
Abstract
Inherited retinal diseases (IRDs) can induce severe sight-threatening retinal degeneration and impose a considerable economic burden on patients and society, making efforts to cure blindness imperative. Transgenic animals mimicking human genetic diseases have long been used as a primary research tool to decipher the underlying pathogenesis, but there are still some obvious limitations. As an alternative strategy, patient-derived induced pluripotent stem cells (iPSCs), particularly three-dimensional (3D) organoid technology, are considered a promising platform for modeling different forms of IRDs, including retinitis pigmentosa, Leber congenital amaurosis, X-linked recessive retinoschisis, Batten disease, achromatopsia, and best vitelliform macular dystrophy. Here, this paper focuses on the status of patient-derived iPSCs and organoids in IRDs in recent years concerning disease modeling and therapeutic exploration, along with potential challenges for translating laboratory research to clinical application. Finally, the importance of human iPSCs and organoids in combination with emerging technologies such as multi-omics integration analysis, 3D bioprinting, or microfluidic chip platform are highlighted. Patient-derived retinal organoids may be a preferred choice for more accurately uncovering the mechanisms of human retinal diseases and will contribute to clinical practice.
Similar content being viewed by others
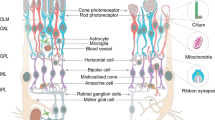

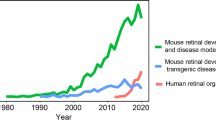
Background
Inherited retinal diseases (IRDs) affect millions globally and have become one of the leading causes of irreversible vision loss in children and the working population in developed countries [1,2,3]. IRDs, a group of disorders with high clinical and genetic heterogeneity, are associated with 317 pathogenic genes, among which 281 have been identified (RetNet: http://sph.uth.edu/retnet/, last accessed 10 June 2023). These genes have been found to play roles in almost all aspects of retinal structure and function, including retinal development, phototransduction, visual cycle, ciliary trafficking, ion channels, phagocytosis, mitochondrial function, protein degradation, outer segment structure, and pre-mRNA splicing [4]. Substantial progress has been made in elucidating the molecular genetic factors involved in IRDs and mutation screening techniques in the past two decades [5, 6]. However, the pathological mechanisms associated with specific genotypes still need to be better understood, owing to the availability of limited treatment options. Establishing accurate and available disease models, categorized by mutation and disease phenotype, is vital for gaining insight into IRDs.
Rodents have been widely employed as experimental models for studying the pathogenesis and treatment of human genetic diseases. However, they suffer the demerits of not altogether representing the occurrence and progression of the disease due to the translation barrier between rodents and humans [7, 8], e.g., mice are deficient in a macula on their retinas. Moreover, many studies reported that IRD mice fail to capture the pathological features of retinal photoreceptor degeneration [9,10,11,12].
Induced pluripotent stem cells (iPSCs) have the inherent merit of unlimited proliferation, self-renewal capacity, and multidirectional differentiation [13]. Human iPSCs can retain the unique genomic information of each individual since they are derived from autologous cells and devoid of limitations posed by embryonic stem cells, like ethical issues and immune rejection after transplantation [14]. Human iPSCs from somatic cell reprogramming have opened up an entirely new perspective for obtaining patient-specific cell lines, which are then differentiated into desired cell types under appropriate conditions, including retinal ganglion cells, vascular endothelial cells, cardiomyocytes, osteoblasts, hematopoietic cells, and neurons [14, 15]. In recent years, the emergence of three-dimensional (3D) organoids capable of forming complex tissue-like structures has gradually transformed our ability to model human development and disease, drug screening, and cell therapy [16,17,18]. Herein, this review systematically summarizes the role of patient-derived iPSCs and organoids in IRDs based on previously published studies.
Application of patient-derived iPSCs in IRDs
Reprogramming technology is considered one of the most important advances in the field of stem cell research and regenerative medicine [14, 19, 20]. Fibroblasts were the first somatic cells to be applied for reprogramming into human and mouse iPSCs [21, 22]. Subsequently, more and more cell types from patients were identified to induce iPSCs, like peripheral blood mononuclear cells (PBMCs), urine cells, and dermal fibroblasts, usually used as cell sources of human iPSCs, as shown in Table 1. In 2011, the first study of IRD patient-derived iPSCs for disease modeling and drug screening was reported by Jin et al. [23].
Although the human dermal fibroblast extraction and culture are convenient, it requires an invasive sampling of donor skin tissue which might translate into permanent scarring. Moreover, DNA variations within the cells are another potential obstacle due to long-term exposure of skin tissue to ultraviolet rays from sunlight [24]. As an alternative source, PBMCs can be isolated from routine blood samples with written informed patient consent and then reprogrammed into iPSCs, since most of the blood withdrawal methods use peripheral venipuncture techniques, which means less trauma and pain for the donor, but it has been reported that blood samples kept at room temperature for a long time without timely processing translate into a decreased number of iPSC colonies; nevertheless, they can be cryopreserved without affecting their reprogramming efficiency [25]. Compared to fibroblasts and PBMCs, urine cell extraction is non-invasive, convenient, simple, reproducible, and discomfort-free, facilitating the willingness of most participants for autologous urine collection. Urine samples are considered an ideal cell source for reprogramming technology because of their prominent advantages. However, poor proliferation and low success rates of urine cells derived from healthy adults and patients have been reported [26].
The preferred somatic cell source for generating iPSCs still needs more consensus since selecting a suitable cell source depends on the actual situation owing to the difference in extraction, culture, and expansion [27]. Notably, patient-specific iPSC cell source is generally required to be consistent with normal control-derived iPSCs. Furthermore, additional measures need to be taken while sampling to avoid contamination, such as skin surface disinfection, sterile disposable gloves and masks, and timely transportation and extraction of somatic cells. Mycoplasma detection of all cell samples was performed regularly. Uncontaminated and well-conditioned iPSCs are more conducive to differentiation into target cells and organoids.
Application of patient-derived organoids in IRDs
More and more evidence indicated that patient-derived retinal organoids (ROs) have the potential to serve as an ideal platform for tissue and organ reconstruction and in vitro disease modeling [28,29,30]. The main reasons are listed as follows: (1) ROs with laminar structure are similar to the natural retina and have a variety of tissue-specific cells, including photoreceptor cells, retinal pigment epithelium (RPE) cells, Müller glial cells, ganglion cells, amacrine cells, and bipolar cells [31]. (2) ROs show high reproducibility and fidelity of retinal development [17, 32, 33]. (3) Human iPSC-derived ROs have the advantages of fewer ethical concerns, easy availability, and large-scale production [34]. An overview of recent advances in patient-derived ROs in IRDs is below and summarized in Table 2.
Retinitis pigmentosa (RP)
RP, the most common type of IRD, is characterized by progressive degeneration of RPE cells and photoreceptors with a prevalence of approximately one in 4000 [35]. It initially manifests as night blindness, visual field constriction, and changes in the fundus, eventually leading to irreversible impairment of central vision [36]. The molecular pathogenesis of RP is not fully understood, and there is still no cure or effective treatment to slow down the disease progression [37, 38]. Patient-derived RPE cells and ROs for modeling RP could recapitulate the genotype–phenotype features of the disease. Due to clinical and genetic heterogeneity, different retinal degeneration phenotypes caused by intra-gene variations and the same phenotype caused by mutations in multiple genes could be presented.
Mutations in pre-mRNA processing factors (PRPFs) are the main cause of autosomal dominant RP related to the formation of the U4/U6.U5 tri-snRNP complex, a core spliceosome component [39]. For instance, RPE cells generated from iPSCs of an RP patient carrying the PRPF8 mutation showed widespread changes in alternative splicing events and dysregulated expression of genes involved in the splicing process and ribosome, indicating loss of spliceosome function [40]. In addition, ROs and RPE models from patient-specific iPSCs with the PRPF31 mutation showed impaired pre-mRNA splicing process as described by Baskin et al. On the other hand, abnormal photoreceptor and RPE changes were also observed, including cell morphology, cilium structure, apical-basal polarity, and phagocytosis function of the photoreceptor outer segment (POS) [41]. Such an aberrant phenotype was also observed in iPSC-RPE cells from the PRPF6-mutated patients [42]. These reports suggested that progressive RPE and photoreceptor degeneration might be attributed to the mis-splicing of genes vital for retinal structure and function. Similarly, cytoplasmic mislocalization of PRPF31 protein in RPE and photoreceptor cells with reduced expression in nuclear localization has been reported lately [43]. Moreover, the effect of PRPF31 mutation on the spliceosome impaired U4/U6.U5 tri-snRNP assembly and decreased splicing activity [43]. The mutant PRPF31 protein causes photoreceptor cell degeneration in organoids, with rods expiring first, followed by cones, which correspond to the results obtained in RP patients [44]. In contrast, some transgenic mice with prpf3T494M/+, prpf8H2309P/+, and prpf31± exhibited unsatisfactory performances in photoreceptor degeneration [9, 45].
The protein trafficking function of connecting cilia in photoreceptors is regulated by the retinitis pigmentosa GTPase regulator (RPGR), which is necessary for photoreceptor development. In the context of disease modeling, patient-derived ROs showed that gelsolin failed to be activated due to disturbed interaction between mutant RPGR protein and gelsolin, resulting in impaired F-actin disassembly of cilia and mislocalization of photoreceptor markers rhodopsin and opsin [46]. Mice with knockout RPGR and Gelsolin showed significant abnormalities in F-actin polymerization and rhodopsin expression [47]. Therefore, using patient-derived ROs could explain the ciliary phenotype of F-actin dysregulation as a unique RPGR mechanism. To explore the effectiveness of in vitro gene editing, Deng et al. performed CRISPR/Cas9-mediated gene correction of RPGR mutation to restore expression levels of target genes and proteins, thereby rescuing ciliary lesions and photoreceptor loss in iPSC-derived organoids from three RP patients [48]. Gene therapy for heterogeneous IRD may be a promising strategy to address the underlying molecular defects, but its development in clinical treatment remains a challenge.
Additionally, early retinal development is impeded in some forms of RP. Mutations in the USH2A gene encoding usherin protein induce autosomal recessive non-syndromic RP and Usher syndrome [49]. The usherin has been known to house several motifs associated with extracellular matrix (ECM) proteins, such as laminin and fibronectin type III, which are essential for supporting the centrosome-cilium interface and the inner segment/outer segment region of photoreceptors [50, 51]. The patient-specific ROs model carrying the USH2A mutation was established a decade ago [52]. Recently, a study revealed defective retinal progenitor cell development and neuroretinal layer formation due to abnormal retinal differentiation and polarization in the USH2A-related ROs, where increased apoptosis was observed in the mutated organoids along with decreased proliferation and laminin expression on day 34 compared to the normal control group [53]. Moreover, multi-omics data analysis showed that the down-regulation of ECM organization promoted patient-derived iPSCs and ROs apoptosis via the PI3K-Akt signaling pathway [37].
The gene IMPG2 encodes interphotoreceptor matrix proteoglycan 2, a protein expressed by cone and rod photoreceptor cells that plays a role in supporting the growth and maintenance of light-sensitive POS [54]. Mutation in IMPG2 is associated with a severe form of autosomal recessive RP. Impg2 knockout mice exhibited a relatively mild and late-onset photoreceptor phenotype compared to human disease [55]. Human iPSC-ROs harboring patient-specific or gene-edited mutations in IMPG2 universally lacked a functional POS layer and interphotoreceptor matrix disruption due to loss of IMPG2 protein or its inability to undergo normal post-translational modification. This POS phenotype was reversed after the correction of the IMPG2 mutation by CRISPR/Cas9 gene editing. Interestingly, transplantation of IMPG2-mutated ROs into the protected subretinal space of immunodeficient rats restored POS growth, suggesting that POS is vulnerable to mechanical stress environment [56].
Leber congenital amaurosis (LCA)
LCA is the most severe form of IRD, leading to congenital or early-onset blindness [57, 58]. Patients typically present with nystagmus, poor pupillary light response, severe retinal degeneration, and nearly disappeared full-field electroretinogram in infancy or childhood [59]. Luxturna was approved by the Food and Drug Administration (FDA) in 2017 as the first gene therapy drug in ophthalmology, but it is only available for biallelic RPE65 mutation-associated LCA with a mounting cost of $850,000 [60]. To date, at least 26 pathogenic genes have been linked to LCA, mainly in an autosomal recessive inheritance pattern (RetNet).
Mutation in the aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) gene is one of the most clinically severe forms of the disease (known as LCA-4 type), accounting for 5%–10% of all LCA cases [57]. AIPL1 protein acts as a photoreceptor-specific cochaperone, interacting with the molecular chaperone heat shock protein 90 (HSP90) to regulate the stability and assembly of phosphodiesterase 6 (PDE6) holoenzyme in the phototransduction cascade, which is responsible for regulating intracellular levels of cyclic guanosine monophosphate (cGMP) in rods and cones [61]. The phenotype of patient iPSC-derived ROs in vitro modeling LCA was similar to LCA-4 rodents as described previously [62]. The loss of AIPL1 protein hindered the PDE6 holoenzyme formation, resulting in increased cGMP levels in photoreceptor cells [63, 64]. Similarly, Leung et al. confirmed these molecular pathological changes in AIPL1-mutated ROs derived from four patients and attempted to investigate the effectiveness of the reagent PTC124, a translational read-through-inducing drug [65]. The results showed a slight increase in full-length AIPL1 protein but failed to completely restore the functional expression of PDE6 and reduce the cGMP levels in photoreceptor cells. However, CRISPR/Cas9-mediated gene editing could rescue the mutant phenotype, as observed in the AIPL1-corrected organoids [65].
In addition, modeling CRX mutation-related LCA using patient iPSC-ROs technique revealed immature photoreceptor cell development and reduced visual opsin expression [66], which were alleviated using CRX gene augmentation therapy mediated by adeno-associated virus (AAV) vectors [67]. The organoid model of the cilia gene CEP290 recapitulated the LCA-10 disease phenotype and exhibited abnormal splicing and ciliary defects [68]. Contrarily, eupatilin, a bioactive flavonoid, has improved cilium formation and length in CEP290-associated ROs [69]. CRISPR/Cas9-mediated gene correction of a nonsense variant in LCA5 rescued lebercilin expression and localization along the ciliary axoneme in patient-derived ROs [70].
X-linked recessive retinoschisis (XLRS)
XLRS, also called RS1-associated IRD, is characterized by a splitting of the neurosensory retina and cystic macular dystrophy affecting the young male population [71]. Retinoschisin encoded by the RS1 gene is assembled by retinal bipolar cells and photoreceptors, followed by its secretion into the extracellular surfaces as a homo-octameric complex [71]. The protein contains an amino-terminal signal peptide, the RS1 domain, and a discoidin domain, a specialized domain found in a family of extracellular surface proteins that plays an important role in retinal cell adhesion and cell–cell interactions [72]. At present, several XLRS mouse models have been constructed to recapitulate the retinoschisis phenotype [73, 74]. A recent study has shown that this specific retinopathy may occur in patient iPSC-derived ROs [75]. PBMCs were extracted from blood samples of two patients diagnosed with XLRS, reprogrammed into iPSCs, and then induced into RO disease models. On day 150 of differentiation, RS1 mutant ROs exhibited cyst/schisis-like features similar to the fundus characteristics of retinal splitting between the inner and outer nuclear layers in XLRS patients and mice. Western blotting and immunofluorescence staining showed that patient-derived ROs had aberrant RS1 protein expression and secretion, resulting in altered paxillin dynamics, photoreceptor development, and retinopathy-related gene expression [75]. Subsequently, CRISPR/Cas9-mediated correction of RS1 deficiency effectively reversed pathological changes in morphological structure and molecular expression, and likewise, introducing this RS1-specific mutation into normal control iPSCs successfully reproduced the disease phenotypes [75].
Other IRDs
Other relatively uncommon types of IRDs have also been studied, such as Batten disease, achromatopsia, and best vitelliform macular dystrophy (BVMD). It has been reported that patient-derived RO models of Batten disease with the CLN3 mutation exhibited altered pre-mRNA splicing, accumulation of mitochondrial ATPase subunit-C, peroxisomes mislocalization, and vacuolization of photoreceptor inner segments [76]. Achromatopsia is characterized by loss of cone photoreceptor function. At the same time, achromatopsia ROs from patients carrying the ATF6 variants exhibited molecular and cellular phenotypes, including cone defects, increased endoplasmic reticulum stress, Müller cell activation, disrupted mitochondrial structure, and elevated mitochondrial respiratory chain activity gene expression [77]. Intervention with AA147, a lead small molecular ATF6 agonist, may enhance cone photoreceptor growth and gene expression in the disease ROs by promoting Class 1 ATF6-regulated transcriptional activity [78]. In addition, impaired bestrophin channel activity was observed in BVMD patient-derived RPE cells with the BEST1 mutation, which was restored by AAV-mediated BEST1 gene augmentation [79, 80].
Taken together, a genotype–phenotype correlation of the disease was corroborated through a series of tests and analysis in patient iPSC-derived RO models, which can accurately reflect instead of mimic the complex clinical and genetic background of human retinal disease, may provide a very favorable experimental tool and platform for launching relevant research, and may also contribute to future drug development and gene therapy strategies. Recently, a clinical trial of a CRISPR/Cas9-mediated gene therapy drug for RP disease was conducted in China (NCT05805007).
Preclinical application
In recent years, stem cell-derived ROs can be prepared into suitable retinal sheets or purified photoreceptor cells for transplantation in animal models of retinal degeneration to restore the structural and functional integrity of the host retina [81,82,83,84,85]. Notably, purified photoreceptors can directly form host-graft synaptic contact but seldom survive for long after transplantation [86]. In contrast, neuroretina-like graft sheets develop a structured layer in the form of a rosette that promotes graft photoreceptor survival and synaptic interaction with host bipolar cells, and retinal ganglion cell responses to light can be detected via multiple electrode arrays in end-stage retinal degeneration models [87, 88]. A protocol for the preparation, quality control, and transplantation of retinal sheets into retinal degeneration rats has been established and validated previously [82]. Based on prior proof-of-concept studies, Kobe City Eye Hospital has launched the first human clinical trial in Japan using retinal sheets from allogeneic iPSC-derived ROs for transplantation in advanced RP patients (jRCTa050200027). Two patients underwent the surgery, and no serious adverse events have been reported for at least one year following transplantation. Since the graft sheet with a tiny area of approximately 0.5 × 1 mm was delivered into a limited area, improving visual function may be insufficient and requires a better version to induce adequate efficacy [89]. Furthermore, the presence of bipolar cells and their established synaptic connections within the graft may impede graft-host neural integration. Yamasaki et al. induced the ISL1 gene deletion to significantly reduce the number of retinal bipolar cells to enhance functional integration after transplantation [90]. Besides RO transplantation therapy, a clinical trial of intestinal organoid transplantation in patients with ulcerative colitis was approved in February 2020 and conducted at Tokyo Medical and Dental University in Japan (jRCTb032190207).
Challenges in the clinical application of iPSCs and organoids
With the rapid progress of regenerative medicine and precision therapy, human iPSCs and 3D organoids play a prominent role in cell transplantation, gene therapy, and drug testing [29, 91,92,93]. Treatment decisions will become multi-faceted and personalized. Using human iPSC derivatives for transplantation is a way to avoid or reduce the risk of autoimmune rejection, as these cells can be derived from patient samples [94, 95]. However, the clinical application of human iPSCs and organoids is contentious, related to the tumorigenicity and heterogeneity of iPSCs, as well as the absence of standardized culture protocols, such as viability and batch effect of iPSC-derived cells or organoids.
Regulatory requirement
The development of iPSCs and organoids is an important step in overcoming a clinical application challenge, which requires the use of bioprocesses that are compliant with quality and regulatory guidelines. Regulatory affairs for new cellular products may vary globally, but they are usually manufactured under current good manufacturing practice (GMP) conditions [96]. The establishment of automated and high-throughput methods, supported by machine learning and advanced robotics, will contribute to product consistency, repeatability, and traceability in future clinical applications [97]. Bohrer et al. recently developed a robotic cell culture platform called Cell X to produce clinical-grade patient-specific iPSCs and ROs [98]. Briefly, iPSC clone generation, picking, expansion, and spontaneous retinal formation were all tasks performed by the robotic system, and single-cell RNA sequencing showed that these organoids generated automatically are comparable to those obtained manually [98]. The incorporation of the Cell X robotic platform into iPSCs production and differentiation enables fine labor and time under GMP standards and reduces batch-to-batch variability caused by human error or protocol drift.
In addition, most methods of iPSC generation and retinal induction rely on animal-derived components (i.e., fetal bovine serum) and/or animal-derived matrix molecules or feeder cells [34, 99,100,101]. However, it is undesirable for cell therapy developers and regulatory agencies to expose clinical-grade cells or organoids to products of animal origin. Recently, Slembrouck-Brec et al. described a defined xeno-free and feeder-free culture condition for the generation of human iPSC-derived ROs and RPE cells [102]. In our previous study, fetal bovine serum was replaced with human platelet lysates to establish a xeno-free ROs culture workflow that facilitates clinical application [103].
Tumorigenicity
The clinical application of human iPSCs and their derivatives raises issues about efficacy and safety. Mandai et al. reported a clinical study of iPSC-derived autologous RPE cell sheets in two patients with advanced neovascular age-related macular degeneration [104]. The first patient underwent surgical removal of the neovascular membrane followed by subretinal transplantation of an autogenous iPSC-derived RPE cell sheet. One year after surgery, there was no sign of graft rejection or recurrence of the neovascular membrane. However, three abnormal DNA copy number mutations were detected in RPE cells from another patient; therefore, surgery was not conducted because it might affect gene expression dysregulation [104]. Many researchers have found that either iPSC-derived grafts may retain undifferentiated stem cells or immature progenitor cells that continue to proliferate [105, 106], or genetic mutations during in vitro culture may drive tumorigenesis [107]. Moreover, if transcription factors used in reprogramming technology are integrated into the cell genome, specifically the c-Myc factor, teratomas or tumors may emerge [108]. However, only some studies have focused on the genetic safety of iPSC-derived autografts; thus, their long-term in vivo safety still needs to be well understood.
Heterogeneity
The iPSCs heterogeneity in differentiating potential is a hurdle for downstream applications, including drug screening, gene therapy, and cell therapy. Human iPSCs and their derivatives vary in efficiency across cell lines, which may be attributed to genetic background, epigenetic variables, passage, and culture protocols. For example, most normal iPSCs induce ROs effectively, but a few indeed exhibit inefficiencies or cannot generate retinal tissues [109]. Recently, an optimized system with the addition of recombinant Dickkopf-related protein 1 (DKK1) could significantly improve ROs' self-organization capacity in specific iPSC lines [110]. Additionally, genetic abnormalities affect the development of patient-derived ROs with distinct diseases or variants. Mahato et al. reported that the retinal forming efficiency of RP disease-specific iPSCs was identical to that of the healthy control cells; however, iPSCs with the RB1−/− mutation failed to form eye field primordial structures [111]. The passage approach employing enzyme and manual purification was more effective than flow cytometry-based sorting for high-yield purification of functional RPE cells from diverse stem cell sources [112]. Although many protocols for generating iPSC-derived ROs have been developed, there are still differences among them. Given this, if iPSCs and their derivatives are to be used clinically, conditions for iPSCs culture and differentiation must be standardized, and regular monitoring of genetic variation throughout the process must be emphasized. In addition, rigorously designed preclinical studies in large animal models are required. Assessing the long-term efficacy and safety of iPSC-based therapies will be meaningful to promote clinical applications in future.
Future trends
Patient-derived ROs have been used to model IRDs, enabling the recapitulation of disease genotype–phenotype features in vitro. Recently, human organoid technology has integrated multi-omics data to deeply analyze the pathogenesis of retinal diseases, or combined with microfluidic chip platform and 3D bioprinting technology to create more mature and complex organoids [113], which may become the development direction in disease research and tissue engineering (Fig. 1).

Future trends in the application of patient iPSC-derived ROs. Human organoid technology can be used for disease modeling, in-depth analysis of retinal pathogenesis in combination with multi-omics data, or biomimetic construction of retinal tissue in combination with 3D bioprinting and microfluidic chips
Multi-omics integration analysis
Although IRDs occur due to mutations in the causative gene, the exact molecular mechanisms remain unclear, and more effective treatment strategies are to be discovered [114, 115]. With advances in high-throughput sequencing technology, genomics, epigenomics, transcriptomics, proteomics, metabolomics, and single cell-omics are frequently used in research to better understand biological processes at the gene, protein, and metabolic levels and discover new biomarkers and therapeutic targets [116]. However, single omics data is insufficient for studying systems biology across multiple levels. Multi-omics analysis, which integrates data from two or more omics, has recently been popular in uncovering mechanistic insights [117,118,119,120].
It was observed that the USH2A mutation dysregulated ECM-related gene expression in patient-derived ROs, which was well validated at transcriptomic and proteomic levels, suggesting an interaction between gene expression and protein synthesis in USH2A-related ROs [37]. The degeneration of photoreceptor cells is the main hallmark of IRDs, although the early molecular and cellular events before photoreceptor death are not fully understood. An integrative multi-omics approach was performed in the Pde6brd1/rd1 mouse model of RP, including temporal transcriptomics of purified rod photoreceptors along with proteomic and metabolomic analysis of the retina [121]. They found that mitochondrial damage and metabolic disruptions are early pathological factors of photoreceptor cell death in retinal degeneration. It was demonstrated for the first time that calcium signaling defects are drivers of mitochondrial and metabolic changes. The molecular mechanisms underlying the onset and early progression of an XLRS mouse model were investigated by combined transcriptomic-proteomic analysis [122]. However, bulk RNA sequencing cannot provide cell-type-specific changes in gene expression. In contrast, single-cell RNA sequencing enables extensive molecular characterization at single-cell resolution and removes the interference caused by diverse cell compositions, making the information obtained more comprehensive [31, 123]. Lee et al. analyzed bulk RNA sequencing data from achromatopsia patient-derived ROs carrying the ATF6 mutation and identified disrupted mitochondrial structure and abnormal respiratory chain activity gene expression [77]. Single-cell RNA sequencing subsequently indicated considerable down-regulation of cone-related and up-regulation of Müller cell-related genes. Thus, the combination of bulk and single-cell RNA sequencing allows us to establish an integrated understanding of transcriptomes in studying human retinal diseases.
Furthermore, studies based on a multi-omics strategy may help identify biomarkers for early diagnosis or potential therapeutic targets. A recent study showed that microRNA-143 expression was significantly downregulated in oxygen-induced retinopathy rats, and intravitreal injection of its mimics inhibited retinal neovascularization [124]. This is possible by regulating endothelial cell–matrix adhesion and mediating the hypoxia-inducible factor-1 signaling pathway; therefore, microRNA-143 can be used as a potential biomarker and therapeutic target. In addition to attenuating retinal angiogenesis, microRNA-143 had a suppressive effect on retinoblastoma [125]. Bioinformatics analysis of multi-omics data also identified TTK, RRM2, and CDK1 as potential retinoblastoma molecular biomarkers [126]. TTK, described as an oncogene that promotes tumor progression, was highly expressed in various cancers [127,128,129], making it a promising therapeutic target.
3D bioprinting technology
3D bioprinting technology is the inclusion of 3D printing into tissue engineering and regenerative medicine applications, allowing the rapid and reproducible fabrication of complex biomimetic tissues or organs in vitro, such as 3D-bioprinted ventricles, corneal stroma, skin, bone, and cartilage tissue [130,131,132,133]. In recent years, the efficacy of 3D tissue or organ structure printing has been markedly improved due to the rapid development of functional bio-inks.
Additionally, 3D bioprinting technology is also used for personalized modeling engineering to flexibly design the external shape and internal structure of an object. Despite advances in self-organizing retinal morphogenesis, patient-derived ROs are not currently optimal for testing candidate drugs or cell therapies. For example, ROs often vary in size and quality, contain some off-target tissues and their development may be inconsistent [82]. We previously used 3D-printed polydimethylsiloxane (PDMS) microwell platform for adherent ROs cultivation [103]. Unlike suspended ROs on ultralow adhesion microwell plates, iPSC-derived ROs on PDMS molds were confined to their respective microcavities but shared the same medium and microenvironment, which could not only avoid the fusion of multiple ROs but also ensure the long-term culture and survival of ROs, resulting in efficient and homogeneous ROs with fewer apoptotic cells (Fig. 2). The PDMS microwell platform using 3D bioprinting is envisaged to improve the robustness of in vitro retinal organogenesis and standardization of ROs. However, guiding the proper spatial arrangement of photoreceptor cells for transplantation remains challenging. In 3D scaffolds, retinal progenitor cells harvested from dissociated ROs formed neuronal processes that extended into and aligned with scaffold vertical pores [134]. To precisely establish tissue structures in vitro, strategies based on biomaterials similar to the extracellular microenvironment have been developed to enhance cell characterization. Shrestha et al. used two-photon polymerization to construct a hyaluronic acid (HA) and gelatin scaffold, enabling ECM-derived molecules to offer cellular support and retain significant vitality and proliferation of rat retinal cells [135]. Furthermore, an immersion bioprinting method produced patient-derived brain tumor organoids using HA and collagen bio-inks, where organoids embedded in the HA bath displayed homogeneous volume and geometry for subsequent anti-cancer drug studies [136].

Self-organization of ROs from human iPSCs on a PDMS microwell platform. A Schematic diagram of manufacturing PDMS microwell molds, including (a) design and fabrication of 3D-printed positive molds, (b) addition of PDMS biomaterials, and (c) fabrication of complementary PDMS molds. B Immunofluorescence staining images of adherent 3D ROs. (a) Ciliary margin domain was stained with RDH10 (red). (b) Neural retina domain was stained with VSX2 (green) and eye field was stained with PAX6 (red). (c, d) RPE domain was stained with ZO-1 (green), MITF (red), and PRE65 (red). Nuclei were labeled with DAPI (blue). Scale bar: 50 µm. [103] Copyright Sun et al. 2023, Biofabrication
Microfluidic chip platform
Organ-on-a-chip, such as microfluidic retina-on-a-chip models, is an emerging technology that allows the development of novel platforms to simulate the complex structure and microenvironment of the retina in artificially controlled perfusion devices. Briefly, organ-on-a-chip includes the different cell types, structural organization, and microenvironment, usually separated by microporous membranes, offering the advantage of controlling cellular and multiorgan interactions exposed to cultural conditions. Multiple organ-on-a-chip systems that model the interface between RPE and photoreceptor cells, microvascular endothelium and RPE, microglia, and cerebral organoids have been described [29, 137,138,139]. For example, a study utilized a microfluidic chip platform to co-culture iPSC-derived RPE cells and ROs, which generated the desired pattern, i.e., an outer retinal morphology with vasculature-like perfusion [29] (Fig. 3). After a week of running the microfluidic retina-on-a-chip, it was revealed that photoreceptor calcium dynamics and digested outer segment-like structure signs, replicating retinal basic activities associated with the visual cycle. It was then evaluated with chloroquine and gentamicin, known to induce retinal damage, and the results revealed cell dysfunction and death [29]. Researchers recently analyzed the efficacy, kinetics, and cell tropism of seven different AAV vectors using the retina-on-a-chip platform with satisfactory results [140]. First, they evaluated the performance of different types of AAV vectors in mouse retinas and human iPSC-ROs. Significantly higher fluorescence expression was detected when delivered with the AAV2.7m8 vector, which is consistent with data reported by Dalkara et al. from AAV2.7m8 with highly efficient transduction in the retina of mice and non-human primates [141]. Subsequently, the same vector panel was applied to the retina-on-a-chip model, and the results showed that the AAV2.7m8 vector had stronger transduction signals and cell tropism compared to other AAV types. In addition, two recently developed second-generation AAV vectors, AAV2.NN and AAV2.GL, were analyzed using the retina-on-a-chip platform and subsequently demonstrated their efficient transduction for rod and cone photoreceptors as well as Müller cells [140]. Many retinal diseases involve the outer layers of the retina, including RPE and photoreceptor cell layers. Thus, we hypothesize that IRD patient-specific retina-on-a-chip can replicate the corresponding physiological tissue or organ microenvironment in vitro and has great potential as a tool for high-throughput pharmacology and drug screening.

Microfluidic retina-on-a-chip. A Photo and B schematic representation of ROs and RPE co-cultured in a microfluidic retina-on-a-chip model. C Immunofluorescence staining of ROM1 (green), phalloidin (white), and rhodopsin (red) was performed after 7 days of co-culture. Scale bar: 40 µm. D Electron microscope image. Scale bar: 5 µm. [29] Copyright Achberger et al. 2019, eLife
In addition to modeling the outer retina, two microfluidic organ-on-a-chip models of the outer blood-retinal barrier were reported [137, 142]. In one of the models, RPE and human umbilical vein endothelial cells were co-cultured in a microfluidic chip with microchannels and an open-top culture chamber separated by a polyester membrane [142]. Upon inducing oxidative stress by treating with hydrogen peroxide, a dose-dependent increase in barrier permeability was observed by using a dynamic assay for fluorescence tracing, analogous to the clinically used fluorescence angiography. This method allows semi-quantitative evaluation of the endothelial barrier by analyzing the slope of the fluorescence increase in the perfusion phase and qualitative assessment of lesions and defects by analyzing local fluorescent dye accumulation in the removal phase. They also found that optical coherence tomography could detect changes in microvessel diameter and quality after imaging 3D vascular structures generated by cells in the collagen I hydrogel chip. Another designed model consisted of an iPSC-derived RPE monolayer in the upper compartment and primary human retinal microvascular endothelial cells and choroidal fibroblasts in a hydrogel scaffold in the lower compartment, respectively [137]. After seven days, retinal endothelial cells' vasculogenic self-assembly developed into a dense network of microvessels approximately 10 – 25 μm in diameter, enhancing the RPE phenotype, including intercellular tight junctions, laminin production and deposition, RPE pigmentation, and RPE65 protein expression.
Conclusions
Human iPSCs and 3D organoid technology play a role in studying human organogenesis and development, disease modeling, drug screening, and preclinical therapies. Recently, the first human clinical trial using intestinal organoids to treat ulcerative colitis patients is ongoing in Japan (jRCTb032190207). In this paper, we reviewed the concept and sources of iPSCs, the recent research advancement of patient-derived iPSCs and organoids in IRDs, and the main challenges that need to be overcome in clinical application. Moreover, multi-omics integration analysis, 3D bioprinting technology, and microfluidic chip platform are further promising patient-derived ROs research avenues.
However, the lack of vascular networks, immune cells, continuous RPE monolayer, and the central nervous system may limit RO generation and development. Co-culture systems for interaction between multi-organoids, or organoids with cells/spheroids, have been studied to address the constraints of traditional organoid cultivation [143,144,145,146,147]. Meanwhile, establishing an organoid culture system with standardized, high-throughput, and undifferentiated operations is required. Homogeneous organoids will simulate the complex organ structure and function, reproduce cell-to-cell communication and molecular features, and explore disease pathogenesis and treatment. Organoids from healthy individuals or patients can also provide a comprehensive evaluation of susceptibility across age, gender, and ethnicity, potentially facilitating the implementation of personalized intervention strategies.
Availability of data and materials
Not applicable.
Abbreviations
- 3D:
-
Three-dimension
- AAV:
-
Adeno-associated virus
- AIPL1:
-
Aryl hydrocarbon receptor-interacting protein-like 1
- ATF6:
-
Activating transcription factor 6
- BVMD:
-
Best vitelliform macular dystrophy
- Cas9:
-
CRISPR-associated protein 9
- CEP290:
-
Centrosomal protein 290
- cGMP:
-
Cyclic guanosine monophosphate
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CRX:
-
Cone-rod homeobox
- DFs:
-
Dermal fibroblasts
- DKK1:
-
Dickkopf-related protein 1
- ECM:
-
Extracellular matrix
- FDA:
-
Food and drug administration
- GMP:
-
Good manufacturing practice
- HA:
-
Hyaluronic acid
- HSP90:
-
Heat shock protein 90
- IMPG2:
-
Interphotoreceptor matrix proteoglycan 2
- iPSCs:
-
Induced pluripotent stem cells
- IRDs:
-
Inherited retinal diseases
- LCA:
-
Leber congenital amaurosis
- PBMCs:
-
Peripheral blood mononuclear cells
- PDE6:
-
Phosphodiesterase 6
- PDMS:
-
Polydimethylsiloxane
- POS:
-
Photoreceptor outer segment
- PRPFs:
-
Pre-mRNA processing factors
- ROs:
-
Retinal organoids
- RP:
-
Retinitis pigmentosa
- RPE:
-
Retinal pigment epithelium
- RPGR:
-
Retinitis pigmentosa GTPase regulator
- RS1:
-
Retinoschisin 1
- snRNP:
-
Small nuclear ribonucleoprotein particle
- UCs:
-
Urine cells
- USH2A:
-
Usherin
- XLRS:
-
X-linked recessive retinoschisis
References
Mawatari G, et al. Clinical and genetic characteristics of 14 patients from 13 Japanese families with RPGR-associated retinal disorder: report of eight novel variants. Hum Genome Var. 2019;6:34.
Gersch J, et al. Investigation of structural alterations in inherited retinal diseases: a quantitative SD-OCT-analysis of retinal layer thicknesses in light of underlying genetic mutations. Int J Mol Sci. 2022;23(24):16007.
Shen RJ, et al. Consanguinity-based analysis of exome sequencing yields likely genetic causes in patients with inherited retinal dystrophy. Orphanet J Rare Dis. 2021;16(1):278.
Wright AF, et al. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11(4):273–84.
Tsipi M, et al. Genomic screening of ABCA4 and array CGH analysis underline the genetic variability of Greek patients with inherited retinal diseases. Meta Gene. 2016;8:37–43.
Villanueva-Mendoza C, et al. The genetic landscape of inherited retinal diseases in a Mexican cohort: genes, mutations and phenotypes. Genes. 2021;12(11):1824.
Veleri S, et al. Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis Model Mech. 2015;8(2):109–29.
Onos KD, et al. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res Bull. 2016;122:1–11.
Graziotto JJ, et al. Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52(1):190–8.
Lu B, et al. Cell transplantation to arrest early changes in an ush2a animal model. Invest Ophthalmol Vis Sci. 2010;51(4):2269–76.
Garanto A, et al. Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for Leber congenital amaurosis. PLoS ONE. 2013;8(11):e79369.
Valdes-Sanchez L, et al. Retinal pigment epithelium degeneration caused by aggregation of PRPF31 and the role of HSP70 family of proteins. Mol Med. 2019;26(1):1.
Wei M, Li S, Le W. Nanomaterials modulate stem cell differentiation: biological interaction and underlying mechanisms. J Nanobiotechnol. 2017;15(1):75.
Shi Y, et al. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–30.
Capetian P, et al. Plasmid-based generation of induced neural stem cells from adult human fibroblasts. Front Cell Neurosci. 2016;10:245.
Li J, et al. Generation and staging of human retinal organoids based on self-formed ectodermal autonomous multi-zone system. Front Cell Dev Biol. 2021;9:732382.
Gonzalez-Cordero A, et al. Recapitulation of human retinal development from human pluripotent stem cells generates transplantable populations of cone photoreceptors. Stem Cell Rep. 2017;9(3):820–37.
Lee CT, et al. 3D brain Organoids derived from pluripotent stem cells: promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci. 2017;24(1):59.
Karagiannis P, et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol Rev. 2019;99(1):79–114.
Li Y, Darabi R. Role of epigenetics in cellular reprogramming; from iPSCs to disease modeling and cell therapy. J Cell Biochem. 2022;123(2):147–54.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Jin ZB, et al. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS ONE. 2011;6(2):e17084.
Saini N, et al. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet. 2016;12(10):e1006385.
Agu CA, et al. Successful generation of human induced pluripotent stem cell lines from blood samples held at room temperature for up to 48 hr. Stem Cell Reports. 2015;5(4):660–71.
Li D, et al. Optimized approaches for generation of integration-free iPSCs from human urine-derived cells with small molecules and autologous feeder. Stem Cell Reports. 2016;6(5):717–28.
Foltz LP, Clegg DO. Patient-derived induced pluripotent stem cells for modelling genetic retinal dystrophies. Prog Retin Eye Res. 2019;68:54–66.
Li M, Izpisua Belmonte JC. Organoids - preclinical models of human disease. N Engl J Med. 2019;380(6):569–79.
Achberger K, et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife. 2019;8:e46188.
Movio MI, et al. Retinal organoids from human-induced pluripotent stem cells: From studying retinal dystrophies to early diagnosis of Alzheimer’s and Parkinson’s disease. Semin Cell Dev Biol. 2023;144:77–86.
Cowan CS, et al. Cell types of the human retina and its organoids at single-cell resolution. Cell. 2020;182(6):1623–40.
Kim S, et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc Natl Acad Sci USA. 2019;116(22):10824–33.
Collin J, et al. Deconstructing retinal organoids: single cell RNA-seq reveals the cellular components of human pluripotent stem cell-derived retina. Stem Cells. 2019;37(5):593–8.
Zerti D, et al. Developing a simple method to enhance the generation of cone and rod photoreceptors in pluripotent stem cell-derived retinal organoids. Stem Cells. 2020;38(1):45–51.
Liu W, et al. Retinitis pigmentosa: progress in molecular pathology and biotherapeutical strategies. Int J Mol Sci. 2022;23(9).
Dias MF, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog Retin Eye Res. 2018;63(107):131.
Su T, et al. Retinal organoids and microfluidic chip-based approaches to explore the retinitis pigmentosa with USH2A mutations. Front Bioeng Biotechnol. 2022;10:939774.
Hassall MM, et al. Analysis of early cone dysfunction in an in vivo model of rod-cone dystrophy. Int J Mol Sci. 2020;21(17):6055.
Wood KA, et al. The role of the U5 snRNP in genetic disorders and cancer. Front Genet. 2021;12:636620.
Arzalluz-Luque A, et al. Mutant PRPF8 causes widespread splicing changes in spliceosome components in retinitis pigmentosa patient iPSC-derived RPE cells. Front Neurosci. 2021;15:636969.
Buskin A, et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat Commun. 2018;9(1):4234.
Liang Y, et al. Aberrant retinal pigment epithelial cells derived from induced pluripotent stem cells of a retinitis pigmentosa patient with the PRPF6 mutation. Int J Mol Sci. 2022;23(16):9049.
Georgiou M, et al. Activation of autophagy reverses progressive and deleterious protein aggregation in PRPF31 patient-induced pluripotent stem cell-derived retinal pigment epithelium cells. Clin Transl Med. 2022;12(3):e759.
Rodrigues A, et al. Modeling PRPF31 retinitis pigmentosa using retinal pigment epithelium and organoids combined with gene augmentation rescue. NPJ Regen Med. 2022;7(1):39.
Farkas MH, et al. Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am J Pathol. 2014;184(10):2641–52.
Chahine Karam F, et al. Human iPSC-derived retinal organoids and retinal pigment epithelium for novel intronic RPGR variant assessment for therapy suitability. J Pers Med. 2022;12(3):502.
Megaw R, et al. Gelsolin dysfunction causes photoreceptor loss in induced pluripotent cell and animal retinitis pigmentosa models. Nat Commun. 2017;8(1):271.
Deng WL, et al. Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 2018;10(4):1267–81.
Toualbi L, Toms M, Moosajee M. USH2A-retinopathy: from genetics to therapeutics. Exp Eye Res. 2020;201:108330.
Stemerdink M, et al. Genetics, pathogenesis and therapeutic developments for Usher syndrome type 2. Hum Genet. 2022;141(3–4):737–58.
Zaw K, et al. Pathogenesis and treatment of usher syndrome type IIA. Asia Pac J Ophthalmol. 2022;11(4):369–79.
Tucker BA, et al. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife. 2013;2:e00824.
Guo Y, et al. Modeling retinitis pigmentosa: retinal organoids generated from the iPSCs of a patient with the USH2A mutation show early developmental abnormalities. Front Cell Neurosci. 2019;13:361.
Ishikawa M, Sawada Y, Yoshitomi T. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp Eye Res. 2015;133:3–18.
Xu H, et al. Deletion of the Impg2 gene causes the degeneration of rod and cone cells in mice. Hum Mol Genet. 2020;29(10):1624–34.
Mayerl SJ, et al. Human retinal organoids harboring IMPG2 mutations exhibit a photoreceptor outer segment phenotype that models advanced retinitis pigmentosa. Stem Cell Rep. 2022;17(11):2409–20.
Sacristan-Reviriego A, van der Spuy J. The leber congenital amaurosis-linked protein AIPL1 and its critical role in photoreceptors. Adv Exp Med Biol. 2018;1074:381–6.
Sato S, et al. Novel mutation identified in Leber congenital amaurosis—a case report. BMC Ophthalmol. 2020;20(1):313.
Kumaran N, et al. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–54.
Apte RS. Gene therapy for retinal degeneration. Cell. 2018;173(1):5.
Sacristan-Reviriego A, et al. The integrity and organization of the human AIPL1 functional domains is critical for its role as a HSP90-dependent co-chaperone for rod PDE6. Hum Mol Genet. 2017;26(22):4465–80.
Ramamurthy V, et al. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004;101(38):13897–902.
Lukovic D, et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA patient maintain cytoarchitecture despite reduced levels of mutant AIPL1. Sci Rep. 2020;10(1):5426.
Perdigao PRL, et al. Retinal organoids from an AIPL1 CRISPR/Cas9 knockout Cell line successfully recapitulate the molecular features of LCA4 disease. Int J Mol Sci. 2023;24(6):5912.
Leung A, et al. Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital amaurosis. Stem Cell Rep. 2022;17(10):2187–202.
Chirco KR, et al. Allele-specific gene editing to rescue dominant CRX-associated LCA7 phenotypes in a retinal organoid model. Stem Cell Rep. 2021;16(11):2690–702.
Kruczek K, et al. Gene therapy of dominant CRX-Leber congenital amaurosis using patient stem cell-derived retinal organoids. Stem Cell Rep. 2021;16(2):252–63.
Parfitt DA, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell. 2016;18(6):769–81.
Corral-Serrano JC, et al. Eupatilin improves cilia defects in human CEP290 ciliopathy models. Cells. 2023;12(12):1575.
Afanasyeva TAV, et al. CRISPR-Cas9 correction of a nonsense mutation in LCA5 rescues lebercilin expression and localization in human retinal organoids. Mol Ther Methods Clin Dev. 2023;29:522–31.
Ku CA, Wei LW, Sieving PA. X-Linked Retinoschisis. Cold Spring Harb Perspect Med. 2023.
Wu WW, et al. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. 2005;280(11):10721–30.
Liu Y, et al. Mouse models of X-linked juvenile retinoschisis have an early onset phenotype, the severity of which varies with genotype. Hum Mol Genet. 2019;28(18):3072–90.
Chen D, et al. Recapitulating X-linked juvenile retinoschisis in mouse model by knock-in patient-specific novel mutation. Front Mol Neurosci. 2017;10:453.
Huang KC, et al. Morphological and molecular defects in human three-dimensional retinal organoid model of X-linked juvenile retinoschisis. Stem Cell Rep. 2019;13(5):906–23.
Zhang X, et al. Gene correction of the CLN3 c.175G>A variant in patient-derived induced pluripotent stem cells prevents pathological changes in retinal organoids. Mol Genet Genom Med. 2021;9(3):1601.
Lee EJ, et al. Mitochondria and endoplasmic reticulum stress in retinal organoids from patients with vision loss. Am J Pathol. 2022;193:1721–39.
Kroeger H, et al. ATF6 is essential for human cone photoreceptor development. Proc Natl Acad Sci USA. 2021;118(39):e2103196118.
Navines-Ferrer A, et al. Impaired bestrophin channel activity in an iPSC-RPE model of best vitelliform macular dystrophy (BVMD) from an early onset patient carrying the P77S dominant mutation. Int J Mol Sci. 2022;23(13):7432.
Ji C, et al. Investigation and restoration of BEST1 activity in patient-derived RPEs with dominant mutations. Sci Rep. 2019;9(1):19026.
Ribeiro J, et al. Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell-derived cone photoreceptors. Cell Rep. 2021;35(3):109022.
Watari K, et al. Self-organization, quality control, and preclinical studies of human iPSC-derived retinal sheets for tissue-transplantation therapy. Commun Biol. 2023;6(1):164.
Thomas BB, et al. Co-grafts of human embryonic stem cell derived retina organoids and retinal pigment epithelium for retinal reconstruction in immunodeficient retinal degenerate royal college of surgeons rats. Front Neurosci. 2021;15:752958.
Gasparini SJ, et al. Transplanted human cones incorporate into the retina and function in a murine cone degeneration model. J Clin Invest. 2022;132(12).
McLelland BT, et al. Transplanted hESC-derived retina organoid sheets differentiate, integrate, and improve visual function in retinal degenerate rats. Invest Ophthalmol Vis Sci. 2018;59(6):2586–603.
Mandai M. Pluripotent stem cell-derived retinal organoid/cells for retinal regeneration therapies: a review. Regen Ther. 2023;22:59–67.
Mandai M, et al. iPSC-derived retina transplants improve vision in rd1 end-stage retinal-degeneration mice. Stem Cell Rep. 2017;8(4):1112–3.
Iraha S, et al. Establishment of immunodeficient retinal degeneration model mice and functional maturation of human ESC-derived retinal sheets after transplantation. Stem Cell Rep. 2018;10(3):1059–74.
Maeda T, et al. Strategies of pluripotent stem cell-based therapy for retinal degeneration: update and challenges. Trends Mol Med. 2022;28(5):388–404.
Yamasaki S, et al. A Genetic modification that reduces ON-bipolar cells in hESC-derived retinas enhances functional integration after transplantation. iScience. 2022;25(1):103657.
West EL, Ribeiro J, Ali RR. Development of stem cell therapies for retinal degeneration. Cold Spring Harb Perspect Biol. 2020;12(8):a035683.
Ebeling MC, et al. Testing mitochondrial-targeted drugs in iPSC-RPE from patients with age-related macular degeneration. Pharmaceuticals (Basel). 2022;15(1):62.
Chen HY, et al. Reserpine maintains photoreceptor survival in retinal ciliopathy by resolving proteostasis imbalance and ciliogenesis defects. Elife. 2023;12:e83205.
van Romunde SHM, et al. Destructive inflammatory reaction after an autologous retinal pigment epithelium and choroid transplantation: no detection of an auto-immune response. J Ophthalmic Inflamm Infect. 2022;12(1):27.
Akiba R, et al. Progress of iPS cell-based transplantation therapy for retinal diseases. Jpn J Ophthalmol. 2023;67(2):119–28.
Kasendra M, et al. Intestinal organoids: roadmap to the clinic. Am J Physiol Gastrointest Liver Physiol. 2021;321(1):G1–10.
Tristan CA, et al. Robotic high-throughput biomanufacturing and functional differentiation of human pluripotent stem cells. Stem Cell Rep. 2021;16(12):3076–92.
Bohrer LR, et al. Automating iPSC generation to enable autologous photoreceptor cell replacement therapy. J Transl Med. 2023;21(1):161.
Kuwahara A, et al. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat Commun. 2015;6:6286.
Wahlin KJ, et al. Photoreceptor outer segment-like structures in long-term 3D retinas from human pluripotent stem cells. Sci Rep. 2017;7(1):766.
Norrie JL, et al. Retinoblastoma from human stem cell-derived retinal organoids. Nat Commun. 2021;12(1):4535.
Slembrouck-Brec A, et al. Defined xeno-free and feeder-free culture conditions for the generation of human iPSC-derived retinal cell models. J Vis Exp. 2018;139:e57795.
Sun X, et al. One-stop assembly of adherent 3D retinal organoids from hiPSCs based on 3D-printed derived PDMS microwell platform. Biofabrication. 2023;15(3):35005.
Mandai M, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038–46.
Regent F, et al. Automation of human pluripotent stem cell differentiation toward retinal pigment epithelial cells for large-scale productions. Sci Rep. 2019;9(1):10646.
Reichman S, et al. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc Natl Acad Sci USA. 2014;111(23):8518–23.
Yamanaka S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell. 2020;27(4):523–31.
Moon H, Park H, Ro SW. c-Myc-driven hepatocarcinogenesis. Anticancer Res. 2021;41(10):4937–46.
Guan Y, Xie B, Zhong X. Retinal Organoid Induction System for Derivation of 3D Retinal Tissues from Human Pluripotent Stem Cells. J Vis Exp. 2021;170:e62435.
Luo Z, et al. An optimized system for effective derivation of three-dimensional retinal tissue via Wnt signaling regulation. Stem Cells. 2018;36(11):1709–22.
Mahato S, et al. Generation of retinal organoids from healthy and retinal disease-specific human-induced pluripotent stem cells. J Vis Exp. 2022;190:e64509.
Regha K, et al. Customized strategies for high-yield purification of retinal pigment epithelial cells differentiated from different stem cell sources. Sci Rep. 2022;12(1):15563.
Shin K. Stem cells, organoids and their applications for human diseases: special issue of BMB reports in 2023. BMB Rep. 2023;56(1):1.
Bennett J. Overview of retinal gene therapy: current status and future challenges. Cold Spring Harb Perspect Med. 2023;13(7):a041278.
Schneider N, et al. Inherited retinal diseases: linking genes, disease-causing variants, and relevant therapeutic modalities. Prog Retin Eye Res. 2022;89:101029.
Lei Y, et al. Essential role of multi-omics approaches in the study of retinal vascular diseases. Cells. 2022;12(1):103.
Lv K, et al. Integrated multi-omics reveals the activated retinal microglia with intracellular metabolic reprogramming contributes to inflammation in STZ-induced early diabetic retinopathy. Front Immunol. 2022;13:942768.
Soundararajan A, et al. Multiomics analysis reveals the mechanical stress-dependent changes in trabecular meshwork cytoskeletal-extracellular matrix interactions. Front Cell Dev Biol. 2022;10:874828.
Boron D, et al. Recent multiomics approaches in endometrial cancer. Int J Mol Sci. 2022;23(3):1237.
Miao Z, et al. Multi-omics integration in the age of million single-cell data. Nat Rev Nephrol. 2021;17(11):710–24.
Jiang K, et al. Multiomics analyses reveal early metabolic imbalance and mitochondrial stress in neonatal photoreceptors leading to cell death in Pde6brd1/rd1 mouse model of retinal degeneration. Hum Mol Genet. 2022;31(13):2137–54.
Jin X, et al. Retinal proteomic alterations and combined transcriptomic-proteomic analysis in the early stages of progression of a mouse model of X-linked retinoschisis. Cells. 2022;11(14):2150.
Sridhar A, et al. Single-cell transcriptomic comparison of human fetal retina, hPSC-derived retinal organoids, and long-term retinal cultures. Cell Rep. 2020;30(5):1644–59.
Wang JH, et al. An Integrative multi-omics analysis reveals MicroRNA-143 as a potential therapeutic to attenuate retinal angiogenesis. Nucleic Acid Ther. 2022;32(4):251–66.
Wang LL, Hu HF, Feng YQ. Suppressive effect of microRNA-143 in retinoblastoma. Int J Ophthalmol. 2016;9(11):1584–90.
Zeng Y, et al. Bioinformatics analysis of multi-omics data identifying molecular biomarker candidates and epigenetically regulatory targets associated with retinoblastoma. Medicine (Baltimore). 2020;99(47):e23314.
Tang J, et al. Overexpression of ASPM, CDC20, and TTK confer a poorer prognosis in breast cancer identified by gene co-expression network analysis. Front Oncol. 2019;9:310.
Yang Q, Yu B, Sun J. TTK, CDC25A, and ESPL1 as prognostic biomarkers for endometrial cancer. Biomed Res Int. 2020;2020:4625123.
Chen S, et al. Silencing TTK expression inhibits the proliferation and progression of prostate cancer. Exp Cell Res. 2019;385(1):111669.
Choi S, et al. Fibre-infused gel scaffolds guide cardiomyocyte alignment in 3D-printed ventricles. Nat Mater. 2023;22:1039.
Moro A, et al. Hyaluronic acid based next generation bioink for 3D bioprinting of human stem cell derived corneal stromal model with innervation. Biofabrication. 2022;15(1):015020.
Zhang D, et al. 3D-bioprinted human lipoaspirate-derived cell-laden skin constructs for healing of full-thickness skin defects. Int J Bioprint. 2023;9(4):718.
Schipani R, et al. Reinforcing interpenetrating network hydrogels with 3D printed polymer networks to engineer cartilage mimetic composites. Biofabrication. 2020;12(3):035011.
Worthington KS, et al. Two-photon polymerization for production of human iPSC-derived retinal cell grafts. Acta Biomater. 2017;55:385–95.
Shrestha A, et al. Development of high-resolution three-dimensional-printed extracellular matrix scaffolds and their compatibility with pluripotent stem cells and early retinal cells. J Ocul Pharmacol Ther. 2020;36(1):42–55.
Clark CC, et al. Immersion bioprinting of hyaluronan and collagen bioink-supported 3D patient-derived brain tumor organoids. Biomed Mater. 2022;18(1):015014.
Paek J, et al. Microphysiological engineering of self-assembled and perfusable microvascular beds for the production of vascularized three-dimensional human microtissues. ACS Nano. 2019;13(7):7627–43.
Salmon I, et al. Engineering neurovascular organoids with 3D printed microfluidic chips. Lab Chip. 2022;22(8):1615–29.
Park J, et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat Neurosci. 2018;21(7):941–51.
Achberger K, et al. Human stem cell-based retina on chip as new translational model for validation of AAV retinal gene therapy vectors. Stem Cell Rep. 2021;16(9):2242–56.
Dalkara D, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5(189):189ra176.
Arik YB, et al. Microfluidic organ-on-a-chip model of the outer blood-retinal barrier with clinically relevant read-outs for tissue permeability and vascular structure. Lab Chip. 2021;21(2):272–83.
Usui-Ouchi A, et al. Integrating human iPSC-derived macrophage progenitors into retinal organoids to generate a mature retinal microglial niche. Glia. 2023;71:2372.
Huang H, et al. Association of placental growth factor and angiopoietin in human retinal endothelial cell-pericyte co-cultures and iPSC-derived vascular organoids. Curr Eye Res. 2023;48(3):297–311.
Kook MG, et al. Generation of cortical brain organoid with vascularization by assembling with vascular spheroid. Int J Stem Cells. 2022;15(1):85–94.
Gao ML, et al. Functional microglia derived from human pluripotent stem cells empower retinal organ. Sci China Life Sci. 2022;65(6):1057–71.
Chichagova V, et al. Incorporating microglia-like cells in human induced pluripotent stem cell-derived retinal organoids. J Cell Mol Med. 2023;27(3):435–45.
Zhou Y, et al. Establishment of non-integrate induced pluripotent stem cell line CSUASOi006-A, from urine-derived cells of a PRPF8-related dominant retinitis pigmentosa patient. Stem Cell Res. 2020;49:102041.
Zhou Y, et al. Establishment of induced pluripotent stem cell line CSUASOi004-A by reprogramming peripheral blood mononuclear cells of a PRPF6-related dominant retinitis pigmentosa patient. Stem Cell Res. 2020;45:101793.
Li YP, Liu H, Jin ZB. Generation of three human iPSC lines from a retinitis pigmentosa family with SLC7A14 mutation. Stem Cell Res. 2020;49:102075.
Zhou Y, et al. Establishment of induced pluripotent stem cell line CSUASOi003—a from an autosomal recessive retinitis pigmentosa patient carrying compound heterozygous mutations in CRB1 gene. Stem Cell Res. 2020;44:101742.
Tang X, et al. Using inducible lentiviral vectors to generate induced pluripotent stem cell line ZOCi001-A from peripheral blood cells of a patient with CRB1(-/-) retinitis pigmentosa. Stem Cell Res. 2020;45:101817.
Zhang X, et al. Generation of an induced pluripotent stem cell line from a patient with retinitis pigmentosa caused by RP1 mutation. Stem Cell Res. 2019;37:101452.
Jin ZB, et al. Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling. Stem Cells Transl Med. 2012;1(6):503–9.
Domingo-Prim J, et al. Establishment of an induced pluripotent stem cell line (FRIMOi005-A) derived from a retinitis pigmentosa patient carrying a dominant mutation in RHO gene. Stem Cell Res. 2019;38:101468.
Riera M, et al. Generation of an induced pluripotent stem cell line (FRIMOi002-A) from a retinitis pigmentosa patient carrying compound heterozygous mutations in USH2A gene. Stem Cell Res. 2019;35:101386.
Park H, et al. Generation of a human induced pluripotent stem cell line from a patient with Leber congenital amaurosis. Stem Cell Res. 2020;43:101725.
Lukovic D, et al. Generation of a human iPSC line from a patient with Leber congenital amaurosis caused by mutation in AIPL1. Stem Cell Res. 2018;33:151–5.
Erkilic N, et al. Generation of a human iPSC line, INMi004-A, with a point mutation in CRX associated with autosomal dominant Leber congenital amaurosis. Stem Cell Res. 2019;38:101476.
Zou X, et al. Generation of a human induced pluripotent stem cell line (PUMCHi018-A) from an early-onset severe retinal dystrophy patient with RDH12 mutations. Stem Cell Res. 2022;59:102655.
Sarkar H, et al. Generation of two human iPSC lines from patients with autosomal dominant retinitis pigmentosa (UCLi014-A) and autosomal recessive Leber congenital amaurosis (UCLi015-A), associated with RDH12 variants. Stem Cell Res. 2021;54:102449.
Peng CH, et al. Generation of induced pluripotent stem cells from a patient with X-linked juvenile retinoschisis. Stem Cell Res. 2018;29:152–6.
Mao S, et al. Establishment of a human induced pluripotent stem cell line (CSUASOi005-A), from peripheral blood mononuclear cells of a patient with X-linked juvenile retinoschisis carrying a novel mutation in RS1 gene. Stem Cell Res. 2020;43:101718.
Yan X, et al. Establishment of CSUASOi001-A, a non-integrated induced pluripotent stem cell line from urine-derived cells of a Chinese patient carrying RS1 gene mutation. Stem Cell Res. 2019;38:101466.
Zhang L, et al. Generation of a X-linked juvenile retinoschisis patient-derived induced pluripotent stem cell line ZOCi004-A. Stem Cell Res. 2022;65:102937.
Liu Y, et al. Generation of induced pluripotent stem cell (iPSC) line ZOCi003-A derived from peripheral blood mononuclear cells of X-linked juvenile retinoschisis harboring a hemizygous mutation in RS1 gene. Stem Cell Res. 2021;57:102595.
Lane A, et al. Modeling and rescue of RP2 retinitis pigmentosa using iPSC-derived retinal organoids. Stem Cell Rep. 2020;15(1):67–79.
Boon N, et al. AAV-mediated gene augmentation therapy of CRB1 patient-derived retinal organoids restores the histological and transcriptional retinal phenotype. Stem Cell Rep. 2023;18(6):1388.
Gao ML, et al. Patient-specific retinal organoids recapitulate disease features of late-onset retinitis pigmentosa. Front Cell Dev Biol. 2020;8:128.
Tagawa M, et al. Deterioration of phagocytosis in induced pluripotent stem cell-derived retinal pigment epithelial cells established from patients with retinitis pigmentosa carrying Mer tyrosine kinase mutations. Exp Eye Res. 2021;205:108503.
Dulla K, et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c2991+1655A>G LCA10 models. Mol Ther Nucleic Acids. 2018;12:730–40.
Kruczek K, et al. In vitro modeling and rescue of ciliopathy associated with IQCB1/NPHP5 mutations using patient-derived cells. Stem Cell Rep. 2022;17(10):2172–86.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC-RGC, 32061160469), the Science Research Grant of Aier Eye Institute (02-202105), and the Research Grant of Key Laboratory of Regenerative Medicine, Ministry of Education, Jinan University (ZSYXM202201).
Author information
Authors and Affiliations
Contributions
YL searched and analyzed the literature, designed the figure, and drafted the manuscript. XS and CD revised the manuscript. ST and JC revised the manuscript, final approval, and funding. All the authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, Y., Sun, X., Duan, C. et al. Application of patient-derived induced pluripotent stem cells and organoids in inherited retinal diseases. Stem Cell Res Ther 14, 340 (2023). https://doi.org/10.1186/s13287-023-03564-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-023-03564-5