the release of nitric oxide, thereby inhibiting the PI3K-Akt pathway and promoting insulin resistance [26]. Therefore, inflammation impairs insulin signaling by activating inflammation-related factors as well as the IKKβ/NF-κB and JNK pathways, thereby increasing the risk of insulin resistance in aging skeletal muscle.
Oxidative stress
Oxidative stress levels increase in aging skeletal muscle
Mitochondria are the main source of skeletal muscle ROS and are involved in the regulation of various physiological functions of skeletal muscle. At normal concentrations, ROS activate mitogen-activated protein kinase (MAPK) and play an important role in redox signaling and normal cell activity. High levels of ROS can damage mtDNA, proteins and lipids, stimulate apoptosis; and induce skeletal muscle oxidative stress and dysfunction [53]. Studies have shown that as the production of skeletal muscle ROS increases, oxidative stress levels increase, and damage skeletal muscle mtDNA, leading to skeletal muscle mitochondrial dysfunction [53]. After treating mouse skeletal muscle cells with H2O2, mitochondrial fragmentation increased and respiratory capacity decreased [40]. In addition, increased skeletal muscle ROS production in elderly individuals can directly affect the ATP synthase involved in the ETC pathway, thereby inhibiting ATP production and further reducing skeletal muscle mitochondrial function [98].
There are many reasons for the increase in ROS; the decreased cellular antioxidant capacity caused by decreased antioxidant enzyme (superoxide dismutase, catalase and glutathione peroxidase) activity [15] is an important factor leading to increased ROS production. Studies have shown that the activity of antioxidant enzymes is gradually reduced during the process of skeletal muscle aging [15]. The overexpression of skeletal muscle catalase in aged mice can improve age-related mitochondrial oxidative stress and dysfunction, and enhance mitochondrial energy metabolism [4]. These data suggest that the activity of antioxidant enzymes during skeletal muscle aging is reduced, which leads to an increase in ROS levels, leading to oxidative stress and dysfunction.
Oxidative stress increases the risk of insulin resistance in aging skeletal muscle
Oxidative stress can increase the risk of insulin resistance in aging skeletal muscle. Biomarkers of skeletal muscle oxidative damage such as malondialdehyde (MDA), protein carbonyl, 4-hydroxy-2-nonenal, hydroperoxide, protein oxidation products, 3-nitrotyrosine, advanced glycosylation products (AGEs), carbohydrate metabolites, and 8-hydroxy-2′-deoxyguanosine reduce skeletal muscle insulin sensitivity and increase the risk of insulin resistance [3, 35]. Studies have shown that after artificially increasing ROS production in myotubes, IRS-1 tyrosine phosphorylation, Akt activation, and GLUT4 translocation to the plasma membrane are impaired [103]. After treatment with losartan, insulin-stimulated IRS-1 phosphorylation, Akt activation, and GLUT4 translocation could be restored [103]. In addition, after treating the soleus muscle with nitric oxide, insulin-stimulated glucose uptake and glycogen synthesis were reduced and the phosphorylation of IRS-1 and Akt was also reduced [43]. These data suggest that increased ROS production in senile skeletal muscle can reduce insulin sensitivity.
Studies of the underlying mechanisms have shown that skeletal muscle oxidative stress can impair insulin signaling and induce insulin resistance [46]. Skeletal muscle oxidative stress activates several serine-threonine kinase pathways such as IKKβ/NF-κB and JNK, leading to IRS-1 degradation [30] and the inhibition of insulin signaling pathways. Moreover, oxidative stress can also inhibit the translocation of GLUT4 to the plasma membrane [49], further reducing the effect of insulin. In addition, oxidative stress can also induce insulin resistance by impairing mitochondrial function. As mentioned earlier, mitochondrial dysfunction can cause a decrease in mitochondrial β-oxidative capacity, leading to IMCL accumulation, inhibiting the activity of PI3K, Akt, and GLUT4; and inducing skeletal muscle insulin resistance. However, ROS can damage skeletal muscle mitochondrial function. Therefore, the increase in oxidative stress levels in senile skeletal muscle can activate the IKKβ/NF-κB and JNK pathways and impair mitochondrial function, thereby impairing the insulin signaling pathway in skeletal muscle and increasing the risk for insulin resistance.
Changes in the activity of enzymes that regulate insulin sensitivity
Protein tyrosine phosphatase 1B (PTP1B) is an enzyme that regulates insulin-sensitivity. PTP1B phosphorylates IRS-1 tyrosine residues, thereby impairing insulin signaling [102] and inducing skeletal muscle insulin resistance. Studies have shown that PTP1B knockout animal models exhibit increased skeletal muscle insulin sensitivity and reduced insulin resistance [60]. However, PTP1B overexpression can promote insulin resistance [29].
The expression level of PTP1B is increased in senile skeletal muscle. Studies have shown that skeletal muscle PTP1B levels are higher and IRS-1 activity is lower in old males (58 years old) than in young males (24 years old) [37]. In addition, PTP1B levels in skeletal muscle were higher, PTP1B interacted with IRS-1 more and insulin resistance was more severe in 28-week-old rats than in 10-week-old rats [5]. These data indicate increased expression of PTP1B in aging skeletal muscle. Therefore, PTP1B increases the risk of insulin resistance in aging skeletal muscle.
ER stress
ER stress levels increases in aging skeletal muscle
The endoplasmic reticulum (ER) is an important organelle of eukaryotic cells that is involved in synthesizing, folding, packaging and transporting proteins. During skeletal muscle aging, ER stress levels increase. Studies have shown that ER stress-related factors and markers (GRP78 and CHOP) in the soleus muscles of 32-month-old rats are significantly upregulated compared with the levels in the soleus muscles of 6-month-old rats [77]. Moreover, the expression of ER stress-related factors and markers (GRP78, PDI and CHOP) in the gastrocnemius muscles of 24-month-old mice was also significantly higher than that in 6-month-old mice [50]. These data indicate an increase in ER stress levels in senile skeletal muscle. Studies on the underlying mechanism have shown that ER function declines during skeletal muscle aging, leading to the accumulation of unfolded or misfolded proteins [11], thereby inducing ER stress. In addition, a high level of mitochondrial ROS can also induce ER stress [66], and the skeletal muscle aging process can produce a large amount of ROS, thereby further promoting ER stress.
ER stress increases the risk of insulin resistance in aging skeletal muscle
ER stress can disrupt protein folding, leading to the accumulation of misfolded protein [8], which can easily induce inflammation and lipid accumulation, thereby impairing insulin signaling and inducing skeletal muscle insulin resistance [69]. Studies have shown that ER stress can reduce the phosphorylation of IRS-1 and Akt, decrease the expression of oxygen-regulated protein 150 (ORP150), which prevents ER stress; and induce insulin resistance [104]. These data suggest that ER stress reduces skeletal muscle insulin sensitivity and induces skeletal muscle insulin resistance [83].
ER stress also promotes skeletal muscle insulin resistance through the JNK pathway. Studies have shown that ER stress activates JNK, thereby phosphorylating IRS-1 serine 307, impairing insulin signaling and inhibiting Akt phosphorylation. As a result, skeletal muscle insulin resistance is promoted [87]. The use of JNK inhibitors reversed the ER stress-induced inhibition of Akt phosphorylation, thereby improving skeletal muscle insulin sensitivity [92]. Therefore, ER stress can increase the risk of insulin resistance in aging skeletal muscle by directly impairing insulin signaling or activating the JNK pathway.
Autophagy
Autophagic ability decreases during skeletal muscle aging
The autophagic ability of skeletal muscle gradually decreases with age. Studies have shown that the levels of the p62, LC3-II and LC3-I autophagy markers in the skeletal muscle of aged rats are elevated, indicating that the autophagic ability of the skeletal muscle is weakened, resulting in impaired skeletal muscle function, which is more obvious with age [7]. In addition, the proteolytic capacity of mouse skeletal muscle [100] and rat skeletal muscle [31] also decreased with age, which may be related to the decreased lysosomal protease activity. Studies have found skeletal muscle lysosomal lipid accumulation in senile rats, which results in impaired lysosomal function [76], l decreased lysosomal protease activity [7], and decreased skeletal muscle autophagic ability. These data indicate that the activation of the autophagy-lysosomal pathway is reduced during skeletal muscle aging, which results in decreased autophagy in senile skeletal muscle.
Decreased autophagy increases the risk of insulin resistance during the skeletal muscle aging process
As mentioned earlier, skeletal muscle oxidative damage increases with age. The autophagy-lysosomal pathway degrades large amounts of skeletal muscle protein, thereby reducing the oxidative damage to the skeletal muscle [58]. Therefore, the decline in skeletal muscle autophagy is not conducive to the prevention of oxidative damage and is closely related to skeletal muscle insulin resistance. Studies have shown that autophagy markers, p62 levels and LC3-II and LC3-I ratios, are significantly increased in insulin-resistant myocytes, and the myocyte autophagic ability is reduced [17]. The expression of skeletal muscle autophagy-related genes (ATG14, RB1CC1/FIP200, GABARAPL1, SQSTM1/p62 and WIPI1) and proteins (LC3BII, SQSTM1/p62 and ATG5) were also significantly decreased in type 2 diabetic patients [72], and skeletal muscle autophagy was decreased. In addition, the insulin-stimulated p-Akt Ser473 levels were decreased and insulin sensitivity was reduced after blocking the autophagy of C2C12 myotubes with the lysosomal inhibitor chloroquine (CLQ) [17]. Insulin resistance was improved after increasing the autophagic ability of the C2C12 myotubes [17] and the L6 myocytes [1]. Therefore, reduced autophagy can increase the risk of insulin resistance during the process of skeletal muscle aging.
Sarcopenia
Older people are prone to sarcopenia
Mainly aging-associated skeletal muscle alternations are muscle atrophy, often accompanied by sarcopenia [36]. Sarcopenia is an age-related progressive decline in skeletal muscle mass and function in the absence of other diseases. A progressive decline in muscle mass begins around the age of 40, and muscle mass declines by approximately 8% every decade. After the age of 70, muscle mass decreases by 15% every decade [57]. Additionally, by the age of 70, the cross-sectional area of skeletal muscle is approximately 30% smaller than it was at 20 years old [73]. Additionally, as age increases, the composition of the skeletal muscle fiber types also changes. The proportion of type II muscle fibers is reduced [32], resulting in the muscle mass of type II muscle fibers becoming lower than that of type I muscle fibers. In addition, motor neurons also change. Due to the decreased number and vitality of senile skeletal muscle motor units [54], the neuromuscular dominance is also weakened, which is coupled with the decline in muscle mass in the aging skeletal muscles, resulting in a significant decrease in muscle strength [10]. Before the age of 70, leg strength is reduced by 10–15% every decade. After the age of 70, leg strength is reduced by 25–40% every decade [57]. By the age of 70, skeletal muscle strength is 20–40% lower than that of young people [79]. In addition, studies on the underlying mechanisms have shown that myostatin is a major regulator of skeletal muscle size and mass and is expressed almost exclusively in skeletal muscle [16]. The overexpression of myostatin can cause muscle atrophy and plays an important role in sarcopenia [91]. These data indicate a progressive decline in muscle mass and strength during skeletal muscle aging, which is associated with myostatin.
Sarcopenia increases the risk of insulin resistance in aging skeletal muscle
Skeletal muscle mass is an important factor in glucose and energy homeostasis [101] and is positively correlated with skeletal muscle insulin sensitivity. Studies have shown that increased muscle mass increases skeletal muscle glucose uptake and improves insulin sensitivity [20]. Sarcopenia can cause skeletal muscle mass and strength to decrease, thereby reducing skeletal muscle insulin sensitivity. Myostatin plays an important role in this process. Studies have shown that elderly mice treated with myostatin inhibitors for 4 weeks exhibited improvements in sarcopenia and increased skeletal muscle insulin sensitivity [16]. Skeletal muscle glucose utilization and insulin sensitivity are also increased in myostatin knockout mice [94]. Therefore, the decreased skeletal muscle mass and strength caused by sarcopenia can increase the risk of insulin resistance in aging skeletal muscle.
The renin-angiotensin system
RAS is activated in aging skeletal muscle
The renin-angiotensin system (RAS) plays pleiotropic roles in regulating mammalian pathophysiology. Angiotensin II (Ang II) is a key molecule of RAS and is produced as a result of sequential cleavage of angiotensinogen by renin and angiotensin-converting enzyme (ACE) [56]. RAS has two axes, one is the ACE/ANG II/AT1 receptor classic axis. Ang II can bind to the Ang II type 1 (AT1) receptor, thereby activating the AT1 receptor [2], and leading to cell proliferation, hypertrophic responses, apoptosis, generation of ROS, and tissue inflammation [56]. The other is the ACE2/ANG (1–7)/Mas receptor non-classical axis. Ang II is cleaved by ACE2 to form another peptide Ang (1–7). This ACE2-Ang (1–7) axis, acting via another G protein-coupled receptor Mas, is involved in vasodilatory, anti-fibrotic, and anti-inflammatory properties [52]. These two axes show different changes in aging skeletal muscle. Studies have shown that the skeletal muscle aging process can activate RAS classic axis and activate the AT1 receptor [55, 56, 64], which induces inflammation and oxidative stress. However, inhibiting the classic axis can prolong the physiological aging process and promotes longevity in rodents [12]. In addition, RAS non-classical axis weakens in aged skeletal muscles [75]. However, activating the RAS non-classical axis can reduce the aging phenotype in aged mice [75]. These data indicate that the RAS classical axis is activated and the RAS non-classical axis is weakened in aging skeletal muscle.
RAS activation increases the risk of insulin resistance in aging skeletal muscle
Excessive activation of RAS is closely related to skeletal muscle insulin resistance. Skeletal muscle ACE/ANG II/AT1 receptor axis activation can promote skeletal muscle insulin resistance and muscle atrophy [13, 34]. Studies have shown that after injecting Ang II into rats, skeletal muscle glucose tolerance and insulin signaling pathway are impaired, and skeletal muscle insulin resistance appears [90]. After inhibiting the ACE/ANG II/AT1 receptor axis, skeletal muscle glucose tolerance and insulin resistance were improved [42]. In addition, the ACE2/ANG (1–7)/Mas receptor axis can inhibit the ACE/ANG II/AT1 receptor axis, activate the insulin signaling pathway, and thereby improve skeletal muscle insulin resistance [42]. Studies have shown that after ACE inhibition, skeletal muscle insulin sensitivity is enhanced, and after the Mas receptor is inhibited, the enhancement effect is eliminated [28]. These data indicate that activation of the RAS classical axis can promote skeletal muscle insulin resistance, while activation of the RAS non-classical axis can inhibit the classical axis, thereby improving skeletal muscle insulin resistance. Therefore, activation of the RAS classical axis and weakening of the RAS non-classical axis in aging skeletal muscle may increase the risk of skeletal muscle insulin resistance.
Integrating these mechanisms
There are also interactions between these mechanisms. Among them, The ER and mitochondria join together at multiple contact sites to form specific domains, termed mitochondria-ER associated membranes (MAMs) [6, 19, 68]. It is closely related to the autophagy process. There are several important autophagy-related proteins in mitochondria, such as ATG5, which is critical for autophagosome formation, translocates to the MAM compartment during phagophore biogenesis and then dissociates from MAMs upon completion of the autophagosome [39]. Therefore, MAM plays an important role in autophagy, while mitochondrial dysfunction and ER stress can decrease autophagy capacity. In addition, increased ROS is an important factor inducing inflammation, while mitochondria and ER are important sources of ROS [67]. Therefore, mitochondrial dysfunction and ER stress can generate a large amount of ROS and induce inflammation and oxidative stress.
In summary, mitochondrial dysfunction and ER stress can decrease autophagy capacity, increased ROS production and IMCL accumulation, and then induce inflammation and oxidative stress. Furthermore, the over-activated renin-angiotensin system also increases inflammation levels and induces oxidative stress. In addition, the occurrence of sarcopenia will exacerbate the above processes. Finally, increased inflammation and oxidative stress can impair mitochondrial function and exacerbate ER stress in turn, thereby further exacerbating the above processes and increasing the risk of insulin resistance.
Prevention and treatment
The aforementioned mechanisms, such as mitochondrial oxidative ability, inflammation, oxidative stress, insulin sensitivity regulating enzymes, ER stress, autophagy ability, and RAS axis, can be used as targets for the prevention and treatment of aging skeletal muscle insulin resistance. In addition, there are non-pharmacological treatments, such as exercise, that can prevent and treat insulin resistance in aging skeletal muscle. Studies have shown that exercise can increase skeletal muscle mass and improve skeletal muscle insulin sensitivity [36, 65, 88]. Exercise can also enhance mitochondrial oxidative capacity, enhance skeletal muscle autophagy and antioxidant capacity, reduce oxidative stress and inflammation levels [36, 74], and improve skeletal muscle insulin resistance. Therefore, both pharmacological treatments targeting these mechanisms and exercise can prevent and treat aging skeletal muscle insulin resistance.
Conclusions
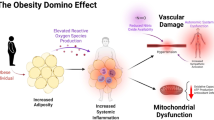
An increased risk of senile skeletal muscle insulin resistance is associated with skeletal muscle dysfunction. During the aging of skeletal muscle, mitochondrial dysfunction, intramyocellular lipid accumulation, increased inflammation, oxidative stress, changes in the activities of enzymes that regulate insulin sensitivity, endoplasmic reticulum stress, decreased autophagy, sarcopenia and over-activated RAS all induce skeletal muscle insulin resistance. These processes can impair skeletal muscle insulin sensitivity and increase the risk of insulin resistance and type 2 diabetes during the skeletal muscle aging process (Fig. 1). Of course, pharmacological treatments targeting these mechanisms and exercise can prevent and treat aging skeletal muscle insulin resistance. Therefore, in view of the above-mentioned aspects closely related to aging skeletal muscle insulin resistance, further exploration of relevant mechanisms and development of related drugs require further research in the future.

Skeletal muscle aging can increase insulin resistance by promoting mitochondrial dysfunction, IMCL accumulation, inflammation, oxidative stress, PTP1B expression, ER stress, decreased autophagy, sarcopenia and over-activated RAS. In addition, skeletal muscle mitochondrial dysfunction promotes IMCL accumulation and induces oxidative stress and ER stress, moreover, IMCL accumulation, oxidative stress, and ER stress can induce inflammation. IMCL intramyocellular lipid, PTP1B protein tyrosine phosphatase 1B, ER endoplasmic reticulum, RAS renin-angiotensin system
Availability of data and materials
All data generated or analysed during this study are included in this published article (Fig. 1).
Abbreviations
- IDF:
-
International diabetes federation
- GLUT4:
-
Glucose transporter 4
- ROS:
-
Reactive oxygen species
- METC:
-
Mitochondrial electron transport chain
- IMCL:
-
Intramyocellular lipid
- CER:
-
Ceramide
- DAG:
-
Diacylglycerol
- TNF-α:
-
Tumor necrosis factor-α
- TLR2:
-
Toll-like receptor 2
- IL-1β:
-
Interleukin-1β
- MCP-1:
-
Monocyte chemoattractant protein-1
- CRP:
-
C-reactive protein
- SOCS1:
-
Cytokine signaling inhibitor 1
- MAPK:
-
Mitogen-activated protein kinase
- AGEs:
-
Advanced glycosylation products
- PTP1B:
-
Protein tyrosine phosphatase 1B
- ER:
-
Endoplasmic reticulum
- ORP150:
-
Oxygen-regulated protein 150
- RAS:
-
Renin-angiotensin system
- ACE:
-
Angiotensin-converting enzyme
- Ang II:
-
Angiotensin II
References
Ahlstrom P, Rai E, Chakma S, Cho HH, Rengasamy P, Sweeney G. Adiponectin improves insulin sensitivity via activation of autophagic flux. J Mol Endocrinol. 2017;59:339–50.
Akazawa H, Yano M, Yabumoto C, Kudo-Sakamoto Y, Komuro I. Angiotensin II type 1 and type 2 receptor-induced cell signaling. Curr Pharm Des. 2013;19:2988–95.
Aldwin Suryo R, Morgan PE, Hawkins CL, Davies MJ. Cellular effects of peptide and protein hydroperoxides. Free Radic Biol Med. 2010;49:S93.
Alisa U, Gaetano S, Wenjun X, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci USA. 2014;111:15250–5.
Aparecida Emiko H, Fernanda AR, Miriam Sterman D, Mario José AS. Modulation of IR/PTP1B interaction and downstream signaling in insulin sensitive tissues of MSG-rats. Life Sci. 2003;73:1369–81.
Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20:1427–35.
Baehr LM, West DWD, George M, Marshall AG, Gustavo DSL, Keith B, Bodine SC. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Milano). 2016;8:127–46.
Bánhegyi G, et al. Endoplasmic reticulum stress. Ann N Y Acad Sci. 2007;1113:58–71.
Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–7.
Baumann CW, Kwak D, Liu H, Thompson LV. Age-induced oxidative stress: how does it influence skeletal muscle quantity and quality? J Appl Physiol. 2016;121:1047–52.
Belaya I, Suwa M, Chen T, Giniatullin R, Kanninen KM, Atalay M, Kumagai S. Long-term exercise protects against cellular stresses in aged mice. Oxid Med Cell Longev. 2018;2018:2894247.
Benigni A, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–30.
Cabello-Verrugio C, Morales MG, Rivera JC, Cabrera D, Simon F. Renin-angiotensin system: an old player with novel functions in skeletal muscle. Med Res Rev. 2015;35:437–63.
Campbell TL, et al. High-fat feeding does not induce an autophagic or apoptotic phenotype in female rat skeletal muscle. Exp Biol Med. 2015;240:657–68.
Campo AD, Jaimovich E, Tevy MF. Mitochondria in the aging muscles of flies and mice: new perspectives for old characters. Oxid Med Cell Longev. 2016;2016:1–10.
Camporez JP, et al. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc Natl Acad Sci USA. 2016;113:2212–7.
Chang YC, Liu HW, Chen YT, Chen YA, Chen YJ, Chang SJ. Resveratrol protects muscle cells against palmitate-induced cellular senescence and insulin resistance through ameliorating autophagic flux. J Food Drug Anal. 2018;26:1066–74.
Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21:589–98.
Chu Q, et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 2019;10:4883.
Cleasby ME, Jarmin S, Eilers W, Elashry M, Andersen DK, Dickson G, Foster K. Local overexpression of the myostatin propeptide increases glucose transporter expression and enhances skeletal muscle glucose disposal. Am J Physiol Endocrinol Metab. 2014;306:E814–23.
Coen PM, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci. 2013;68:447–55.
Cowie CC, et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care. 2009;32:287–94.
da Costa JP, Vitorino R, Silva GM, Vogel C, Duarte AC, Rocha-Santos T. A synopsis on aging-theories, mechanisms and future prospects. Ageing Res Rev. 2016;29:90–112.
Dagdeviren S, et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017;31:701–10.
David S, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–8.
De Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105.
Donato AR, et al. Increased ceramide content and NFκB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males. J Appl Physiol. 2012;113:1727–36.
Echeverria-Rodriguez O, Del Valle-Mondragon L, Hong E. Angiotensin 1-7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides. 2014;51:26–30.
Elchebly M, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–8.
Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–52.
Ferrington DA, Husom AD, Thompson LDV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–6.
Fielding RA, et al. Sarcopenia: an undiagnosed condition in older adults. current consensus definition: prevalence, etiology, and consequences. International Working Group on Sarcopenia. J Am Med Dir Assoc. 2011;12:249–56.
Francis BS, Carolyn C, Benjamin TW, Andrew JM, Chris ES, van Loon LJ, Kostas T. Lipid-induced insulin resistance is associated with an impaired skeletal muscle protein synthetic response to amino acid ingestion in healthy young men. Diabetes. 2015;64:1615–20.
Frantz EDC, Prodel E, Braz ID, Giori IG, Bargut TCL, Magliano DC, Nobrega ACL. Modulation of the renin-angiotensin system in white adipose tissue and skeletal muscle: focus on exercise training. Clin Sci (Lond). 2018;132:1487–507.
Furukawa S, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2017;114:1752–61.
Gomes MJ, et al. Skeletal muscle aging: influence of oxidative stress and physical exercise. Oncotarget. 2017;8:20428–40.
Guadalupe-Grau A, Larsen S, Guerra B, Calbet JAL, Dela F, Helge JW. Influence of age on leptin induced skeletal muscle signalling. Acta Physiol. 2014;211:214–28.
Haiyan X, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30.
Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–93.
Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–67.
Haran PH, Rivas DA, Fielding RA. Role and potential mechanisms of anabolic resistance in sarcopenia. J Cachexia Sarcopenia Muscle. 2012;3:157–62.
Henriksen EJ, Prasannarong M. The role of the renin-angiotensin system in the development of insulin resistance in skeletal muscle. Mol Cell Endocrinol. 2013;378:15–22.
Holappa M, Vapaatalo H, Vaajanen A. Many faces of renin-angiotensin system—focus on eye. Open Ophthalmol J. 2017;11:122–42.
Holland WL, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–70.
Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79.
Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–8.
Huffman DM, Barzilai N. Role of visceral adipose tissue in aging. BBA General Subjects. 2009;1790:1117–23.
Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Silvio I, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–6.
Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J. 2017;40:257–62.
Hwee DT, Baehr LM, Philp A, Baar K, Bodine SC. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell. 2013;13:92–101.
Jeannette A, Chris P, Esther J, Anoop M, Teresa T, Elizabeth BS. IDF DIABETES ATLAS eighth edition 2017. Brussels: IDF; 2017.
Jiang F, et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol. 2014;11:413–26.
Johnson ML, Robinson MM, Sreekumaran NK. Skeletal muscle aging and the mitochondrion. Trends Endocrinol Metab. 2013;24:247–56.
Joseph AM, Adhihetty PJ, Leeuwenburgh C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J Physiol. 2016;594:8695716.
Kamo T, Akazawa H, Komuro I. Pleiotropic effects of angiotensin II receptor signaling in cardiovascular homeostasis and aging. Int Heart J. 2015;56:249–54.
Kamo T, Akazawa H, Suzuki JI, Komuro I. Roles of renin-angiotensin system and Wnt pathway in aging-related phenotypes. Inflamm Regen. 2016;36:12.
Kim TN, Choi KM. Sarcopenia: definition, epidemiology, and pathophysiology. J Bone Metab. 2013;20:1–10.
Kim Y, Triolo M, Hood DA. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev. 2017;2017:1–16.
Kitt Falk P, et al. Effect of aging on muscle mitochondrial substrate utilization in humans. Proc Natl Acad Sci USA. 2015;112:11330–4.
Klaman LD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–89.
Kurek K, Mikłosz A, Łukaszuk B, Chabowski A, Górski J, Żendzian-Piotrowska M. Inhibition of ceramide de novo synthesis ameliorates diet induced skeletal muscles insulin resistance. J Diabetes Res. 2015;2015:1–9.
Legrand D, Adriaensen W, Vaes B, Mathe Iuml C, Wallemacq P, Degryse J. The relationship between grip strength and muscle mass (MM), inflammatory biomarkers and physical performance in community-dwelling very old persons. Arch Gerontol Geriatr. 2013;57:345–51.
Ljubicic V, Hood DA. Diminished contraction-induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell. 2009;8:394–404.
Lory V, Balazova L, Krskova K, Horvathova L, Olszanecki R, Suski M, Zorad S. Obesity and aging affects skeletal muscle renin-angiotensin system and myosin heavy chain proportions in pre-diabetic Zucker rats. J Physiol Biochem. 2019;75:351–65.
Luan X, Tian X, Zhang H, Huang R, Li N, Chen P, Wang R. Exercise as a prescription for patients with various diseases. J Sport Health Sci. 2019;8:422–41.
Ly LD, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017;49:e291.
Marchi S, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635.
Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837:461–9.
Mélissa F, Eric H, Pascal F, Fabienne F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–90.
Meneilly GS, Elliott T, Tessier D, Hards L, Tildesley H. NIDDM in the elderly. Diabetes Care. 1996;19:1320–5.
Michot C, et al. Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim Biophys Acta. 2013;1832:2103–14.
Møller AB, et al. Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Sci Rep. 2017;7:43775.
Muscaritoli M, et al. Consensus definition of sarcopenia, cachexia and pre-cachexia: joint document elaborated by Special Interest Groups (SIG) “cachexia-anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin Nutr. 2010;29:154–9.
Nieman DC, Wentz LM. The compelling link between physical activity and the body’s defense system. J Sport Health Sci. 2019;8:201–17.
Nozato S, et al. Angiotensin 1–7 alleviates aging-associated muscle weakness and bone loss, but is not associated with accelerated aging in ACE2-knockout mice. Clin Sci (Lond). 2019;133:2005–18.
O’Leary MF, Vainshtein A, Iqbal S, Ostojic O, Hood DA. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304:C422–30.
Ogata T, Machida S, Oishi Y, Higuchi M, Muraoka I. Differential cell death regulation between adult-unloaded and aged rat soleus muscle. Mech Ageing Dev. 2009;130:328–36.
Paresh D, Ahmad A, Arindam B. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25:4–7.
Porter MM, Vandervoort AA, Lexell J. Aging of human muscle: structure, function and adaptability. Scand J Med Sci Sports. 2010;5:129–42.
Proctor DN, Joyner MJ. Skeletal muscle mass and the reduction of VO2max in trained older subjects. J Appl Physiol. 1997;82:1411–5.
Rivas DA, Mcdonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol. 2016;310:R561–9.
Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci USA. 1996;93:15364–9.
Salvadó L, Palomer X, Barroso E, Vázquez-Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26:438–48.
Seo E, Kim S, Lee SJ, Oh BC, Jun HS. Ginseng berry extract supplementation improves age-related decline of insulin signaling in mice. Nutrients. 2015;7:3038–53.
Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA. 2005;102:5618–23.
Short KR, Vittone JL, Bigelow ML, Proctor DN, Nair KS. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am J Physiol Endocrinol Metab. 2004;286:E92–101.
Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6:174–84.
Song G, Chen C, Zhang J, Chang L, Zhu D, Wang X. Association of traditional Chinese exercises with glycemic responses in people with type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. J Sport Health Sci. 2018;7:442–52.
St-Jean-Pelletier F, et al. The impact of ageing, physical activity, and pre-frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J Cachexia Sarcopenia Muscle. 2017;8:213–28.
Surapongchai J, Prasannarong M, Bupha-Intr T, Saengsirisuwan V. Angiotensin II induces differential insulin action in rat skeletal muscle. J Endocrinol. 2017;232:547–60.
Teresa AZ, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8.
Thoudam T, et al. PDK4 Augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes. 2018;68:571–86.
Tilg H, Moschen A. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008;14:222–31.
Tingqing G, William J, Tatyana C, Jennifer P, Oksana G, Mcpherron AC. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE. 2009;4:e4937.
Turner N, Cooney GJ, Kraegen EW, Bruce CR. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J Endocrinol. 2014;220:T61–79.
Ussher JR, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–64.
Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-α-mediated insulin resistance in genetic obesity. Endocrinology. 1998;139:4832–8.
Wagatsuma A, Sakuma K. Molecular mechanisms for age-associated mitochondrial deficiency in skeletal muscle. J Aging Res. 2012;2012:768304.
Westermeier F, et al. Defective insulin signaling and mitochondrial dynamics in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1853:1113–8.
Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2010;45:138–48.
Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr. 2006;84:475–82.
Yip SC, Saha S, Chernoff J. PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci. 2010;35:442–9.
Yongzhong W, et al. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2006;281:35137–46.
Yoshihisa N, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–51.
Acknowledgements
Not applicable.
Funding
National Natural Science Foundation of China (31300975), Natural Science Foundation of Shanghai (18ZR1437100), Shanghai Key Lab of Human Performance (Shanghai University of sport) (11DZ2261100).
Author information
Authors and Affiliations
Contributions
JS wrote the full text, and WX is the correspondent author. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Iagree to publish all the data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shou, J., Chen, PJ. & Xiao, WH. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr 12, 14 (2020). https://doi.org/10.1186/s13098-020-0523-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13098-020-0523-x