
Abstract
In drug discovery, rapid and accurate prediction of protein–ligand binding affinities is a pivotal task for lead optimization with acceptable on-target potency as well as pharmacological efficacy. Furthermore, researchers hope for a high correlation between docking score and pose with key interactive residues, although scoring functions as free energy surrogates of protein–ligand complexes have failed to provide collinearity. Recently, various machine learning or deep learning methods have been proposed to overcome the drawbacks of scoring functions. Despite being highly accurate, their featurization process is complex and the meaning of the embedded features cannot directly be interpreted by human recognition without an additional feature analysis. Here, we propose SMPLIP-Score (Substructural Molecular and Protein–Ligand Interaction Pattern Score), a direct interpretable predictor of absolute binding affinity. Our simple featurization embeds the interaction fingerprint pattern on the ligand-binding site environment and molecular fragments of ligands into an input vectorized matrix for learning layers (random forest or deep neural network). Despite their less complex features than other state-of-the-art models, SMPLIP-Score achieved comparable performance, a Pearson’s correlation coefficient up to 0.80, and a root mean square error up to 1.18 in pK units with several benchmark datasets (PDBbind v.2015, Astex Diverse Set, CSAR NRC HiQ, FEP, PDBbind NMR, and CASF-2016). For this model, generality, predictive power, ranking power, and robustness were examined using direct interpretation of feature matrices for specific targets.

Similar content being viewed by others


Introduction
Protein–ligand binding in living organisms is a biological phenomenon that involves comprehensive processes such as molecular recognition and changes in protein conformation [1]. During drug development, any new molecules are evaluated empirically by measuring their binding strength to a protein target in vivo or in vitro. In contrast, computational ligand-based and target-based approaches are being used to predict the binding strengths of ligands [2,3,4]. In recent years, with advances in computational power, FEP (free-energy perturbation) methods [5, 6], MM–GBSA/MM-PBSA (molecular-mechanics–generalized Born surface area/molecular mechanics-Poisson–Boltzmann surface area) approaches [7,8,9], and molecular docking methods [10,11,12,13] have been widely used to accurately or relatively predict ligand binding poses and binding strengths with varying computational costs. Notably, for these predictions, physics-based, empirical, knowledge-based, and descriptor-based scoring functions have been used [14, 15]. These scoring functions are predetermined additive functional forms and are implemented in popular molecular docking programs such as AutoDock Vina [13], GlideScore [10], and Surflex-Dock [16]. Although these scoring functions were conveniently and widely used, they sometimes failed to discriminate binders from non-binders. Furthermore, although scoring functions as free energy surrogates for protein–ligand complexes have failed to provide collinearity, researchers hope for high correlations between docking scores and poses with key interactive residues. Thus, additional parameters have been included in the scoring functions of docking programs [17,18,19].
In recent years, machine learning (ML) and deep learning (DL) methods have achieved remarkable success in image and speech recognition, medical diagnosis, learning associations, classification, and regression analysis [20, 21]. ML methods have also been used to predict ligand binding strength by replacing linear scoring functions. These methods can be characterized by explicit and implicit features derived from protein, ligands, or protein–ligand pairs [22]. First, ligand binding strength depends on the vector summation of intermolecular interaction features such as hydrophobic, H-bond, π–π, cation–π, and charge interactions. Thus, several methods have been developed for extracting these features in different ways in the featurization process [23,24,25]. These features are either derived from an atom-centered or grid-based approach. For featurization, Gomes et al. [23] represented the structure of proteins and ligands as a combination of neighbor lists and atom types in their atom-centered approach for DL. Wallach et al. [26] and Ragoza et al. [27] represented the protein–ligand complex in a 3D-grid box to extract various interactions for the classification task. AtomNet [26], Pafnucy [28], KDEEP [24], and RosENet [29] are some recent examples using an atom-based or grid-based approach to extract the features to build a convolutional neural network (CNN) model. Although the state-of-the-art DL predictors showed statistically significant and robust performance in their tested protein–ligand databases, interpreting the models is a challenge and a problem hampering further progress. Obviously, the features for affinity prediction is complex for describing atomic information in 3D space and the dimensions of 3D features is higher than data dimension such as other drug discovery prediction models (eg. 3D QSAR). Notably, the features embedded through a featurization process tend to show inscrutable patterns for human and the models fail to show how to understand their prediction for the decision-making of drug design (especially, preferred substructure of a ligand, its desirable binding pose, and the correlation with binding affinity). Therefore, a simple and interpretable featurization is required to explain an effective binding mode together with its predictive model that has reliable predictive power.
Second, diverse representations of protein–ligand interactions have been generated. Examples include algebraic graph theory (AGL-Score) [30], multiple layers of specific element pairs (OnionNet) [31] that shows local and non-local interactions (distance-dependent), protein–ligand extended connectivity fingerprint (PLEC-NN) [32], docking features (in ΔvinaRF20) [33], and predefined protein–ligand-interactions (ID-Score) [14]. Furthermore, molecular fingerprints, which are popular features in ligand-based virtual screening, have been applied to encode protein–ligand interactions. The fingerprint pattern can help to annotate protein families and their bound ligands. Recently, versatile tools, which capture protein–ligand binding interaction information as a fingerprint pattern with a binary string of 1 (if an interaction is present) or 0 (if an interaction is absent), have been developed, such as PLIP (Protein–Ligand Interaction Profiler) [34], IFP (Interaction Fingerprint) [35], SIFt (Structural Interaction Fingerprint) [36], and APIF (atom-pair-based interaction fingerprint) [37]. Among these tools, IFP has gained considerable popularity and suitability in drug-discovery experiments and has been used for (i) post-processing the docking result [38], (ii) prioritizing the scaffold pose [39], (iii) predicting the ligand pose [40], (iv) selecting the virtual hits [41], (v) comparing binding sites [42], and (v) designing target-oriented libraries [43]. The notable merit of IFP is its on-the-fly calculation of the interactions based on a certain set of rules (atom-types) and geometric relationships (distances and angles) between the interacting atoms of proteins and ligands [35]. Based on IFP, Chupakhin et al. built a neural network model to predict ligand-binding modes for three chosen targets (CDK2, p38-α, and HSP90-α) [42]. Unfortunately, their model was limited to these three target proteins.
Third, in addition to protein–ligand interaction features, some scoring functions use features from ligand structures (e.g., AutoDock [12], AutoDock Vina [13], and NNScore 2.0 [44]). Lin et al. reported that ligand features can reveal effective polypharmacological relationships between target proteins [45]. Boyles et al. predicted ligand-binding affinity using combined ligand features (derived from RDKit) and different scoring function (RF-Score, NNScore, and Vina) features [3]. Notably, various ligand shapes, from linear to multiple ring systems, can exhibit different binding affinity strengths even within one homologous protein class, suggesting the importance of ligand features in binding affinity prediction models [46, 47]. Thus, taking these into account, the combination of ligand features and interaction-based features can further improve performance of the scoring function.
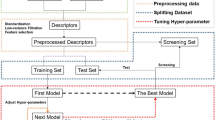
Based on these reported characteristics and drawbacks, we were motivated how to simplify usage, provide more interpretable features to explain protein–ligand binding directly, and provide ligand features for capturing polypharmacology. For this purpose, protein–ligand interaction-based fingerprint and ligand features were generated using IChem and SMF (substructural molecular fragment) tools, respectively. From these features, our best prediction model was realized in SMPLIP-Score (Substructural Molecular and Protein–Ligand Interaction Pattern-Score), as shown in Fig. 1. This work aimed to investigate three points: (a) the reliability of the predictive models that can be built from the IFP features of the protein–ligand complex and SMF of the ligand; (b) the efficiency of this featurization method based on complexity comparison within the SMPLIP features and between SMPLIP and the state of the art, and (c) the robustness or effectiveness of our models determined by comparing the predictive performances between simulated docking poses and experimental crystal poses.

SMPLIP-Score workflow for binding affinity prediction. Publically available protein–ligand binding datasets were used to extract the information encoding the interaction fingerprint pattern (IFP), interaction distances (Int-Dist) from protein–ligand complex, and ligand fragments to which machine learning or deep learning method may be applied for affinity prediction
Materials and methods
Dataset for learning
The protein–ligand database was downloaded from http://www.pdbbind-cn.org (PDBbind version 2015), which includes proteins and ligands in *.pdb and.mol2/.sdf file formats, respectively, each assigned with a PDB ID. The PDBbind database is a standard dataset that has previously been used to develop ligand-binding affinity prediction models [24, 25, 29, 31]. This dataset was categorized into three overlapping sets (i.e., general, refined, and core sets) with the total number of compounds for each set comprised of 11,908, 3706, and 195 proteins and ligands, respectively. These compounds were resolved by either X-ray or NMR methods with resolutions ranging from 0.75 to 4.60 Å, except for NMR solutions. The binding strength of each ligand to proteins was measured in IC50, Kd, and Ki and reported in mM, μM, and nM, respectively. In the present work, we used the refined and core sets with the binding strength in only Kd or Ki. The overlapping complexes between the refined and core sets were removed from the refined set. The refined set was randomly partitioned with a ratio of 80:20 into a train and valid set (a total of six subsets of the train and valid sets was created with a different random seed). Herein, the validation set was used to evaluate the robustness of the model's fit based on different random seeds. The training and validation were performed on a refined set and core set used to test the prediction performance of the developed models.
Dataset preprocessing
The PDBbind dataset was cleaned and processed using the KNIME analytic platform [48]. The silent features of the KNIME analytic platform allow any user to perform several programming tasks using several nodes, without any previous background knowledge of programming languages. We created datasets cleaning the KNIME workflow that included nodes from the Community and Schrödinger suite [49], considering the protein path as input, iteratively reading the input PDB structure, adding H-atoms, correcting the bond order, removing water molecules from the protein files, and converting them into *.mol2 files. During preprocessing, the protein file with a resolution of < 2.5 Å was retained, so that a well-resolved protein structure could be used for feature construction (Additional file 1: Figure S1). After the preprocessing steps, a total of 3481 and 180 datasets were retained for featurization from the refined and core sets, respectively. Additional file 1: Figure S2 shows the characterization of the input PDBbind dataset after preprocessing.
Feature construction
We used two types of features that represent the ligand's active-binding site environment; one was based on the interaction pattern observed between a ligand and the protein's binding site amino acid residues, and the second utilized ligand fragments based on atoms and neighboring atoms. The IFP between each protein–ligand complex was calculated using the IChem tool, which is based on OEChem TK [35]. Using this tool, we extracted only the on/off information of the IFP and then created seven-bit-string values for interactive amino acid residues without any additional geometrical parameters (e.g., angle, dihedral angle, or distance) or location information of atoms/residues. These seven-bit-string values represented hydrophobic, aromatic face-to-face, aromatic edge-to-face, H-bond accepted by ligand, hydrogen bond donated by ligand, ionic bond with ligand negatively charged, and ionic bond with ligand positively charged under standard geometric rules. Therefore, if any amino acid residues within the binding pocket formed any interaction with the ligand atoms, then the respective interaction was assigned a value of 1; otherwise, it was assigned a value of 0. Moreover, if two arginine residues respectively were hydrogen bonded with the ligand in a protein–ligand complex, the ‘ARG_HBond_LIGAND’ feature was assigned the integer 2. Considering the 20 standard amino acids in biological systems and the 7 bits of interaction information, a matrix of 20 × 7 = 140 was constructed. Notably, favorable interactions were only formed when two interacting atoms from proteins and ligands were in close proximity to each other. These interactions were distance-dependent and governed by spatial and geometric rules. Thus, the interaction distance information from the protein–ligand interaction pair was also extracted and combined with an interaction fingerprint pattern that equaled a total of 280 lengths (see detailed feature constructions in Additional file 1). The refined and core sets of the PDBbind database (release 2015) contain more than three thousand co-crystal ligands, which are structurally diverse and vary in shape and size with ligand lengths up to 47 Å. Thus, considering the different shapes and sizes of ligands, we used the SMF program to calculate the substructural fragment descriptors for the ligands [50]. Two types of substructural fragment descriptors were calculated: the sequence of atoms with a path length of up to six atoms and atoms with their neighbors. Both types contributed a total of 2282 substructural fragment descriptors (see detailed feature constructions in Additional file 1). The feature construction method used in this study is shown in Fig. 2.

a Interaction fingerprint pattern (IFP) generation. The interaction information (seven type) for each amino acid was extracted and interaction types from the same amino acid was concatenation in a matrix with a fixed size of 140. Similarly, the observed interaction distances were measured. b The ligand fragmentation pattern (FG1, FG2, …, FGN) for the input ligand, which corresponds to atoms sequence and augmented atoms were calculated. (Refer feature constructions in Additional file 1 for detailed information)
Machine learning and deep learning methods
In recent years, the various branches of artificial intelligence (i.e., ML and DL) have gained wide applicability in drug design and discovery, which includes predicting the numerous properties of a set of ligands or predicting affinity of the bound ligand in the protein binding pocket. Based on successful prediction performances, in the present study, we used random forest (RF) and deep neural network (DNN) as the ensemble learning method and DL method, respectively.
Ensemble learning
Ensemble learning-based methods combine several models, which were built individually to improve prediction performance. The general ensemble learning methods can be divided into two categories: bagging and boosting. Bagging is also called bootstrap aggregation, where multiple sample sets are produced and these sets are trained by individual learners. The main advantage of using bagging algorithms is that they decrease the prediction variance of the model and improve the accuracy of the ensemble. In this study, we used the RF as a bagging algorithm to build the regression model. RF is an ensemble of the decision tree (B) \(\{ T_{1} (X),....,T_{B} (X)\}\), as a base learning model, where \(X = \{ x_{1} ,...,x_{p} \}\) is a p-dimensional vector of molecular properties. Ensemble learning produces output \(\{ \widehat{Y}_{1} = T_{1} (X),....,\widehat{{Y_{B} }} = T_{B} (X)\}\), where \(\widehat{{Y_{b} }},b = 1,...,B,\) is the prediction for a molecule by the bth tree. The outputs obtained from all trees are aggregated to produce one final prediction for each molecule [51]. In regression modeling, this is the average of the individual tree predictions. The scikit-learn package (version 0.22) [52] was used to train and build the RF model. To build the models, different parameters from the RF were used (n_estimators = 100, 200, 300, 400, 500 and max_features = ‘auto’ and ‘sqrt’). The random_state parameters were fixed to different seed numbers during training to reproduce the statistical results (Additional file 1: Table S1).
Deep neural network
In recent decades, DL has been used for image classification, video processing, speech recognition, and natural language processing. In addition, these methods have been used in drug design and discovery applications over the last few years [53, 54]. A typical DNN method uses an artificial neural network (ANN) to make a decision or solve the problem. The standard DNN architecture includes the input layer, hidden layers, and output layers. In this study, we used a DNN to build the model and perform the predictions. The DNN model was trained using Keras (version 2.2.4) with the TensorFlow backend module [55]. The DNN training utilized a sequential model, which was initialized followed by four dense layers with 400, 200, and 100 units. Finally, the output layer was connected to one neuron to produce the predicted pKd. During the DNN training, early stopping criteria (Δloss), dropout, batch normalization, and L2 regularization were adopted to avoid over-fitting of the DNN model. The DNN model was trained with tunable parameters that included dropout regularization (0.1 to 0.6), alpha (0.1 to 1.0), and batch sizes of 64, 128, and 256. During learning, the best model was obtained as the learning entered an over-fitting stage, which was based on a modified loss formula (LOSS) adopted from Zheng et al. [31].
where PCC is the Pearson correlation coefficient and RMSE is the root mean square error. Furthermore, some additional parameters, such as a learning rate of 0.001, decay constant of 1e−6, and momentum of 0.9, were kept constant during the learning. Rectified linear units (ReLUs) were used at each layer as activation functions, and the stochastic gradient descent (SGD) optimizer was selected to search for optimal weights in the model. The DNN script was adopted from the work of Zheng et al. [31].
Evaluation metrics
The quality and performance of each ML or DL model were assessed using various evaluation metrics, including RMSE, mean absolute error (MAE), and PCC. The detailed information is as follows:
where RMSE measures the average magnitude of the error and represents the square root of the average of squared differences between the predicted and experimental values.
The MAE is another evaluation metric that differs from RMSE, as MAE is the average of the summed absolute differences of the predicted and experimental values.
The PCC was used to estimate the linear relationship between the predicted and experimental values. This metric also assesses the scoring function ability of the model.
Benchmark datasets for evaluation
We used five different benchmark datasets to assess the accuracy and efficiency of SMPLIP-Score. Previously, these datasets were used by many researchers to measure the quality and performance of their ML/DL/CNN models.
Astex diverse datasets
These datasets comprised diverse and high-quality protein–ligand pairs. Mooij et al. [56] manually curated these datasets to validate the protein–ligand docking program. After checking and comparing the overlapped protein–ligand pairs, 15 protein–ligand pairs remained for further processing.
Community structure–activity resource (CSAR) datasets
These in-house datasets were collected and managed by the University of Michigan. Among the CSAR datasets, we used the CSAR-HiQ-NRC (Set01 and Set02) benchmark dataset from http://csardock.org. The original input dataset contained 176 and 167 protein–ligand pairs for Set01 and Set02, respectively, with binding affinity data in Kd/Ki. After comparing and excluding the overlapped protein–ligand pairs from the refined set, a total of 56 and 64 pairs remained in Set01 and Set02 for further processing, respectively.
Comparative assessment of scoring functions (CASF) datasets
The CASF datasets are part of the PDBbind dataset and consist of a collection of high-quality protein–ligand complexes that are provided to assess scoring functions. We used CASF-2016 (http://www.pdbbind-cn.org/casf.php), which is comprised of 285 protein–ligand pairs with their experimental activity in Kd/Ki. A total of 122 protein–ligand pairs were selected after excluding overlapped pairs from the training set.
FEP dataset
This dataset is comprised of eight targets (BACE, CDK2, JNK1, MCL1, p38, PTP1B, thrombin, and Tyk2) and contains 199 compounds selected from the literature by Wang et al. [6] in order to predict relative ligand-binding affinities using the FEP method. While there are 199 compounds for eight targets, binding affinities in Ki have only been reported for five targets (BACE, MCL1, PTP1B, thrombin, and Tyk2). Therefore, in our work, we excluded CDK2, JNK1, and p38 from our FEP dataset. The remaining five targets were not part of the refined set, so all the reported compounds were selected for the BACE, MCL1, PTP1B, thrombin, and Tyk2 targets (36, 42, 22, 11, and 16 compounds, respectively).
NMR PDBbind dataset
We also tested the performance of the SMPLIP-Score on protein–ligand pairs that were resolved by NMR. The refined and core sets lacked NMR-resolved structures; thus, these structures were obtained from the general set. A total of 191 protein–ligand pairs were selected from the general set.
Results and discussion
We built predictive models using IFP and SMF features, optimized the models, and evaluated their predictive power on the benchmark dataset. We further tested the robustness and effectiveness of our best models using the poses (input features) derived from the molecular docking simulations. We used the RF as an ensemble method and DNN as a DL method to build the predictive models, SMPLIP-RF and SMPLIP-DNN.
SMPLIP-RF model
We first investigated the use of a single feature or combined features on the prediction performance of the model using the RF method. We trained six different models (with a different random seed) and different combinations of feature(s), n_estimators, and max_features were used for each model. All the parameters were set to default in the RF models, except max_features (‘auto’ or ‘sqrt’) and n_estimators (100, 200, 300, 400, or 500). Additional file 1: Table S1 and Figures S3–S10 report the statistical results, and the best model (based on the lowest RMSE reported on the test data) was selected for each feature or combined features after comparing all the statistical results for the different sets (Table 1 and Fig. 3).

The bar plot for statistical comparison from different feature (s) combination. The PCC (Pearson-correlation-coefficient) and RMSE (root-mean-squared-error) were compared from the train, valid, and test data
Comparing the predictive power of the test data (the RF models with different combinations of features), the features from IFP + Frag at n_estimators = 100 and max_features = ‘auto’ showed the best performance with the lowest RMSE (1.489) and high PCC (0.771) at a significance value of 8.71E−37 (refer to Additional file 1 for a detailed discussion). This model (IFP + Frag: 0.771) had a higher PCC and comparable performance to some models reported by Boyles et al. (Vina + RDKit: 0.749; RF-Score + RDKit: 0.778), but slightly lower than that from other models (RF-Score-v3 + RDKit: 0.780; NNScore 2.0 + RDKit: 0.786) [3]. Notably, this result suggests that facile, recognizable SMPLIP features (especially IFP + Frag) can show sufficient predictive power that is never inferior to known scoring using complex features such as atom-centered or grid-based features. Moreover, we built a null model and compared its statistics with the IFP and IFP + Frag features (Additional file 1: Table S2). The statistical metrics revealed that the addition of IFP features significantly reduced the overall prediction error.
SMPLIP-DNN model
We further evaluated the prediction performance of SMPLIP features using the DNN method. For the SMPLIP-DNN model, we used the same set of training, validation, and test data that had been used to compare the statistical performance of the RF model. The predictive model was built in our DNN training with screened hyperparameter values for dropout, alpha, and batch size. The models for each feature or combined features with optimized hyperparameter values are shown in Additional file 1: Table S3 and Figures S11–S14. In Additional file 1: Table S3, the best model at each epoch was based on the modified loss formula, which prevents overfitting of the model. The best model for each feature combination is shown in Table 2.
Notably, in the RF model, the IFP + Frag features possess high predictive power for the test data, while in the DNN model, these features did not dramatically improve the test data (PCC: 0.733; RMSE: 1.530). For the IFP + Frag features, the best model was obtained with a batch size of 64, dropout of 0.1, and alpha value of 0.7 at 129 epochs (Additional file 1: Figure S15).
Comparison of predictive performance with known models
We built the SMPLIP-RF and SMPLIP-DNN models to predict ligand-binding affinities. Under our investigated conditions (different partitioning of the dataset, different hyperparameter options, and chosen features), all models presented statistical reliability with the distribution of PCC and RMSE values from independently trained models (Additional file 1: Tables S1 and S2) and with their variance analysis (Additional file 1: Table S4). Although the IFP + Frag feature-based model outperformed on predicting binding affinity in both the RF and DNN methods, the statistical performance (PCC and RMSE) of the RF model on the test data was better than the DNN model. Figure 4 shows the correlation of predicted and experimental binding affinities from SMPLIP-RF (IFP + Frag) as a scatter plot for the training, valid, and test data.

2D-Scatter plot between predicted and experimental of SMPLIP-RF (IFP + Frag) for a training, b valid, and c test data
Consequently, the best SMPLIP-Score motivated us to assess its scoring and ranking power on other predictive models with different featurization methods. For this purpose, the performance of the SMPLIP-Score was further compared with that of other state-of-the-art models (Table 3). First, the SMPLIP-Score was compared with the featurizer methods of Gomes et al. [23], specifically atomic convolutional neural network (ACNN), GRID-RF, GRID-NN, GCNN, extended connectivity fingerprint (ECFP)-RF, and ECFP-NN. Their featurizer performs a 3D spatial convolution operation to learn atomic-level chemical interactions from a protein–ligand pair. Using these featurization methods, they performed poorly on the core set. Statistical comparison revealed that our SMPILP-Score performed better than the GRID-RF and ACNN models (Table 3). Second, the algebraic topology featurization of Cang et al. [57] was compared with the SMPLIP-Score. Despite their slightly higher PCC values than those of the SMPLIP-Score, the RMSE of the SMPLIP-Score was distinctly lower than their RMSE, suggesting better performance. Third, the rigidity index score (RI-Score) of Nguyen et al. [58] produced a higher RMSE using core data as the test set. Dziubinska et al.’s 3D grid-based CNN model, the Pafnucy model [28], was also inferior to our SMPLIP-Score model.
In turn, Wójcikowski et al.’s [32] circular fingerprint featurization (PLEC-linear and PLEC-NN) with different depth levels (protein depth of 5 and a ligand depth of 1) and the SMPLIP-Score produced comparable performances despite the fact that the latter uses fewer features (2422) than the PLEC (65,536) models do. Furthermore, the SMPLIP-Score is readily understandable based on the interpretability of the embedded feature matrix because SMPLIP feature is mimetic to human’s recognition on binding poses, which didn’t use either any geometrical or topological information. In detail, only existence information of residue types (20 amino acids), interactions (seven types), and ligand substructures (2282 fragments) were directly embedded in SMPLIP while the location numbers of bits (of the radial fingerprints) were embedded in PLEC [32]. Moreover, OnionNet [31], a popular method based on a feature matrix derived from rotation-free element pair-specific contacts between protein and ligand, achieved a slightly higher RMSE than the SMPLIP-Score based on the core set. Consistently, the SMPLIP-Score even outperformed popular ML scoring functions, such as RF-Score-v3 [59], X-Score [60], AutoDock Vina [61], and AutoDock [61] using the PDBbind v.2013 core set. Notably, all the models/methods compared here are different in terms of their featurization processes and require state-of-the-art architecture (ML/DL/CNN) to achieve predictive power.
Complexity comparison of SMPLIP with information cost
Obviously, time complexity can be determined by the dimensions of features (d), the number of data points (n), and learning parameters such as the number of bagged trees (t) of RF, and the number of layers (l) and number of nodes of DNN. As shown in the big O in Table 4, feature size (d) can be dominant in some model, which shows the dimensions of features are larger than the number of data points. To our best knowledge, almost all binding affinity prediction models also show d >> n for respective benchmark datasets. In the cases, an efficient feature size need to be considered for the complexity. Similarly, \({O}_{RF}\), can depend on the dimensions of sampled features (dʹ) under the conditions of similar data (nʹ) and hyperparameters (t). Thus, because the optimal number of PLEC features was 65,536 and 27-fold larger than the best SMPLIP feature, the PLEC-linear is 27-fold more complex than SMPLIP-linear under the same learning condition in Table 4. Expectedly, this difference in complexity between PLEC-RF and SMPLIP-RF is reduced from 27 folds into 5.2 folds (\({d}^{^{\prime}}: sqrt\)). In cases of DNN models, the number of learning parameters is overwhelmingly d or n, as shown in the \({O}_{DNN}.\) When we measured run times, the run times followed the big O. ‘IFP’, ‘IFP + Int-Dist’, ‘IFP + Frag’, and ‘IFP + Int-Dist + Frag’ showed their run times proportional to the dimensions of features under RF method. In the case of space complexity, the dimension of features didn’t make an effect on space complexity and different learning parameters show also trivial difference.
In addition to computational complexity, information cost (or information complexity) [62] of SMPLIP could be considered through the comparison of used information quantity. At this time, we considered Shanon information cost, H(X): = Ex∼X[− log2 PX(x)]. Because the expected value, E is the summation of each probability of each feature (\(-\sum_{i=1}^{d}{p}_{i}{\mathrm{log}}_{2}{p}_{i}\)), the information cost, H increases according to the dimensions of features. Thus, low dimensions of features show low information cost. Similarly, ‘IFP’ feature of SMPLIP is less complex than ‘IFP + Frag’ and ‘IFP + Frag’ of SMPLIP also is less complex than PLEC in the view of information cost. Notably, both IFP and Frag in SMPLIP only captured on/off information without either geometric parameters (e.g., angle, dihedral angle, or distance) or location information of atoms/residues. Despite of low information cost, SMPLIP showed comparative predictive performance to state-of-the art methods and SMPLIP-RF was the most cost-effective among tested learning conditions. Furthermore, the extraction of interesting features through simple counting approximates human recognition and data treatment. In other words, SMPLIP can provide direct interpretation, which does not require additional feature importance analysis such as the distribution of weights in learning architectures [28] or visualized contour functions (Ragoza et al.) [27]. When specific targets or chemical scaffolds need to be understood for drug design, the data matrix of SMPLIP can directly compare docking poses based on the simple counting. In brief, SMPLIP-RF demonstrated its cost-effectiveness based on the following views: (1) low computational complexity, (2) low information cost (presence or absence), (3) direct interpretation from embedded feature matrix to docking pose without additional analysis, and (4) predictive power comparable to state-of-the-art models.
Generalization of SMPLIP-Score evaluated through benchmark datasets
The generalization of the SMPLIP-Score was tested using additional benchmark datasets. These datasets were previously used by other researchers to evaluate the performance of their ML/DL/CNN models. The benchmark datasets used here belong to the Astex Diverse, CSAR NRC HiQ, CASF-2016, FEP, and PDBbind NMR datasets. SMPLIP-RF based on IFP + Frag features with n_estimators = 100 and max_features = ‘auto’ was used for generalized evaluation of the benchmark datasets. Table 5 lists the comparative assessment of the calculated scoring metrics (PCC, RMSE, and Spearman's rank correlation coefficient (Sp)) for these datasets. The scatter plot for the predicted binding affinity against the experimental binding affinity for these benchmark datasets is shown in Additional file 1: Figure S16. The original Astex Diverse dataset consisted of 93 protein–ligand pairs and after removing the overlapped pairs from the PDBbind refined set, a small set of 15 protein–ligand pairs remained for prediction measurements. We achieved PCC, Sp, and RMSE values of 0.724, 0.764, and 1.177 for this dataset, respectively. The SMPLIP-Score achieved a slightly lower scoring than DeepAtom but better scoring and ranking than RF-Score, Pafnucy, Res4HTMD, and RosENet using the Astex Diverse dataset with previously reported models [25, 28, 29, 63].
The second selected benchmark (the CSAR NRC HiQ dataset) consists of both Set01 and Set02. These sets were also used for docking program validation. After checking the overlapped protein–ligand pairs, 56 and 64 pairs were left in Set01 and Set02, respectively, for predictive evaluation. Compared with KDEEP, a CNN model, the SMPLIP-Score has better performance. However, a slightly lower performance was observed in Sp when compared to models from Res4HTMD and RosENet in predicting the binding affinity of the CSAR NRC HiQ sets [24, 29].
Another benchmark dataset, popularly known as the FEP dataset, was selected from the work of Wang et al. [6]. This dataset was used to predict the relative binding potency using a modern free-energy calculation protocol and forcefield. This dataset was comprised of ligands from BACE, MCL1, PTP1B, thrombin, and Tyk2 targets. Notably, except for thrombin, all prediction performances from SMPLIP-Score were positive, and the prediction ranking was PTP1B > Tyk2 > BACE > MCL1 > thrombin. Comparison of the prediction results with those of other methods (i.e., KDEEP and Res4HTMD) revealed that for all of the FEP targets, SMPLIP-Score performed better than the KDEEP model, while for BACE and PTP1B targets, Res4HTMD performed better [24, 29].
Finally, we further predicted the ligand-binding affinity of the dataset for which different experimental techniques have been used. A total of 191 protein–ligand pairs derived from the NMR method were selected and predicted for their ligand-binding affinities. Compared with the RosENet model, our model did not predict well the ligand-binding affinity derived from the NMR method [29]. Overall, SMPLIP-Score performed very well in benchmark evaluations in predicting the ligand-binding affinities of most cases, which affirms the reliability of our model for binding affinity predictions of diversified datasets, and it can be used further for virtual drug screening.
Ranking power on the benchmark dataset
SMPLIP-Score was further assessed for its ranking power, as indicated by the value of Sp for the CASF-2016 benchmark dataset. The CASF benchmark dataset consists of high, medium, and low active crystal and locally optimized poses from each protein target with a cluster number. Here, using these poses, two types of rankings were calculated; first, we calculated rankings of all Sp for the reduced set, and second, we calculated the ranking of individual Sp for each cluster, followed by an average of all clusters. The calculated PCC and Sp values for the CASF benchmark dataset are listed in Table 6. For this dataset, the PCC on the crystal pose and locally optimized pose remained the same, while there was a small increase in RMSE. Although the differences in root-mean-square deviations (RMSDs) for crystal and locally optimized poses were not large, such small differences in prediction/error were expected because any ML/DL model is sensitive to input features. Notably, the prediction results for the crystal pose and minimized pose were not even > 0.1, indicating that our model was less sensitive to the minimized pose than to the crystal pose. Additionally, the ranking metrics indicated that the average value of Sp for all the clusters was lower than the overall Sp. This suggests that, while each cluster has high, medium, and low active compounds, the difference in activity in some clusters is not large, rendering the ranking of compounds prone to change by prediction error. We further compared the prediction performance of the SMPLIP-Score on other ML/DL models. The statistical results from models based on the AutoDock Vina, ΔSAS (buried percentage of the solvent-accessible surface) [64], and ΔVinaRF20 [33] functions are listed in Table 6, which shows that the input features used in all three ML/DL methods are different from our features. Nevertheless, the performance of our model was greater than the AutoDock Vina and ΔSAS functions, but not ΔVinaRF20. Notably, ΔVinaRF20 uses descriptors derived from the AutoDock Vina interaction, ligand-dependent, and bSASA terms, which can result in superior performance with the benchmark dataset. Nevertheless, our prediction model uses protein–ligand interaction fingerprint and ligand-dependent features for predicting ligand-binding affinities, and we expect future additional interaction terms, such as desolvation, entropy effects, and surface and shape matching properties, may further improve the prediction performance.
Robustness and effectiveness of SMPLIP-Score
Ligand-based or target-based discovery depends on the reliability of predicted binding poses either through experimental or computational methods. In particular, molecular docking simulations are the more popular method to generate such poses with a typical RMSD criterion (< 2.0 Å) than X-ray crystal poses. In addition, because every predictive model for binding affinity relies on features generated from input poses, the robustness of such a model is also affected by the input poses. Thus, we studied the robustness of SMPLIP-Score according to the change in input poses. To measure the robustness of SMPLIP-Score, we conducted docking simulations using selected datasets (PDBbind-Core Set, Astex Diverse Set, and CASF-2016) to generate docking poses. The molecular docking simulations were performed using the AutoDock Vina program [13] (Exhaustiveness: 32; Num_modes: 50), and after docking, only poses with RMSD < 2.0 Å from the crystal pose were selected as reliable poses. For the PDBbind Core Set, Astex Diverse Set, and CASF-2016 Set, a total of 163, 15, and 100 poses had RMSD < 2.0 Å, respectively. Then, SMPLIP features (IFP + Frag) were extracted from the molecular docking poses and the ligand-binding affinities were predicted. The prediction results are shown in Table 7 and the scatter plot is shown in Fig. 5.

Thee Scatter plots of the predicted binding affinity against the experimental activity from the a PDBbind Core Set; b Astex Diverse Set; c CASF-2016 Set
When comparing the statistical performance of models built on docked poses (Table 7) with those built on crystal poses (Tables 1 and 5), we found that reasonable poses (RMSD < 2.0 Å) rarely made an effect on the affinity prediction and our result corroborated earlier work [31]. A slight variation in PCC and RMSE was observed between Tables 7 and 1 (or Table 5). In addition, feature matrices encoded by IChem and SMF can be directly compared between docked poses and crystal poses to reveal the interpretability of SMPLIP featurization. Thus, to analyze such changes, we randomly selected a few poses (crystal and docked) for comparison of interactions from these datasets. When two feature matrices were identical for both crystal and docked poses, they were excluded from this discussion. From the PDBbind Core Set, the superimposed crystal and docked poses for PDBs 2JDM and 3VH9 and IFP are shown in Additional file 1: Figure S17(a, b) and Tables S5, S6, respectively. The first selected 2JDM represents the protein–ligand pair from Pseudomonas aeruginosa lectin II (PA-IIL) in complex with methyl-a-l-fucopyranoside with the reported experimental binding affinity, pKd = 5.4 M [65]. Both the crystal pose and docked pose with RMSD of 0.376 Å showed similar interactions (Additional file 1: Table S5) and thus had the same predicted affinity of 5.739 M. Similarly, another selected PDB, 3VH9, belongs to an Aeromonas proteolytica aminopeptidase enzyme bound with 8-quinolinol, with an experimentally determined binding affinity of 6.2 M [66]. The IFP in Additional file 1: Table S6 shows that most of the interactions from crystal and docked poses were common; however, additional interactions such as H-bonds (Asp117, Asp179) and hydrophobic interactions (Cys223) have been observed, resulting in an improvement in the predicted binding affinity from 4.362 M (crystal pose) to 4.579 M (docked pose). Furthermore, the binding interactions of the superimposed crystal and docked poses for the selected PDBs 1TT1 and 1SQN from the Astex Diverse Set are shown in Additional file 1: Figure S18(a, b) and those from IFP are shown in Additional file 1: Tables S7, S8, respectively. PDB 1TT1 is a GluR6 kainate receptor subunit bound to 3-(carboxymethyl)-4-isopropenylproline with 4.19 M experimental binding affinity [67]. The binding interaction revealed that its docked pose had almost the same orientations of the groups. Interestingly, the crystal and docked poses had the same interactions with the binding site residues, even with an RMSD of 1.176 Å, and both poses were predicted at 6.488 M. Similarly, another selected PDB was 1SQN, which represents the progesterone receptor in complex with norethindrone with a reported affinity of 9.4 M [68]. Notably, most of the interactions were hydrophobic, and the crystal and docked poses had almost identical IFPs except for additional hydrophobic (Leu763, Phe778) interactions for the docked pose. While the docked pose had additional interactions, similar predicted values of 8.086 M and 8.002 M were observed for the crystal and docked poses, respectively. Lastly, we selected PDBs 2Y5H and 1W4O from the CASF-2016 Set to study the IFP. The first selected PDB, 2Y5H, contains factor Xa, a serine protease from the blood coagulation cascade, crystallized with derivatives of pyrrolo[3,4-a]pyrrolizine [69]. The superimposed crystal and docked poses and IFP are shown in Additional file 1: Figure S19(a) and Table S9, respectively. The calculated IFP showed identical interactions for the crystal and docked poses with the same predicted values of 7.459 M. Another selected PDB, 1W4O, represents ribonuclease-A protein in bound form with non-natural 3′-nucleotides [70] (Additional file 1: Figure S19(b)). The calculated IFP (Additional file 1: Table S10) for the docked pose shows that most of the interactions are shared by the crystal pose. However, a few interactions change their types while interacting with binding site residues. For example, the Phe120 residue formed H-bonds with the ligand (HBond_LIG) in the docked pose, whereas in the crystal pose, the backbone of the same residue formed HBond_PROT with the ligand. Similarly, in the crystal pose, Lys41 formed ionic (Ionic_PROT) interactions, while in the docked pose it formed H-bond (HBond_PROT) interactions. These trivial changes were reflected in the predicted affinity of the crystal pose (4.460) and docked pose (4.621).
Despite there being a close relationship between the predicted values for crystal and docked poses, we further evaluated the prediction values for poses with high RMSD values and checked the effectiveness of the SMPLIP-Score. For this purpose, we selected some poses with high RMSDs and predicted the binding affinity values (Additional file 1: Figures S20–S21 and Tables S11–S12). Additional file 1: Figure S20, Tables S11, S13 show the comparison of the binding pose (docked) and IFP for PDB 2JDM. This revealed that the docked pose with an RMSD of 3.53 Å (Additional file 1: Figure S20(b)) retained most of the hydrophobic interactions (Asp99, Ser23, and Thr98), but lost most hydrogen bond interactions, when compared with the crystal pose; this pose was predicted at pKd = 4.869 M. Likewise, another docked pose with an RMSD of 3.043 Å (Additional file 1: Figure S20(c)) also lost most of the hydrophobic and hydrogen bond interactions and predicted an affinity of 4.846 M. Notably, both predicted values for the docked poses were lower than the experimental binding affinity (5.4 M), while the most preferable docked pose with an RMSD of 0.376 Å (Additional file 1: Figure S20(a)) predicted 5.703 M, closer to the experimental value. This suggests that a reliable pose was required for an accurate prediction. Moreover, Additional file 1: Figure S21 and Table S12 show the comparison of the binding pose and IFP for docked poses from PDB 3VH9. Notably, the SMPLIP-Score predicted the crystal pose at 4.362 M, while its docked pose predicted 4.579 M (RMSD: 0.531 Å), 3.963 M (RMSD: 2.621 Å), and 4.199 M (RMSD: 4.27 Å) (Additional file 1: Table S13). Remarkably, the docked pose that predicted a value of 3.963 M showed only hydrophobic (Met180 and Ile255) interactions, but it did not make any hydrogen bond interactions due to flipping of the pose in the binding pocket. The prediction result further rationalizes the reliable pose requirement for accurate binding affinity predictions.
Overall, the comparison of IFP results from both docked and crystal poses indicates that, during the virtual screening (molecular docking) experiments, the identified small molecules must have complementary interactions with the crystal pose to be predicted accurately. Nonetheless, the additional interactions observed in the protein–ligand complexes may lead to changes in the observed prediction of binding affinity.
Conclusions
Herein, we report SMPLIP-Score as a robust and effective predictor and compared it with state-of-the-art featurization processes/methods. SMPLIP features, originating from protein–ligand interaction patterns and ligand features, showed cost-effectiveness as well as interpretability of the feature matrix embedded for learning. Most notably, the best SMPLIP features (IFP + Frag) demonstrated scoring power, ranking power, and robustness using various benchmark datasets. Obviously, the comparison between crystal and docked poses verified the robustness of SMPLIP-Score against input poses. Their interpretable feature matrices can be used directly to provide insight into ligand binding to a protein and the integrated description of binding mode with predicted affinity (having high accuracy) is a replaceable predictor of current scoring function.
Availability of data and materials
Knime workflow, python code, and refined data will be available in GitHub. https://github.com/college-of-pharmacy-gachon-university/SMPLIP-Score.
References
Gilson MK, Zhou H-X (2007) Calculation of protein-ligand binding affinities. Ann Rev Biophys Biomol Structure 36:21–42
Bajusz D, Ferenczy GG, Keseru MG (2017) Structure-based virtual screening approaches in kinase-directed drug discovery. Curr Top Med Chem 17(20):2235–2259
Boyles F, Deane CM, Morris GM (2020) Learning from the ligand: using ligand-based features to improve binding affinity prediction. Bioinformatics 36(3):758–764
Ripphausen P, Stumpfe D, Bajorath J (2012) Analysis of structure-based virtual screening studies and characterization of identified active compounds. Future Med Chem 4(5):603–613
Huang N, Kalyanaraman C, Bernacki K, Jacobson MP (2006) Molecular mechanics methods for predicting protein-ligand binding. Phys Chem Chem Phys 8(44):5166–5177
Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137(7):2695–2703
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10(5):449–461
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51(1):69–82
Lyne PD, Lamb ML, Saeh JC (2006) Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem 49(16):4805–4808
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749
Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267(3):727–748
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Li G-B, Yang L-L, Wang W-J, Li L-L, Yang S-Y (2013) ID-Score: a new empirical scoring function based on a comprehensive set of descriptors related to protein-ligand interactions. J Chem Inf Model 53(3):592–600
Liu J, Wang R (2015) Classification of current scoring functions. J Chem Inf Model 55(3):475–482
Jain AN (2003) Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem 46(4):499–511
Elokely KM, Doerksen RJ (2013) Docking challenge: protein sampling and molecular docking performance. J Chem Inf Model 53(8):1934–1945
Huang S-Y, Grinter SZ, Zou X (2010) Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys 12(40):12899–12908
Sousa SF, Fernandes PA, Ramos MJ (2006) Protein–ligand docking: current status and future challenges. Proteins Struct Funct Bioinform 65(1):15–26
Loh W-Y (2014) Classification and regression tree methods. Wiley StatsRef: Statistics Reference Online
Zhang Q, Yang LT, Chen Z, Li P (2018) A survey on deep learning for Big Data. Inf Fusion 42:146–157
Ellingson SR, Davis B, Allen J (2020) Machine learning and ligand binding predictions: a review of data, methods, and obstacles. Biochim Biophys Acta Gen Subj 1864(6):129545
Gomes J, Ramsundar B, Feinberg EN, Pande VS (2017) Atomic convolutional networks for predicting protein-ligand binding affinity. arXiv preprint arXiv:1703.10603
Jiménez J, Skalic M, Martinez-Rosell G, De Fabritiis G (2018) K Deep: protein-ligand absolute binding affinity prediction via 3d-convolutional neural networks. J Chem Inf Model 58(2):287–296
Li Y, Rezaei MA, Li C, Li X (2019) DeepAtom: a framework for protein-ligand binding affinity prediction, IEEE, pp 303–310
Wallach I, Dzamba M, Heifets A (2015) AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv preprint arXiv:1510.02855
Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR (2017) Protein-ligand scoring with convolutional neural networks. J Chem Inf Model 57(4):942–957
Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P (2018) Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 34(21):3666–3674
Hassan-Harrirou H, Zhang C, Lemmin T (2020) RosENet: improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. J Chem Inf Model 60(6):2791–2802
Nguyen DD, Wei G-W (2019) Agl-Score: algebraic graph learning score for protein-ligand binding scoring, ranking, docking, and screening. J Chem Inf Model 59(7):3291–3304
Zheng L, Fan J, Mu Y (2019) OnionNet: a multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction. ACS Omega 4(14):15956–15965
Wójcikowski M, Kukiełka M, Stepniewska-Dziubinska MM, Siedlecki P (2019) Development of a protein-ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 35(8):1334–1341
Wang C, Zhang Y (2017) Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J Comput Chem 38(3):169–177
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M (2015) PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res 43(W1):W443–W447
Da Silva F, Desaphy J, Rognan D (2018) IChem: a versatile toolkit for detecting, comparing, and predicting protein–ligand interactions. ChemMedChem 13(6):507–510
Deng Z, Chuaqui C, Singh J (2004) Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein−ligand binding interactions. J Med Chem 47(2):337–344
Pérez-Nueno VI, Rabal O, Borrell JI, Teixidó J (2009) APIF: a new interaction fingerprint based on atom pairs and its application to virtual screening. J Chem Inf Model 49(5):1245–1260
Chuaqui C, Deng Z, Singh J (2005) Interaction profiles of protein kinase—inhibitor complexes and their application to virtual screening. J Med Chem 48(1):121–133
Marcou G, Rognan D (2007) Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model 47(1):195–207
Chalopin M, Tesse A, Martínez MC, Rognan D, Arnal J-F, Andriantsitohaina R (2010) Estrogen receptor alpha as a key target of red wine polyphenols action on the endothelium. PLoS ONE 5(1):e8554
de Graaf C, Rognan D (2008) Selective structure-based virtual screening for full and partial agonists of the Β2 adrenergic receptor. J Med Chem 51(16):4978–4985
Chupakhin V, Marcou G, Baskin I, Varnek A, Rognan D (2013) Predicting ligand binding modes from neural networks trained on protein–ligand interaction fingerprints. J Chem Inf Model 53(4):763–772
Deng Z, Chuaqui C, Singh J (2006) Knowledge-based design of target-focused libraries using protein−ligand interaction constraints. J Med Chem 49(2):490–500
Durrant JD, McCammon JA (2011) NNScore 2.0: a neural-network receptor-ligand scoring function. J Chem Inf Model 51(11):2897–2903
Lin H, Sassano MF, Roth BL, Shoichet BK (2013) A pharmacological organization of G protein-coupled receptors. Nat Methods 10(2):140
Biessen EA, Bakkeren HF, Beuting DM, Kuiper J, Van Berkel TJ (1994) Ligand size is a major determinant of high-affinity binding of fucose-and galactose-exposing (Lipo) proteins by the hepatic fucose receptor. Biochem J 299(1):291–296
Smith RD, Engdahl AL, Dunbar JB Jr, Carlson HA (2012) Biophysical limits of protein-ligand binding. J Chem Inf Model 52(8):2098–2106
Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Thiel K, Wiswedel B (2009) KNIME-the Konstanz Information Miner: Version 2.0 and Beyond. AcM SIGKDD Explor Newsl 11(1):26–31
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3):221–234
Varnek A, Fourches D, Hoonakker F, Solov’ev VP (2005) Substructural fragments: an universal language to encode reactions, molecular and supramolecular structures. J Comput-Aided mol Des 19(9–10):693–703
Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP (2003) Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Comput Sci 43(6):1947–1958
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Cao C, Liu F, Tan H, Song D, Shu W, Li W, Zhou Y, Bo X, Xie Z (2018) Deep learning and its applications in biomedicine. Genom Proteom Bioinform 16(1):17–32
Jing Y, Bian Y, Hu Z, Wang L, Xie X-QS (2018) Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the Big Data Era. AAPS J 20(3):58
Chollet, F. Keras, 2015, GitHub. https://github.com/fchollet/keras
Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, Murray CW (2007) Diverse, high-quality test set for the validation of protein−ligand docking performance. J Med Chem 50(4):726–741
Cang Z, Mu L, Wei G-W (2018) Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput Biol 14(1):e1005929
Nguyen DD, Xiao T, Wang M, Wei G-W (2017) Rigidity strengthening: a mechanism for protein-ligand binding. J Chem Inf Model 57(7):1715–1721
Wójcikowski M, Zielenkiewicz P, Siedlecki P (2015) Open Drug Discovery Toolkit (ODDT): a new open-source player in the drug discovery field. J Cheminform 7(1):1–6
Khamis MA, Gomaa W (2015) Comparative assessment of machine-learning scoring functions on PDBbind 2013. Eng Appl Artif Intell 45:136–151
Gaillard T (2018) Evaluation of AutoDock and AutoDock Vina on the CASF-2013 benchmark. J Chem Inf Model 58(8):1697–1706
Braverman M (2015) Interactive information complexity. SIAM Journal on Computing 44(6):1698–1739
Ballester PJ, Schreyer A, Blundell TL (2014) Does a more precise chemical description of protein–ligand complexes lead to more accurate prediction of binding affinity? J Chem Inf Model 54(3):944–955
Su M, Yang Q, Du Y, Feng G, Liu Z, Li Y, Wang R (2018) Comparative assessment of scoring functions: the CASF-2016 update. J Chem Inf Model 59(2):895–913
Adam J, Pokorná M, Sabin C, Mitchell EP, Imberty A, Wimmerová M (2007) Engineering of PA-IIL lectin from Pseudomonas Aeruginosa-unravelling the role of the specificity loop for sugar preference. BMC Struct Biol 7(1):36
Hanaya K, Suetsugu M, Saijo S, Yamato I, Aoki S (2012) Potent inhibition of dinuclear zinc (II) peptidase, an aminopeptidase from aeromonas proteolytica, by 8-quinolinol derivatives: inhibitor design based on Zn 2+ fluorophores, kinetic, and X-ray crystallographic study. J Biol Inorg Chem 17(4):517–529
Mayer ML (2005) Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron 45(4):539–552
Madauss KP, Deng S-J, Austin RJ, Lambert MH, McLay I, Pritchard J, Short SA, Stewart EL, Uings IJ, Williams SP (2004) Progesterone receptor ligand binding pocket flexibility: crystal structures of the norethindrone and mometasone furoate complexes. J Med Chem 47(13):3381–3387
Salonen LM, Holland MC, Kaib PS, Haap W, Benz J, Mary J-L, Kuster O, Schweizer WB, Banner DW, Diederich F (2012) Molecular recognition at the active site of factor Xa: cation–π interactions, stacking on planar peptide surfaces, and replacement of structural water. Chem Eur J 18(1):213–222
Jenkins CL, Thiyagarajan N, Sweeney RY, Guy MP, Kelemen BR, Acharya KR, Raines RT (2005) Binding of non-natural 3′-nucleotides to ribonuclease A. FEBS J 272(3):744–755
Acknowledgements
The authors would like to thank OpenEye Scientific Software for providing an academic free license.
Funding
This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science and Technology (No.:2017R1E1A1A01076642, 2020R1I1A1A01074750).
Author information
Authors and Affiliations
Contributions
MK and SK conceived and designed the study. SK carried out all modeling and data work. MK and SK analyzed results, wrote the manuscript, and revised it. MK provided every research work facility. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors confirm that this article content has no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Additional tables, figures, and methods are available.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kumar, S., Kim, Mh. SMPLIP-Score: predicting ligand binding affinity from simple and interpretable on-the-fly interaction fingerprint pattern descriptors. J Cheminform 13, 28 (2021). https://doi.org/10.1186/s13321-021-00507-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-021-00507-1