
Abstract
Introduction
Mantle cell lymphoma is a rare lymphoma of the gastrointestinal tract that may present as multiple lymphomatous polyposis. We report a case of lymphomatous polyposis with a review of the literature.
Case report
A 56-year-old man of Black ethnicity and Ivorian nationality with no relevant past medical history, consulted for a sudden onset symptoms of gastrointestinal obstruction, which evolved over 2 days. Macroscopic examination revealed the presence of multiple polyploid formations of the colonic mucosa. Histology showed diffuse lymphomatous proliferation of submucosa consisting off small lymphoid cells with a hyperchromatic crenelated nucleus, suggesting lymphomatous polyposis. Immunohistochemical examination showed expression by the tumor cells of antibodies to CD20, CD5, Bcl2, and cyclin D1. They did not express antibodies to CD10 and CD23. The Ki67 proliferation index was 25%. We have thus retained the diagnosis of mantle cell lymphomatous polyposis.
Conclusion
Multiple lymphomatous polyposis is a rare entity characterized by the presence of numerous gastrointestinal polyploid lesions sometimes involving several segments of the gastrointestinal tract. Typical lymphoma presenting as lymphomatous polyposis is mantle cell lymphoma; although, other tumors may have this aspect.
Similar content being viewed by others

Introduction
Non-Hodgkin lymphoma (NHL) of the digestive tract is the most common extraganglionic lymphoma. Approximately 15–30% of primary extraganglionic lymphomas occur in the gastrointestinal tract, accounting for 1–10% of all gastrointestinal cancers [1,2,3]. Mucosa-associated lymphoid tissue (MALT) lymphoma is the most common histological subtype in the stomach, mantle cell lymphoma (MCL) in the terminal ileum, jejunum, and colon, T cell lymphoma is associated with enteropathy in the jejunum, and follicular lymphoma in the duodenum with geographic variation in distribution [4].
Mantle cell lymphoma is an aggressive B cell neoplasm characterized primarily by a monotonous proliferation of small-to-medium-sized lymphocytes coexpressing CD5, CD20, and cyclin D1 epitopes and frequently presents chromosomal translocations, t(11 14) (q13; q32) [5, 6]. Mantle cell lymphoma occurs more frequently in older males, representing approximately 4% of all lymphomas in Western countries [7]. Mantle cell lymphoma usually affects the gastrointestinal tract and may present as multiple areas of lymphomatous polyposis [8]. It is a rare type of gastrointestinal lymphoma, which infiltrates extensively into the intestine [9] and is characterized by the presence of several lymphomatous polyps along one or more segments of the gastrointestinal tract [2, 10]. Since the first description in 1961, only a few cases have been reported [11].
We report an unusual case of a patient with multiple areas of lymphomatous polyposis with extensive colorectal involvement and acute colonic obstruction, an atypical complication of this rare disease. On the basis of this case study, the pitfalls in the diagnosis and prognosis of gastrointestinal tract disorders of these polyposis presentations as well as the treatment options are discussed.
Case report/observation
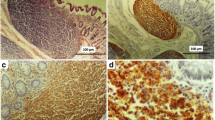
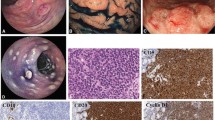
This was a 56-year-old man of Black ethnicity and Ivorian nationality without any relevant past medical history, who consulted for abdominal pain with an abrupt obstipation of solids and gas for 2 days. The physical examination displayed a patient in good general condition. There was abdominal distension associated with tympanism. The rectal examination was normal, and examination of the other organs was normal. The lymph node areas were normal. Abdominal X-rays revealed dilated bowel loops with fluid levels consistent with descending colonic obstruction. An exploratory laparotomy was indicated. Numerous polyploid formations of the left colon and sigmoid were palpated, and the largest obstructed the colon lumen. A subtotal colectomy was performed with reconstruction via an ileocolostomy. There were no apparent signs of polyposis in the distal sigmoid and rectum. The surgical specimen was sent to the anatomic and cytologic pathology department of the teaching hospital of Treichville (Ivory Coast). Macroscopy of the surgical specimen showed multiple polyploid formations in the colonic mucosa (Fig. 1). Microscopic examination after standard paraffin embedding techniques identified a diffuse lymphoid proliferation of the submucosa, made of small lymphoid cells with a hyperchromatic crenelated nucleus. (Fig. 2A, B). These features suggested a lymphomatous polyposis of MALT lymphoma type, either lymphocytic lymphoma type or mantle cell lymphoma type. Immunohistochemical analysis showed the positivity of the tumor to CD20, CD5, Bcl2, and cyclin D1, while it was negative to CD10 and CD23. The Ki67 proliferation index was 25%. (Fig. 2C–E). In view of the morphological and immunohistochemical results, we made the diagnosis of primary mantle cell lymphomatous polyposis. The clinic-radiological assessment classified our patient at stage II according to the Ann Arbor classification modified by Cheson et al. (Table 1) [12]. The patient received chemotherapy according to the cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) protocol associated with rituximab. After 1 year of follow-up, the patient has maintained a good general condition without recurrence.

Multiple polypoid formations in the colonic lumen

A Colon submucosa with infiltration by neoplastic lymphocytes (hematoxylin and eosin ×40). B Lymphoid cells with hyperchromatic nuclei (hematoxylin and eosin ×100). C The neoplastic lymphocytes are immunoreactive for CD20 (immunohistochemistry ×400). D The neoplastic lymphocytes are immunoreactive for CD5 (immunohistochemistry ×400). E The neoplastic lymphocytes are immunoreactive for cyclin D, which supports the diagnosis of mantle cell lymphoma (immunohistochemistry ×100)
Discussion
Epidemiological features
Multiple lymphomatous polyposis is a rare and heterogeneous group of small B cell lymphomas. B cell polyposis neoplasms include mantle cell lymphoma, follicular lymphoma, and MALT lymphoma [3]. Initially described by Cornes et al. in 1961, multiple lymphomatous polyposis is characterized by the presence of diffuse proliferation of atypical lymphocytes presenting as multiple polypoid lesions in different segments of the gastrointestinal tract [13]. B cell lymphomas of the gastrointestinal tract are more common than T cell lymphomas because these polyps histologically originate from the mantle area of the lymphoid follicle of mucosal associated lymphoid tissue (MALT) [14]. Therefore, the typical lymphoma presenting as lymphomatous polyposis is mantle cell lymphoma, which has been reported with a frequency of up to 9% of all B cell lymphomas of the gastrointestinal tract [14]. The colon and the rectum are generally the most affected segments (90% of mantle cell lymphoma cases), followed by the small intestine, stomach, and duodenum [2, 15]. Regional lymph node involvement is common. Other possible extradigestive sites are the bone marrow, the peripheral lymph nodes, Waldeyer’s ring, the spleen, and the liver [14]. The patient was 56 years old. Lymphomatous polyposis frequently occurs in elderly patients, with a male predominance [16,17,18]. The etiopathogenesis is unknown, but a genetic background, exposure to previous chemotherapy, and exposure to ionizing radiation could contribute to the development of gastrointestinal mantle cell lymphomatous polyposis [13, 19].
Clinical aspects
The clinical profile of mantle cell lymphomatous polyposis is heterogeneous. On the basis of the largest series of studies done by Ruskone-Fourmestraux et al., the main symptoms are abdominal pain, diarrhea, and hematochezia [2, 10, 20]; intestinal obstruction, exudative enteropathy, intestinal malabsorption, chylous ascites, and surgical abdomen are less frequent [2, 10]. Our patient was admitted for bowel obstruction.
Histopathological aspects
Lymphomatous polyposis is more often mantle cell lymphoma. However, lymphomatous polyposis can originate from a follicular lymphoma or a MALT lymphoma [9]. Grossly, it is characterized by the presence of numerous polypoid lesions sometimes involving several segments of the gastrointestinal tract [14]. We found multiple polyploid formations in the colonic mucosa. Histologically, it is a monotonous proliferation of small-to-medium-sized lymphocytes with a hyperchromatic crenelated nucleus [5]. Other entities should be considered for differential diagnosis, including chronic lymphocytic leukemia, peripheral T cell lymphoma, and subtype of lymphocytic lymphoma (diffuse large B cell lymphoma and immune proliferative small intestinal disease) [10, 21,22,23]. Immunohistochemical examination confirmed the diagnosis of mantle cell lymphomatous polyposis. Mantle cell lymphoma expresses B cell markers (CD20 and CD79a), T cell markers (CD5), and cyclin D1. These markers are used in routine immunohistochemical testing to diagnose mantle cell lymphoma. However, CD10 is a marker for diffuse large B cell lymphoma, and CD23 is a marker for follicular lymphoma. Both are generally negative in mantle cell lymphoma [5, 6]. In our patient, immunohistochemical analysis revealed positive immunostaining for anti-CD20, anti-CD5, anti-Bcl2, and anti-cyclin D1 antibodies. In contrast, there was no immunostaining for anti-CD10 and anti-CD23 antibodies. The Ki67 was estimated to be 25%. Thus, positivity of CD20, CD5, Bcl2, and cyclin D1 led to the diagnosis of mantle cell lymphomatous polyposis.
Therapeutic and prognostic aspects
Patients under 65 years of age with good general condition initially benefit from an intensive immunochemotherapy regimen (R-CHOP) combining rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone [24,25,26]. Rituximab monotherapy, a chimeric monoclonal antibody that specifically binds to the CD20 antigen, has response rates of 30%. When it is combined with anthracycline, the response rate can increase to more than 90% [26,27,28]. Standard immunochemotherapy with R-CHOP reduces lesions, and the initial response to treatment is 94% [24,25,26]. Unlike most B cell lymphomas, remissions of mantle cell lymphomatous polyposis are short [26, 27].
Mantle cell lymphoma has one of the poorest prognoses of all types of non-Hodgkin lymphomas, with a median survival time less than 3 years [2, 10, 22] and frequent relapses [3, 29]. Recent studies have suggested a role for bendamustine and rituximab, particularly if the patient’s functional status precludes a more aggressive chemotherapy regimen [30, 31]. The main treatment for mantle cell lymphoma is chemotherapy. Other treatments may include targeted therapy, biological therapy, radiotherapy, and stem cell transplantation [31]. Our patient received cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy in combination with rituximab. After 1 year of follow-up, the patient has maintained a good general condition, without recurrence.
Conclusion
Multiple lymphomatous polyposis is a rare and heterogeneous group of small B cell lymphomas, including mantle cell lymphoma, follicular lymphoma, and MALT lymphoma. It is defined by the presence of diffuse proliferation of atypical lymphocytes presenting as multiple polyploid lesions in different segments of the gastrointestinal tract. The typical lymphoma with lymphomatous polyposis is mantle cell lymphoma, described by overexpression of the anti-cyclin D1 antibody. It mostly occurs in elderly male patients.
Data availability
All data supporting the conclusions of this article are included in the manuscript.
Abbreviations
- NHL:
-
Non-Hodgkin lymphoma
- MCL:
-
Mantle cell lymphoma
- MALT:
-
Mucosa-associated lymphoid tissue
- R-CHOP:
-
Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
References
Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer. 2003;97:2462–73.
Williams ME, Bernstein SH, Jares P, Kahl BS, Witzig TE, Gordon LI. Recent advances in mantle cell lymphoma: report of the 2012 mantle Cell Lymphoma Consortium Workshop. Leuk Lymphoma. 2013;54:1882–90.
Kodama T, Ohshima K, Nomura K, Taniwaki M, Nakamura N, Nakamura S, et al. Lymphomatous polyposis of the gastrointestinal tract, including mantle cell lymphoma, follicular lymphoma and mucosa-associated lymphoid tissue lymphoma. Histopathology. 2005;47:467–78.
Rizvi MA, Evens AM, Tallman MS, Nelson BP, Rosen ST. T-cell non-Hodgkin lymphoma. Blood. 2006;107(4):1255–64.
Cestaro G, De Rosa M, Vitiello C, Galloro G, Gentile M, Cestaro G, et al. Multiple lymphomatous polyposis with diffuse involvement of the gastrointestinal tract: case report. G Chir. 2013;34:173–5.
Inamdar AA, Goy A, Ayoub NM, Attia C, Oton L, Taruvai V, et al. Mantle cell lymphoma in the era of precision medicine-diagnosis, biomarkers and therapeutic agents. Oncotarget. 2016;7:48692–731.
Schmidt C, Dreyling M. Therapy of mantle cell lymphoma: current standards and future strategies. HematolOncolClin North Am. 2008;22:953–63.
Kim JH, Jung HW, Kang KJ, Min BH, Lee JH, Chang DK, et al. Endoscopic findings in mantle cell lymphoma with gastrointestinal tract involvement. Acta Haematol. 2012;127:129–34.
Waisberg J, Anderi ADV, Cardoso PAS, Borducchi JHM, Germini DE, Fares Franco MI, et al. Extensive colorectal lymphomatous polyposis complicated by acute intestinal obstruction: a case report. J Med Case Rep. 2017;11:190.
Dasappa L, Suresh Babu MC, Sirsath NT, Suresh TM, GovindBabu K, Sathyanarayna V, et al. Primary gastrointestinal mantle cell lymphoma: a retrospective study. J Gastrointest Cancer. 2014;45:481–6.
Kahl BS, Dreyling M, Gordon LI, Martin P, Quintanilla-Martinez L, Sotomayor EM. Recent advances and future directions in mantle cell lymphoma research: report of the 2018 mantle cell lymphoma consortium workshop. Leuk Lymphoma. 2019;60:1853–65.
Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of hodgkin and non-hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–67.
Cornes JS. Multiple lymphomatous polyposis of the gastrointestinal tract. Cancer. 1961;14:249–57.
Lee JM, Kim ES, Choi HS, Keum B, Jeen YT, Lee HS, et al. Mantle cell lymphoma with multiple lymphomatous polyposis and intussusception: a case report. Oncol Lett. 2016;11(1):654–6.
Xu XJ, Wu SM. Multiple lymphomatous polyposis of the gastrointestinal tract: report of three cases and literature review. J Dig Dis. 2012;13:649–53.
Remes-Troche JM, De-Anda J, Ochoa V, Barreto-Zuñiga R, Arista-Nasr J, Valdovinos MA. A rare case of multiple lymphomatous polyposis with widespread involvement of the gastrointestinal tract. Arch Pathol Lab Med. 2003;127:1028–30.
The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood. 1997;89:3909–18.
Ruskoné-Fourmestraux A, Audouin J. Primary gastrointestinal tract mantle cell lymphoma as multiple lymphomatous polyposis. Best Pract Res ClinGastroenterol. 2010;24(1):35–42.
Karmali R, Switchenko JM, Goyal S, Shanmugasundaram K, Churnetski MC, Kolla B, et al. Multi-center analysis of practice patterns and outcomes of younger and older patients with mantle cell lymphoma in the rituximab era. Am J Hematol. 2021;96:1374–84.
Till KJ, Abdullah M, Alnassfan T, Janet GZ, Marks T, Coma S, et al. Roles of PI3Kγ and PI3Kδ in mantle cell lymphoma proliferation and migration contributing to efficacy of the PI3Kγ/δ inhibitor duvelisib. Sci Rep. 2023;13:3793.
Samaha H, Dumontet C, Ketterer N, Moullet I, Thieblemont C, Bouafia F, et al. Mantle cell lymphoma: a retrospective study of 121 cases. Leukemia. 1998;12:1281–7.
Li M, Zhang S, Gu F, Xiao W, Yao J, Chao K, et al. Clinicopathological characteristics and prognostic factors of primary gastrointestinal lymphoma: a 22-year experience from South China. Int J Clin ExpPathol. 2014;7:2718–28.
Salar A, Juanpere N, Bellosillo B, Domingo-Domenech E, Espinet B, Seoane A, et al. Gastrointestinal involvement in mantle cell lymphoma: a prospective clinic, endoscopic, and pathologic study. Am J SurgPathol. 2006;30:1274–80.
Tallarico M, Smith SM. From minimal residual disease to maintenance therapy: optimizing tools for treatment of mantle cell lymphoma. Oncology (Williston Park). 2016;30:361–2.
Nakajima Y, Tomita N, Itabashi M, Miyashita K, Watanabe R, Miyazaki T, et al. Analysis of outcomes in patients with supra-diaphragmatic vs infra-diaphragmatic diffuse large B cell lymphoma treated with R-CHOP therapy. LeukRes. 2015;39:198–203.
Lenz G, Dreyling M, Hoster E, Wörmann B, Dührsen U, Metzner B, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23:1984–92.
Determann O, Hoster E, Ott G, Bernd HW, Loddenkemper C, Hansmann ML, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood. 2008;111:2385–7.
Delarue R, Haioun C, Ribrag V, Brice P, Delmer A, Tilly H, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Grouped’Etude des Lymphomes de l’Adulte. Blood. 2013;121:48–53. https://doi.org/10.1182/blood-2011-09-370320.
Abrahamsson A, Albertsson-Lindblad A, Brown PN, Baumgartner Wennerholm S, Pedersen LM, et al. Real world data on primary treatment for mantle cell lymphoma: a Nordic Lymphoma Group observational study. Blood. 2014;124:1288–95.
Flinn IW, van der Jagt R, Kahl B, Wood P, Hawkins T, MacDonald D, et al. First-line treatment of patients with indolent non-Hodgkin lymphoma or mantle-cell lymphoma with bendamustine plus rituximab versus R-CHOP or R-CVP: results of the BRIGHT 5-year follow-up study. J Clin Oncol. 2019;37:984–91.
Nassri R, Nassri A, Alkhasawneh A, de Souza RB, Schey R. Colonic mantle cell lymphoma with multiple lymphomatous polyposis. GE Port J Gastroenterol. 2020;27:296–8.
Acknowledgements
Not applicable.
Funding
The authors did not receive funding from any source to conduct this study.
Author information
Authors and Affiliations
Contributions
BBSK, NAA, and JTD performed laboratory analyses, interpreted the data, and drafted the manuscript. ZIC, MK, and KEK participated in the management of the specimens and reviewed the manuscript. All authors have read and approved the final version of the manuscript to be submitted for publication.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The study received approval from the head of the laboratory to be conducted. We have obtained oral consent from the patient. The manuscript was not submitted to more than one journal for simultaneous review and has not been previously published. This was a single study, not divided into multiple parts, and no data were generated.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Conflict of interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Djiwa, T., Koui, B.B.S., Aman, N.A. et al. Colonic lymphomatous polyposis mantle cell lymphoma: a case report and review of literature. J Med Case Reports 18, 219 (2024). https://doi.org/10.1186/s13256-024-04533-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-024-04533-z