
Abstract
The effects of nicotine and cigarette smoke in many diseases, notably COVID-19 infection, are being debated more frequently. The current basic data for COVID-19 is increasing and indicating the higher risk of COVID-19 infections in smokers due to the overexpression of corresponding host receptors to viral entry. However, current multi-national epidemiological reports indicate a lower incidence of COVID-19 disease in smokers. Current data indicates that smokers are more susceptible to some diseases and more protective of some other. Interestingly, nicotine is also reported to play a dual role, being both inflammatory and anti-inflammatory. In the present study, we tried to investigate the effect of pure nicotine on various cells involved in COVID-19 infection. We followed an organ-based systematic approach to decipher the effect of nicotine in damaged organs corresponding to COVID-19 pathogenesis (12 related diseases). Considering that the effects of nicotine and cigarette smoke are different from each other, it is necessary to be careful in generalizing the effects of nicotine and cigarette to each other in the conducted researches. The generalization and the undifferentiation of nicotine from smoke is a significant bias. Moreover, different doses of nicotine stimulate different effects (dose-dependent response). In addition to further assessing the role of nicotine in COVID-19 infection and any other cases, a clever assessment of underlying diseases should also be considered to achieve a guideline for health providers and a personalized approach to treatment.
Similar content being viewed by others


Implication.
COVID-19 infection is currently the most prominent health challenge, and health decision-makers are looking for indicators to reduce the mortality and morbidity of the infection. Meanwhile, smokers make a considerable population, and current molecular data suggest a higher risk of COVID-19 infection due to the overexpression of Angiotensin-converting enzyme 2 (ACE2), the receptor for viral binding and entry. On the other hand, epidemiological studies show the controversial reports. According to previous investigations, we have systematically and comprehensively assessed nicotine’s effects on different cells and diseases, mainly in COVID-19. As the main result, we have recommended a research roadmap to identify a personalized approach for nicotine therapy (who, when, which dose, and how) for healthcare providers and researchers.
Introduction
Coronaviruses are a large family of viruses in humans and animals, including Middle East Respiratory Syndrome (MERS-CoV), Severe Acute Respiratory Syndrome (SARS-CoV), and coronavirus disease 2019 (SARS-CoV-2 or COVID-19). Last two decades, several outbreaks of coronaviruses are observed. The SARS outbreak was reported in 2003 with about 8000 cases and more than 700 deaths with an estimated case fatality rate (CFR) of 10% [1]. According to WHO (Jan 2020), MERS has infected about 2500 cases with more than 800 deaths (CFR ≈ 34.3%) in different countries since 2012 [2]. The report of WHO showed the novel coronavirus pneumonia, COVID-19, has infected about 177,108,695 confirmed cases with 3,840,223 deaths until 18 June 2021 [3], and any protective or treatment factor is demanding for health managers.
Currently, there are contradictory reports about the effect of smoking on COVID-19 infections. If smokers are protective of COVID-19, new studies could be designed to explore the mechanism of protection. Nevertheless, if smokers are at higher risk, it will be a wake-up call for health providers to adopt separate policies to protect smokers. Currently, we know that smoking is one of the main risk factors for about 20 types and subtypes of cancers, cardiovascular diseases, Chronic Obstructive Pulmonary Disease (COPD), and diabetes. Among the various body parts, the most involvement is directly connected with the respiratory system, digestive system, and nervous system [4]. According to the 2025 estimation of the World Health Organization (WHO), there will be about 1.1 billion smokers worldwide [5]. International statistics show that there are about 8 million deaths each year, mostly (80%) in low- and middle-income countries, equivalent to a total economic cost of US$1.4 trillion each year [6].
Currently, it is clear that current and former smokers had a higher percentage among COVID-19 patients in ICU (~ 2.4 times) and mechanical ventilation support, and they had higher mortality rates and more severe cases (~ 1.4 times) [7,8,9]. Even after treatment, exacerbations of COVID-19 are associated with tobacco smoking in the smokers’ group [10]. Hospitalized smokers have more severe infections and complications, especially pulmonary fibrosis, as the central pathology of SARS, MERS, and COVID-19 [11]. Like MERS, a higher CFR in hospitalized patients with smoking history is also reported for COVID-19 [12].
Moreover, tobacco smoking is involved in more severe complications of COVID-19 infections [9]. It has been shown that cigarette smoke upregulates ACE2 in a dose-dependent manner in lung epithelium, including the goblet, club, Clara cells, and alveolar type 2 (AT2) cells of human and rodent lungs, and quitting smoking decreases ACE2 expression (reversible) [13]. So, smoking history is a predictor of ACE2 expression in the lung cells. The expression of Cathepsin B, which is a protease that activates the spike protein of the virus, is also increased in mice and human cells exposed to cigarette smoke [14]. So, it seems that the patients’ positive smoking history (current and former smokers) can make them more susceptible to COVID-19. Also, results regarding the effect of smoking on the expression of ACE2 are opposite; besides up-regulation (preprint) [15], down-regulation [16] is reported [17].
On the other hand, based on the current epidemiological studies on COVID-19, China [7, 18], Italia [19], Europe, and the United States [20] have reported lower incidence of infected smokers. Tajlil et al. had performed a meta-analysis on 12 reports of COVID-19 epidemiologic data (11,382 cases, including 3,827 cases in 10 Chinese reports and 7,555 cases in two USA reports). They had concluded the significantly lower proportion of hospitalized COVID-19 patients with smoking history [21]. It should be added that all the current smokers, when entering the hospital or the ICU, are forced to stop nicotine uptake. Besides, a large meta-analysis of over 17,278,392 COVID-19 infected adults showed a lower incidence of infection in smokers [22], which is concordant to former findings in China [7], France [23], and the United States [24]. However, several studies showed adverse effects of smoking on COVID-19 outcomes such as studies in Kuwait (1096 cases), England (3179 cases), and USA (12,347 cases) [25,26,27]. There is no solid evidence about the protective effect (prevention or treatment) of smoking on COVID-19 infection. The important thing to remember about epidemiological studies is that they have limitations, especially in this field. These limitations include things like the heterogeneity of COPD disease, the heterogeneity of COVID-19 disease, and the restrictions connected to it discussed the characteristics of epidemiological investigations, such as sample sizes and ascertainment bias [28].
In this study, we first tried to systematically show that cigarette smoke effects, although similar in some effects, are different from pure nicotine. Therefore, many experiments on the effect of smoke conclude their results as a probable effect of nicotine as an equivalent univalent. These conclusions can be misleading. Additionally, different cell responses to nicotine are dose-dependent. Considering the prevalence of the COVID-19 epidemic and different published results in this field and the existing judgments, an attempt was made to investigate the role of cigarettes and nicotine in this epidemic disease. Failure to assign this issue can cause misleading and diversion of people and health providers. Therefore, in this study, we have tried to take a closer basic-clinical look at this issue.
Our approach and methodology
In the first step, articles containing the MeSH terms of “smoke” and “COVID-19” and “meta-analysis” were investigated only in PubMed (16 Jul 2021) (Fig. 1-a). Meta-analysis studies with more than 10 studies were summarized in Tables 1 and 2.

PRISMA table for (a) smoke and COVID-19 and (b) nicotine and COVID-19.
Also, articles containing the MeSH terms of “nicotine” and “COVID-19” were systematically searched in the online databases of PubMed, Google Scholar (Fig. 1-b). Then, all the articles were initially evaluated, and the preliminary design for the signaling and metabolic pathway involved in coronavirus infection and smoking/nicotine responses was performed. We performed a systematic search on (SARS-CoV-2 OR COVID-19) in PubMed and manually reviewed all the publications. Subsequently, all related articles were reviewed and included in this review (2021, June). Since the articles related to the study of the effect of nicotine in COVID-19 were very sparse, two other searching strategies regarding the effect of pure nicotine on different (A) cells, including epithelial, fibroblast, endothelial, and dendritic, macrophages, T, and B cells, and (2) organs, including respiratory, nervous, metabolic, cardiovascular, and urogenital systems. Generally, 12 diseases were candidate according to their similarity with the pulmonary and extrapulmonary complications of COVID-19. Also, Finally, the effect of smoking/nicotine on other common coronaviruses (SARS and MERS) was also evaluated and added to the study.
COVID-19
As a current health-threatening pandemic infection of the respiratory system, COVID-19 is accompanied by respiratory pneumonia symptoms (e.g., dry cough, fatigue, myalgia, fever, and dyspnea) and extrapulmonary symptoms mainly in the heart, liver, gut, kidney, and brain (Fig. 2) [29,30,31]. COVID-19 is an upper respiratory disease with systemic symptoms to distant organs/tissues due to distribution of diverse signaling molecules as a distant signaling mechanism. Chest tightness, breath shortness, and fatigue are severe symptoms of the disease. Patients with original comorbidities, including cardiovascular disease, cancer, kidney and liver diseases, diabetes mellitus, hypertension, tuberculosis, venous thromboembolism, and older adults, are high-risk cases for COVID-19 infection [32].

The multi-organ injury (A) and functioning of different cells (B) in SARS-CoV-2 infection
The renin-angiotensin system (RAS), which regulates water metabolism, endocrine secretion, and blood pressure in the body, is dysregulated during coronaviral infections. As an essential member of RAS, ACE2 is the common receptor for the attachment of SARS-CoV-2 and SARS-CoV. Coronaviruses identify a virus-binding hotspot region on ACE2 protein via spike protein [33]. ACE2 is a transmembrane protein that regulates the vascular system’s tuning and secretion of hormones within the RAS system, especially in the lung, heart, gastrointestinal, and kidney [34]. According to the current understanding, the expression of ACE2 was not different based on age, sex, or different racial groups [35]. Additionally, ACE2 downregulation after the SARS-CoV-2 infection is reported [36].
Of note, the ACE2 receptor is not adequate for viral entry into host cells. Recent evidence has shown that TMPRSS2 (transmembrane serine protease 2), a serine protease, may cleave the SARS-CoV-2 S protein [37]. Furthermore, the SARS-CoV-2 S protein has a furin cleavage site with the RPPA (Reverse Phase Protein Array) sequence [38]. Furin is a ubiquitous protease that is activated when exposed to an acidic pH [39]. Ubiquitous expressions of furin-like proteases may explain the increased pathogenesis of SARS-CoV-2. Studies have shown that RAS activation and ACE2 downregulation are implicated in the pathogenesis of lung damage following the SARS-CoV infection [40]. Renin, a protease secreted by the kidneys, converts angiotensinogen into angiotensin I (Ang I), which is metabolized by ACE into Ang II. Then Ang II is converted into Ang (1–7) by ACE2 [41]. SARS-CoV-2 causes downregulation of ACE2 because of enhanced levels of ACE2 shedding, which results in increased Ang II and reduced Ang (1–7) levels [40]. Ang II can bind to two classes of receptors, the Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R) [42]. The binding of Ang II to AT1R activates protein kinase C (PKC), and induces pulmonary vasoconstriction and increased vascular permeability [43, 44]. PKC activation induces phosphorylation of the ADAM17 (ADAM Metallopeptidase Domain 17) tail and, subsequently, the phosphorylated ADAM17 tail enhances ACE2 shedding, assists with viral entry and tissue injury [45, 46]. PKC activation reduces amiloride-sensitive epithelial sodium channel (ENaC) expression, which is important for alveolar fluid clearance (AFC) and results in enhanced lung edema and worsened lung function [47]. Ji and colleagues reported that the S and E proteins of SARS-CoV diminish both activation and expression of ENaC by activating PKC isoforms [48]. ENaC-ɑ must be activated by proteases, particularly furin, in order for it to function. Anand et al. reported that SARS-CoV-2 hijacks proteases and these proteases subsequently activate ENaC-ɑ for viral activation. Thus, reduced ENaC function could be a reason for manifested pulmonary edema in COVID-19 infections [49]. Besides, activation of PKC downregulates AT2R gene expression [50]. Ang II via AT2R has the opposite effect, it activates the bradykinin-nitric oxide (NO)-cGMP cascade, and, subsequently, vasodilation occurs. Notably, Ang II and Ang (1–7) have the greatest relative affinities for AT1R and AT2R [42].
Besides, most of the patients with COVID-19 show lymphopenia (low level of lymphocytes) and hypercytokinemia (cytokine storm), which is intensified in ICU than non-ICU patients were also evident in SARS-CoV cases in 2003. These two dysregulated systemic responses are associated with increased severity and mortality of the patients [51]. Most of the patients with lymphopenia contained reactive lymphocytes, which was not observed in 2003 epidemy. After the viral entrance, the absolute number of T-lymphocytes and the frequency of CD4+ and CD8+ T cells, and the CD4+/CD8+ ratio are decreased in COVID-19 patients. Also, interferon-gamma )IFNγ( producing CD4+ T cells from severe COVID-19 patients are lower than moderate cases [52]. In COVID-19 patients, M2 macrophages dominate M1 types in BAL samples of cases, indicating their crucial involvement in inflammation and fibrogenesis [53]. The serum amount of IL1, IL6, IL8, IL10, and IL2R, and high-sensitivity C-reactive protein (hs-CRP) are upregulated in cases at the admission. Also, ICU cases express more cytokines than non-ICU patients [53]. SARS-CoV nucleocapsid protein (N protein) activates IL6 expression in a concentration-dependent manner, which requires NF-κB binding cis-acting element at the IL6 promoter region [54]. Further, SARS-CoV activates NLRP3 inflammasome, which upregulates the expression of IL1β and IL18 proinflammatory cytokines in lipopolysaccharide (LPS)-primed macrophages via 3a protein (also known as 3a-NLRP3-IL1b) signaling axis [55]. Generally, the platelet count is normal in COVID-19 patients, but some patients show mild thrombocytopenia (low platelets) [18]. Endothelial dysfunction and organ failure are associated with SARS-CoV-2 infections [56] (Fig. 2). The previous evidence showed that increased levels of pro-inflammatory cytokines may promote hypercoagulative factors, which induce vascular thrombosis [57, 58]. ACE2 down-regulation might be the other etiology of hypercoagulable state in patients with COVID-19, it is a negative feedback regulator of ACE2 expression via activating MAPK1 and MAPK3. Besides, elevated level of Ang II or ACE/Ang II/AT1 axis after ACE2 down-regulation resulting in extreme pro-inflammatory and pro-thrombotic cytokines [57, 59].
Nicotine
Nicotine
Tobacco constitutes a varied mixture of > 8000 chemicals, including nicotine, carbon monoxide, nitrogen oxides, pro-oxidants (e.g. free radicals), aromatic amines, catechols, inorganics (e.g. nickel, chromium, and cadmium) [60, 61]. Although the exact mechanisms by which smoking causes diseases are unclear, chronic inflammation is the main factor in the development of diseases relevant to smoking such as lung cancer, heart disease, COPD, and asthma [61, 62]. Nicotine (C10H14N2), a potent parasympathomimetic alkaloid, is the primary addictive tobacco smoke component. It was named after Jean Nicot, the French ambassador to Portugal, who introduced tobacco seed in Paris in 1550 [63]. A single cigarette tar contains about 10 to 12 mg nicotine, and when a cigarette is smoked, between 1 and 2 mg is inhaled into the lungs [64]. Nicotine interferes with the natural functioning of acetylcholine and over-stimulates acetylcholine receptors. In addition to the activation of nicotinic receptors, nicotine can be absorbed from cellular membranes, especially through the epithelium of the lower respiratory system and alveolus [65]. After absorption through the lungs, nicotine is metabolized in the liver by CYP2A6, UDP glucuronosyltransferase (UGT), and flavin-containing monooxygenase (FMO) enzymes. Cotinine is the principal (70–80%) product of nicotine metabolization and is used as a biomarker in smokers, second-hand smokers, and children exposed to smoke (detectable in blood, urine, saliva, hair, and nail) [66, 67].
Nicotine is only one of 8,000 tobacco components in cigarettes; therefore the consequences of smoking cannot be attributed only to nicotine [68]. In one study, the cytotoxicity of cigarette smoke extract (CSE) containing nicotine (CSE-N) and nicotine-free portion (CSE-O) on human bronchial epithelial cells was studied (BEAS-2B). CSE and CSE-O were shown to be hazardous to BEAS-2B cells, but CSE-N exhibited substantially lower cytotoxicity. CSE-O, but not CSE or CSE-N, enhanced apoptosis in cells stably expressing CYP2A13 (B-2A13). Cytochrome P450 2A13 (CYP2A13), an extrahepatic enzyme mostly expressed in the human respiratory system, has been discovered to mediate cigarette smoke metabolism and toxicity. These findings show that the nicotine component reduces the metabolic activation of CYP2A13 to CSE. As a result, it may be useful in lowering the toxicity produced by other tobacco components [69]. In fact, much more research is required to establish whether pure nicotine has detrimental or good effects, although it can be stated that many of the consequences induced by cigarette smoke, particularly in respiratory problems, are caused by other components in cigarette smoke.
Nicotine-responsive receptors
Nicotine imitates the function of acetylcholine, the principal neurotransmitter in the central nervous system (CNS), by binding to the various acetylcholine receptors. Acetylcholine can bind to two different receptors, including nicotinic (nAChRs) and muscarinic (mAChRs) cholinergic receptors, activated by specific nicotine and muscarine agonists, respectively. The competitive binding of nicotine to nicotinic cholinergic receptors induces a rapid transient allosteric conformational change to form a selective ionic channel. Activation of the receptors in the CNS activates dopamine, the pleasure hormone [70].
After stimulation of the cells with nicotine, calcium internalization increases (~ 100 fold), associated with loss of membrane integrity [71]. Also, in the case of continuous exposure of the receptor to an agonist, long-term inactivation happens. The constant stimulation of the receptor downregulates its expression to provide the tolerance effect [72]. Therefore, smokers need to increase their smoke to take a constant effect. Prolonged nicotine consumption/smoking reduces active receptors (unstimulated receptors) and dopamine release that stimulates the increase of smoking packs to recover their pleasure [72]. After a sleeping period, nicotine concentration is decreased, and more receptors are in the unoccupied status, and more dopamine is accumulated within the cells, which can induce maximal pleasure during the first-smoke of the day. Inactivation of the receptors and their downregulation and decreased dopamine release is called tolerance, which makes the dependency on smokers and addiction [73].
The nAChRs are divided into two different groups of neuronal and non-neuronal receptors based on their expression in various tissues. Structurally, this receptor family is a homo(hetero) pentamer composed of a combination of α(1–10), β(1–5), ε/γ, and δ subunits [74]. There is a specific neuronal and non-neuronal pentamer combination of subunits distributing throughout the body. Among them, α7 pentamer is the most studies homo-pentamer, which interacts with nicotine [75]. Non-neuronal nAChRs are expressed in lung epithelial, endothelial, fibroblast cells, and also in muscles. Seemingly, bronchial epithelial cells express more muscle-type nAChRs [76, 77]. Also, fibroblasts are enriched for more nAChRs subunits than epithelial cells.
There are many publications investigating the interplay between nicotine and nAChRs [78, 79]. Nicotine exposure changes the expression of nAChRs in active smokers compared to non-smokers [71]. In vivo and in vitro studies on nicotine exposure demonstrated the activation of several downstream signaling pathways in fibroblast and airway epithelial cells [80, 81]. In active smokers, bronchial epithelial cells express less the β4 subunit of nAChRs than ex-smokers and never-smokers [82]. Chronic nicotine exposure increases the expression of the α5 subunit of nAChRs in epithelial cells. The α3 subunit is also decreased in airway fibroblast cells of smokers [83]. Besides the epithelial and fibroblast cells, many immune cells also express nicotinic receptors. More specific experiments showed that blockade of muscle-type nAChRs effectively prevents nicotine-mediated calcium internalization through PKC and p38, but not p42/44 signaling. P38 is immediately activated after nicotine treatment. The effect of nicotine on different cells is covered in the following sections in more detail. Furthermore, in line with the direct interaction with nAChRs, nicotine indirectly affects the renin-angiotensin system. Exposure to nicotine increases the expression of ACE2 receptor in airway cells. This upregulation, along with the immune system’s misregulation, is discussed in hypersensitivity pneumonia and coronavirus sections.
Cellular physiology in response to nicotine
Following inhalation of tobacco smoke, nicotine is absorbed mainly by small airways and alveoli of the lungs, and the residue is distributed throughout the body by the circulatory system and alters the physiological processes of cells that express nAChRs. In addition to lungs, nicotine has profound systemic effects on many organs such as kidneys, heart, liver, and gastrointestinal tract, mediated through damage to epithelial, endothelial, and fibroblast cells [84]. Besides, nicotine can massively affect the function of the immune system [85]. As the first line of defense against pathogens, the innate immune system constitutes chemical and physical barriers (such as skin and other epithelial surfaces) and cellular defenses. Dendritic cells (DCs), monocytes and macrophages, Langerhans cells, neutrophils, natural killer (NK) cells, basophils, eosinophils, and mast cells are the cells of the innate immune system. Having the ability of processing and presentation of antigens to specific types of lymphocytes makes macrophages and DCs as two professional antigen-presenting cells (APCs). Likewise, the adaptive (acquired) immune system provides a specific immune response with an immunological memory against infectious agents. T cells and B cells play essential roles in the adaptive immune defense [86].
Depending on the cell type and combination of the various subunits of nAChRs, nicotine contributes to releasing growth factors, modification of extracellular matrix, dysregulated growth, and angiogenesis. Herein, nicotine’s effect on its primary targets includes epithelial, endothelial, fibroblast, and immune cells like DCs, macrophages, T, and B cells are covered (Fig. 3).

An illustration of the effects of nicotine in suppressing or activation of different signaling pathways. An abstract signaling pathway indicating the effect of nicotine via the nAChRs receptor is also provided
Epithelial cells
Epithelial cells form a mechanical barrier that prevents pathogenic substances and secrete mucus and antimicrobial peptides in host defense. Nicotine changes the viability, morphology, and motility of epithelium in various organs, including the respiratory system and oral mucosa [87,88,89]. Nicotine increases the expression of kidney injury molecule-1, the classic epithelial-mesenchymal transition (EMT) markers, vimentin, fibronectin, and production of α-smooth muscle actin (α-SMA), TGF-β, MCP-1, and ROS in the renal epithelium. Nicotine contributes to renal injury via STAT3, JNK, and AP-1 [90, 91]. In addition to the increased frequency of eosinophils in lamina propria (a thin layer of connective tissue), nicotine enhances the expression of ICAM1 on the submucosal connective tissue of the intestine and trachea [92]. Upon exposure to nicotine, the release of granulocyte-macrophage colony-stimulating factor (GM-CSF) and the activation of Akt are also stimulated in airway epithelial cells (AECs) [93, 94]. In response to chronic nicotine exposure, the upregulation of nAChRs occurs in bronchial epithelial cells (BECs), leading to the activation of PKC and MAPK p38. Similarly, the p42/44 signaling pathway is activated in airway fibroblasts [71]. The presence of nicotine also suppresses inflammatory factors such as MUC5AC in airway epithelial cells [95]. The bronchial epithelial cell showed apoptosis and senescence via ROS mediated autophagy-impairment mediated by nicotine [96]. Same to macrophages derived from human monocytes (MDMs) and type II pneumocytes, nicotine decreases the expression of the TLR-2, TLR-4, and NOD-2 on epithelial cells, which happens without alteration of the viability of the cells. Nicotine also reduces the expression of the surfactant protein (SP)-D in type II pneumocytes. During Mycobacterium tuberculosis infection, nicotine decreases IL6 and CCL5 in epithelial cells (EpCs), whereas in MDMs, there is a decrease in IL6, IL8, IL10, and TNF-α, and CCL2, CXCL9, and CXCL10. The authors pointed that nicotine probably modulates the expression of these molecules by an independent nAChRs pathway [97]. IL22 cytokine is a vital link between immunity and barrier function. By increasing transepithelial resistance and epithelial cell regeneration and repair, IL22 pathway maintenances epithelial cell health and the lung function [98]. Nicotine also downregulates the expression of IL-22Rα1 which is followed by suppression of AECs response to IL22 [99].
Fibroblasts
As the primary source of the extracellular matrix (ECM), fibroblasts provide the structural framework for tissues and play a critical role in wound healing [100]. Over the past decades, many studies have provided evidence for nicotine involvement in the modulation of fibroblast activation and function in various systems, such as the heart, lung, gingiva, prostate, and joints. However, gene expression changes may not be identical. Also, nicotine interrupts the airway epithelium’s barrier function, leading to the release of inflammatory proteins into the systemic circulation and causes multi-organ fibrosis [101]. TGF-β is described as the key molecule responsible for nicotine-induced fibrosis in various organs [102,103,104].
Further, nicotine administration results in increased fibronectin (FN1) gene expression in rat hearts [105]. Nicotine also alters the deposition and phagocytosis of collagen in fibroblast cells [106]. In a dose-dependent manner, nicotine raises nerve growth factor (NGF) secretion by lung fibroblasts and stimulated NF-κB nuclear translocation and transcriptional activity [107]. Both fibroblast proliferation via MEK-1/ERK and their collagen type I expression in the lung via α7nAChRs are also induced by nicotine [108]. Also, nicotine tempers fibroblast proliferation via modulation of growth factors and microRNA, particularly by targeting miR-24 [109].
Endothelial cells
The endothelium is a thin layer of cells between the interior surface of blood vessels and lymphatic vessels. They have various tasks like; permeability, leukocyte trafficking, vascular tone regulation, angiogenesis, and immunity. Hence, the impaired endothelial function leads to inflammatory diseases, remarkably vascular disorders (i.e., atherosclerosis) [110]. Nicotine is one of the main components of cigarettes that can cause endothelial dysfunction. Nicotine exposure gives rise to differential gene expression of endothelial cells, growth of atherosclerotic plaques, and angiogenesis [111, 112]. Human coronary endothelial cells treated with nicotine showed increased expression of ACEI, nitric oxide synthase, and vascular cell adhesion molecule-1 (VCAM1) [113]. Angiotensin II Type I Receptor Antagonism (AT1) was found to diminish the adverse effects of nicotine on cardiac structure and function [105]. nAChRs are also expressed on the upper respiratory endothelial cells, neighboring to ACE-2 receptors [114].
Dendritic cells
Since DCs facilitate the initiation of cell-mediated adaptive immune responses, they are pivotal cells of the innate immunity system. Dendritic cells are well-known as the most professional APCs with high expression of major histocompatibility complex (MHC) class II molecules (MHC-II), and they can uptake, process, and present antigens to naïve T cells and promote their polarization into different T cell subsets [115]. Dendritic cells can uptake antigens via several mechanisms such as receptor-mediated endocytosis (using mannose receptor, FcγRI, and FcγRII), macropinocytosis, and phagocytosis (via CD36 and αvβ3 or αvβ5 integrins) [116]. Although rare in human blood, DCs are heterogeneous cell populations with different subsets, including plasmacytoid DC (pDC) and classical DC or myeloid DC (cDC). The latter can further be divided into two subpopulations of cDC1 and cDC2. The DCs subsets have mouse counterparts as well [117].
Aicher et al. showed that nicotine could enhance HLA-DR expression, costimulatory molecules (CD86 and CD40), and adhesion molecules (LFA-1 and its ligand, CD54) in human DCs. Therefore, nicotine increases the production of IL12 in DCs and stimulates the activation of T cells, which is followed by augmented IL2 production and increased CD40L expression. They proposed that ERK1/2, p38 MAPK, and Akt signaling pathways may mediate the effects of nicotine on DCs [118]. For the first time, Nouri-Shirazi et al. provide evidence of the immunosuppressive effect of nicotine on human DCs. They could demonstrate that nicotine exposure alters the function of monocyte-derived DCs, including (1) antigen uptake: no significant change was observed in macropinocytosis, but endocytic and phagocytic properties of immature DCs were reduced. In this context, they observed downregulation of mannose receptor without a change in CD36 expression; (2) maturation and cytokine production: DCs matured in response to lipopolysaccharide; however, they produced a lower level of pro-inflammatory cytokines, including IL1β, IL10, IL12, and TNF-α; and (3) T cell responses: in the presence of nicotine, DCs showed an anti-proliferative effect on T cells which finally led to the inhibition of Th1 polarization and IFN-γ production. The latter was linked to the downregulation of IL12 as well as CD86 (B7.1) and CD80 (B7.2) in human DCs [119, 120]. Correspondingly, Nouri-Shirazi et al. provided similar evidence for nicotine’s effect on mouse DCs [121]. Later, the association of nicotinic environment and dose-dependent functioning of DCs was described; The pro-apoptotic activity in DCs in high doses of nicotine and anti-apoptotic effect in the lower doses [122]. In a recent study, nicotine-treated DCs showed increased expression of CD40 and CD197 and decreased expression of CD86, MHC-II, and CCP3 (an apoptotic molecule). Interestingly, nicotine did not alter CD80 levels in DCs. While the phagocytosis property of DCs was attenuated in the presence of nicotine, no change was seen in its endocytosis function [123].
Macrophages
Macrophages, as mononuclear phagocytes, are highly heterogeneous cells that differentiated from monocytes. They have multiple functions, such as phagocytosis, antigen presentation, and the production of different types of cytokines [124]. Based on their role, several distinct subsets of macrophages have been defined: (1) classically activated macrophages (M1) which mediate antimicrobial defense via secretion of pro-inflammatory cytokines, (2) alternatively activated macrophages (M2) with anti-inflammatory function, (3) regulatory macrophages (Mregs) which secrete large amounts of IL10, (4) tumor-associated macrophages (TAMs) which suppress anti-tumor immunity, and finally (5) myeloid-derived suppressor cells (MDSCs) as a group of immature cells linked to TAMs [125].
It has been reported that exposure of human macrophages to nicotine increases the intracellular ROS through the activation of Src/PI3K/Akt cascade and NADPH oxidase, which activates AMPK/MAPKs and NF-κB signaling. Flowingly, the activation of these signaling pathways upregulates the expression of IL8, a pro-inflammatory chemokine [126]. While nicotine upregulates the expression of IL8, the expression of IL1β, IL6, TNFα, MIP-1α, MIP-1β, and MCP-1 are downregulated. It also attenuates the phagocytosis of macrophages by affecting phagocytic recognition molecules, SR-A1 and TLR-2. [127]. However, macrophages treated with nicotine exhibited upregulated expression of CD36, possibly via the ROS/PKCδ/PPARγ signaling pathway [128]. The exposure of mice to nicotine also showed a rise in macrophages’ infiltration into the bronchoalveolar lavage fluid [129]. Besides the induction of inflammatory macrophages, nicotine treatments promote the M2 polarization, reduces the secretion of several proinflammatory cytokines in macrophages, and suppresses the alveolar macrophages to present the inhaled antigen to lymphocytes [130, 131].
In human monocytes, activation of α7nAChR inhibits IκB phosphorylation, followed by inhibition of nuclear localization and transcriptional activation of NF-κB [132]. Likewise, in murine macrophages, the activated α7nAChR can mediate Jak2-STAT3 activation and then can induce the phosphorylation of STAT3, which subsequently downregulates pro-inflammatory cytokines (e.g., IL6, IL12, and TNF-α) [133]. Similarly, overexpression of IRAK-M (a negative regulator of TLR-mediated immune responses) in human macrophages through JAK2/STAT3/PI3K not only mediates the anti-inflammatory effect of nicotine via α7nAChRs but also triggers the hyperresponsivity [134]. Altogether, these data suggest that nicotine exerts a dual role in macrophages. In a recent study, it has been shown that the effect of nicotine on infected macrophages is contradictory to uninfected macrophages; (1) uninfected macrophages express anti-inflammatory responses, including polarization of CD206+ M2 macrophages and increased expression of IL10, whereas (2) infected macrophages show inflammatory responses, including differentiation to M1 macrophages and increased expression of iNOS, TNF-α, and IL6. Additionally, they have identified a dose-dependent effect of nicotine on macrophages through α7nAChRs [135].
T cells
Once developed in the thymus, T cells circulate throughout the body to participate in the cell-mediated adaptive immune response. Overall, there are two specific T cells, including CD4+ T cells (helper T cells; Th) and CD8+ T cells (cytotoxic T cells; Tc). The CD4+ Th subset includes Th1, Th2, Th9, Th17, Th22, Treg, and Tfh categorized according to their surface molecules, transcription factors, and functions. In parallel, there are such subsets for CD8+ T cells [136].
Many studies have been performed to show how nicotine alters T cell-mediated immune responses. With a dose-dependent response, the serum level of cotinine, the primary metabolite of nicotine catabolism, was correlated with increased naive (CD45RA+) and decreased memory (CD45RO+) CD3+ CD4+ T cells. However, the CD8+ T cell population showed no significant difference in passive smokers than controls [137]. Besides α7nAChRs, nicotine appeared to increment Th2 cells via the activation of α4nAChRs induced through the Gprin1/CDC42 signaling pathway. The α4nAChRs-expressing Th lymphocytes were found in the circulation system, spleen, bone marrow, and thymus [138]. Additionally, nicotine-treated mice showed a higher frequency of PD-1- IL7R+ CD8+ T cells (non-exhausted phenotype associated with loss of tolerance) in bone marrow and spleen. These cells were promoted to produce survivin. In parallel with higher expression and activation of α4/α7nAChRs, the transcription factors of T-bet and Blimp-1 were downregulated in CD8 + T cells [139].
In the context of nicotine’s immunosuppressive properties, a pivotal role has been attributed to the expression and activation of α7 nAChRs on CD4+ T cells. Nicotine skewed polarization from Th1 and Th17 cells to Th2 cells. Although both Th1 (IFN-γ and TNF-α) and Th17 (IL17, IL21, and IL22) cytokines were downregulated, the Th2 cytokine (IL4) was upregulated. This effect was further accompanied by reducing T-bet and augmentation of GATA-3 and lower NF-κB-mediated transcription of I-κB and IL-2 [140]. According to a recent study, the co-culture of CD4+ T cells and DCs in a nicotinic environment reduced the differentiation to Th1 (IFN-γ), increased the differentiation to Treg (FOXP3) and Th2 (IL6, IL10, and IL13) cells [123]. Nicotine stimulation also reduced the proportion of IL-22-producing PBMCs, particularly CD4+ T cells. By targeting IL-22Rα1 expression, the nicotine impaired IL-22/IL-22R signaling axis [99].
B cells
B cells are well-known for their ability to support the antibody-mediated immune responses along with their immunoregulatory functions. Typically, B cells are characterized as CD19+ and B220/CD45R+ cells. There are various types of B cell populations, including (1) B1 cells (CD19+ and B220low/-), which can be detected in most tissues, (2) follicular B cells (FO or B2), (3) marginal zone B cells (MZ), (4) transitional B cells, and (5) regulatory B cells (Bregs); Breg-derived IL10 has a pivotal role in tolerance and differentiation of Treg cells [141,142,143].
Based on a preliminary study, the lack of nicotine-binding sites was shown on B lymphocytes [144]. Later, as the first demonstration, both α4- and α7nAChRs were identified on nicotine treated B lymphocyte-derived cell lines. The authors concluded that long-term exposure with nicotine led to the upregulation of nicotinic receptors coupled with increased cell proliferation and suppressed antibody production. The nAChRs regulated B lymphocyte activation and immune response through CD40 signaling pathway [145, 146]. Skok et al. further confirmed a higher count of B cells (lymphopoiesis) in nicotine-treated mice. Nicotine-treated B cells develop from their early precursor, pre-B cells (B220+ IgM- CD43-) [147]. They have recently reported that not only CD5+ and Foxp3+ B lymphocytes are enriched with a high amount of α7nAChR, even it is necessary for the formation, induction, and functioning of regulatory B lymphocytes [148].
Overall, nicotine influences on immune cell responses seem to be profoundly dependent on experimental design, nicotine concentration, and exposure to nicotine. As discussed below, it is plausible that environmental signals, including autoimmune and infectious disease models, also implicate in the fate of nicotine treated cells. More studies are required to assess both inflammatory and anti-inflammatory effects of nicotine on cells of the immune system.
Neutrophils
Neutrophils are the most abundant type of leukocytes in blood circulation in humans. They are known as a main part of the innate immune system and are the first cells that migrate toward inflamed or infected sites [149]. There are few studies on the effect of nicotine on neutrophils. In the study of Iho et al. the blood level of IL-8 elevated in smokers compared with non-smokers. Increased production of IL-8 was observed in nicotine-stimulated neutrophils in a time- and concentration-dependent manners. Nicotine-induced IL-8 formation is mediated via nAChR, depends on peroxynitrite generation and following NF-κB activation [150]. Also, nicotine stimulates neutrophils to release neutrophil extracellular traps (NETs) in a dose-dependent manner. nAChRs through activation of Akt and PAD4 and without activation of Nox2 produce nicotine-induced NET. These results show that nicotine can be involved in smoking-related diseases [151].
Mast cells
Mast cells play a key role in inflammation caused by allergic reactions [152]. They express receptors for IgE (FcεRI) which bind to allergens in vivo and induce the release of Th2 cytokines and cysteinyl leukotrienes (cysLTs) [152,153,154,155,156]. Activated mast cells are involved in pathologies of some pulmonary diseases such as COPD, emphysema, asthma, and COVID-19 [27, 157,158,159].
Several studies investigated the mechanism by which nicotine may affect mast cells. An in vitro study showed nicotine could suppress the delayed phase of activated mast cells via α7/α9/α10 nAChRs and could inhibit the cytosolic phospholipase A2/MAP kinase pathway [160]. Another in vitro study demonstrated cigarette smoke medium (CSM) suppressed c-kit and FcεRI expression in bone-marrow-derived mast cells (BMMCs) and inhibited the development of mast cells in a toll-like receptor 4 (TLR4)-independent manner [161]. TLR4 a protein that play the role in the maturation of DC and B cells [162, 163]. Exposure of RBL-2H3, as a mast cell model, to cigarette smoke condensate (CSC) stimulated MCPTs, especially tryptases, secretion. In line with this finding, several in vivo investigations showed there is an adverse correlation between MCPTs levels and airway function, suggesting the contribution of MCPTs in airway remodeling in smokers [164]. Increases in mucosal mast cells (MMCs) were shown in the colonic mucosa from mice with food allergy [165]. nicotine inhibited the activation of MMCs via a7 nAChRs and induced the expression of cytokines (Th1 and Th2 types), subsequently ameliorated food allergy mice [166].
Nicotine in diseases
Smoking inserts a high amount of xenobiotics, which in turn can alkylate DNA, activate specific (anti)inflammatory signals, and oxidative stress in active smokers and also second-hand smokers [167, 168]. This makes smoking a major risk factor for some diseases such as COPD, lung cancer, and diabetes. As a challenging concept, while there is a constant inflammation in smokers, there is protection against particular inflammatory diseases, like obesity and uncertain colitis [169]. Whereas ulcerative colitis is reversed after smoking and is worsened after smoking cessation, Crohn’s disease is exacerbated following smoking [170,171,172]. Consequently, smoking can be a double-sided sword. As we have mentioned earlier, the effects of cigarette smoke, although similar in some effects, can be different from pure nicotine [173,174,175,176]. The receptor of α7nAChR is one of the primary candidates to role in this process. Generally, activation of the acetylcholine-α7nAChR signaling axis by acetylcholine or nicotine inhibits NF-κB, the critical transcription factor in the activation of inflammation in different immune cells [174, 175, 177].
In the following sections, we discuss nicotine’s influence and, to some extent, smoking on various model inflammatory/autoimmune diseases affecting different organs, including respiratory, nervous, metabolic, digestive, cardiovascular, and urogenital systems. Mainly, we will review the effects of nicotine on coronaviruses (Fig. 4).

Nicotine shows a dual effect in different diseases. A graphical abstract of major pathogenesis of 13 diseases in several organs (left) and the effect of nicotine (right), in amelioration (light red) or worsening (light green), of the diseases are shown
Smoking and Post-COVID − 19 symptoms
According to a WHO report based on the monitoring of patients with COVID-19 after recovery in 2022, these people, particularly those with co-morbidities, displayed many symptoms for months after recovery [178]. Post-acute COVID-19 syndrome, also known as ‘post-COVID syndrome,‘ ‘long COVID,‘ ‘persistent long COVID,‘ or ‘post-acute COVID sequelae (PACS),‘ is a pathological disease characterized by persistent physical, medical, and cognitive sequelae after acute COVID-19 [179]. Post-acute COVID-19 syndrome, like COVID-19, can impact various systems and organs, including the respiratory, cardiovascular, neurological, gastrointestinal, and musculoskeletal systems [180]. There is significant evidence that post-acute COVID-19 syndrome can impact the whole COVID-19 patient population, and that around 20% and 10% of SARS-CoV-2 positive patients have symptoms that last at least 5 or 12 weeks, respectively [181].
The number of research on the influence of smoking on Post-COVID-19 symptoms is limited, and we will discuss the scant information that exists in this area in this section. According to the results of a 2022 study done in France, smoking can contribute to a rise in Post-Covid-19 symptoms such as cutaneous disorders, tachycardia and/or hypertension. In fact, this study discovered a link between smoking in women and the occurrence of these symptoms following recovery [178]. Another study found that the majority of COVID-19 recovered patients have varying degrees of functional impairments ranging from insignificant to severe based on Post-COVID-19 Functional Status (PCFS). Age, gender, periodic influenza vaccination, smoking, length since symptoms began, requirement for oxygen or ICU admission, and lastly the existence of concomitant comorbidities all had an impact on these limits [182]. Based on the findings of a 2022 study, smoking and vaping are not only risk factors for a more severe clinical form and slower progression of COVID-19, but they are also risk factors for the development of lengthy and persistent post-COVID-19 symptoms [183]. Finally, it is crucial to note that all of the above cases are associated to smoking and post-COVID-19 symptoms, and these cases cannot be attributed to nicotine alone; additional research in this area is required.
Nicotine in Diseases of Respiratory System
Hypersensitivity pneumonia
Hypersensitivity pneumonia (HP), also known as extrinsic allergic alveolitis, is an inflammatory lung disease associated with lymphocytes’ accumulation. Numerous predisposing factors such as genetics and environmental stimuli and lifestyle are involved in developing the disease. Chemical, bacterial, fungal, proteogenic factors, and animal antigens are among HP’s critical risk factors [184].
HP is classified into acute, subacute, and chronic categories and Th1 polarization plays a significant role in developing acute/subacute HP and stimulation of reversible granuloma formation. Afterward, this response continues to Th2 response in chronic HP and promotes inflammatory responses and fibrosis [185]. Increased CD4 + T cells and increased CD4+/CD8 + ratio along with decreased CD8 + are observed in chronic HP. Fibrotic HP is very similar to idiopathic pulmonary fibrosis (IPF) and is probably responsible for recruiting fibrocytes and the activation of fibroblasts, and therefore, the uncontrolled deposition of ECM-related proteins [186]. The CD80 and CD86 molecules are expressed on the APC cells (macrophages and dendritic cells, monocytes) and bind to CD28 and CTL4 molecules on T cells. The CD28 marker is expressed on resting T cells, and its stimulation causes the activation and proliferation of T cells. The CTLA4 marker is also expressed on T cells after stimulation [187]. In HP, changes in T cells (CD28 + and CTLA4 + T cells) and macrophages surface markers are reported. These molecules play essential roles in the accumulation and proliferation of lymphocytes and the expression of TNF-α, IL10, and INF-γ in HP patients [187]. While Alveolar macrophages naturally show a limited expression of B7 and have little capacity to act as APCs, macrophages show higher APC capacity in HP. Interestingly, viruses play a pivotal role in HP pathogenesis acting via B7 in alveolar macrophages [188]. So, inhibition of the costimulatory molecules on macrophage and T cells prevents the development of HP.
Although smokers with chronic HP develop more recurrent periods and have lower survival rates, it has been shown that HP is less common in smokers than non-smokers. The mechanism by which smokers are protected against HP is unclear [189, 190]. Animal studies indicate the anti-inflammatory effects of short-term nicotine exposure, while its long-term effects can cause inflammation and fibrosis [190]. Nicotine also suppresses monocytes and macrophages’ APC activity to display the inhaled antigen to lymphocytes, leading to reduced immune reaction toward HP [189].
Nicotine in Diseases of Nervous System
While neurodegenerative diseases appeared to be more prevalent in smokers than non-smokers, epidemiological studies during the early 1960s provide the first evidence of more smoking is used, less incidence of Parkinson’s disease is observed [191, 192]. Since then, several studies have proven the critical role of nAChRs activation in modulating neuro-immune pathways, and nicotine has been investigated as an effective therapeutic in neurodegeneration and neuroinflammatory diseases [193, 194]. Although nAChRs are widely distributed in different nervous system regions, nicotine neuroprotection in the brain and spinal cord is mainly mediated via α7nAChRs and α4β2 [195]. Below we will review the impact of nicotine in Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS), which are among the major cause of neurological impairment in middle and late life [196].
Alzheimer’s disease
Alzheimer’s disease is the most common progressive neurodegenerative disease, which accounts for 60 to 80% of the world’s 47 million dementia cases [197]. Patients typically display with neuronal loss, memory, and cognitive impairment at the disease onset. According to the degree of cognitive impairment, there are different types of AD [198]. Aggregation of misfolded proteins is a key feature of AD. In the case of amyloid b-protein (Aβ) aggregates and tau hyper-phosphorylation (p-tau) induced pathogenesis, neuronal damage occurred through proinflammatory cytokines (IL1β, IL6, and TNF-α) or increased activity of CDK5 and P38 [199]. Among environmental components, smoking is a significant risk factor for AD, and smoking-related oxidative stress influences the Aβ and tau aggregation [200]. However, nicotine extracted from tobacco may exert neuroprotective effects in AD [201].
Transdermal and intravenous nicotine treatment has been shown to improve cognitive dysfunction in AD patients [202, 203]. In an ongoing Phase II clinical trial (MIND), a nicotine skin patch is examined for memory performance in older adults (ClinicalTrials.gov identifier NCT02720445). Nicotine reduces the Aβ-induced neurotoxicity relevant to oxidative stress through regulating metal homeostasis [204]. Recently, it has been shown that even at a 10:1 ratio, nicotine does not have a significant effect on Aβ and its aggregation pathway and may affect Aβ amyloid burden by affecting Aβ production, degradation, or/and localization [205]. Furthermore, “increased expression of BAG2 shifts the effects of nicotine toward a reduction in levels of p-tau protein, possibly as a consequence of BAG2 inhibition of ERK1/2 via association with Hsp90, which is required for ERK1/2 activity, and/or via BAG2-mediated degradation of p-tau as a consequence of BAG2 phosphorylation by p38/MAPKAPK2” [206].
Parkinson’s disease
As the second most common neurodegenerative disease following AD, Parkinson’s disease (PD) affects older individuals. Parkinson’s disease has both slow and rapid progressive pattern. Tremor, cognitive deficits, bradykinesia, and anxiety are among PD’s main symptoms [207]. Unfortunately, the disease follows a fast increasing trend over the past generations [208]. It has been proposed that genetic and environmental interactions followed by the reactive oxygen species (ROS) production, mitochondrial dysfunction, and formation of Lewy bodies may result in the dopaminergic system degeneration observed in PD. The Lewy bodies mainly include misfolded proteins like alpha-synuclein (SNCA), phosphorylated tau, and Aβ protein [209]. In addition to altered T cells subpopulation (significantly more Th1 and Th17 cells), increased proinflammatory cytokines such as IFN-γ, IL2, IL6, TNF-α, MCP-1, and MIP-1α also point to the role of an autoimmune reaction in PD pathogenesis [210, 211].
Regarding PD, cumulative evidence has provided clues about nicotine’s neuroprotective role, which suggests the implication of nicotine and some of its derivatives for PD therapy. Indeed, some epidemiological findings show a decreased risk of PD in smokers [212, 213]. Previously, nicotine patches and gums have been shown beneficial effects in PD patients [214, 215]. However, the use of nicotine patches has shown conflicting results [216]. Recently, PD patients prescribed by an oral formulation of nicotine (NC001) (ClinicalTrials.gov identifier NCT00957918) have shown improvement in falls and freezing of gait (FOG) [217]. An early clinical trial (ClinicalTrials.gov identifier NCT03865121) also proposed nasal administration as the optimal nicotinic therapy in PD patients. Various mechanisms of action have been attributed to the protection of nicotine against PD, mediated through both nicotinic, mainly α7nAChRs, and non-nicotinic receptors [218, 219]. Nicotine treatment significantly suppresses proteins α-synuclein fibrillation [220]. Enhancement of fibroblast growth factor-2 (FGF-2) and brain-derived neurotrophic factor is another neuroprotective mechanism of nicotine in PD [221]. According to recent research, nicotine has been assumed to protect from PD by suppressing manganese and iron-induced oxidative stress [222]. Interestingly, by inhibiting SIRT6, nicotine downregulates the pro-apoptotic TNFα pathway and activates the pro-survival AKT signaling, which ultimately promotes neuron survival in PD [223].
Multiple sclerosis
Multiple sclerosis is the most common autoimmune-mediated demyelination and disabling disease of the CNS in young adults between 20 and 40 years. More than 2.5 million people worldwide are diagnosed with MS, and it follows an increasing pattern of prevalence [224]. MS is characterized by moderate-to-severe spasticity, paresthesia, dysesthesias, diplopia, ataxia, vertigo, and bladder disturbances [225]. Activation and differentiation of T cell subsets (IFN-γ and IL17 producing CD4 + and CD8 + T cells) and antibody production of B cells have a pivotal role in disease initiation and progression [226]. Through inducing an anti-inflammatory effect, nicotine has been explored to attenuate MS and experimental autoimmune encephalomyelitis (EAE), a mouse model for MS.
There is spars literature about the effect of nicotine on MS. Overall, the studies show an inverse correlation between snuff and the risk of MS [227, 228]. Under the EAE condition, nicotine has been shown to suppress the production of Th1 and Th17 cytokines and increase anti-inflammatory Th2 and immunosuppressive Treg cell responses in the CNS [140, 229]. Nicotine treated mice also showed delayed onset and a significant reduction in the severity of EAE coupled with less inflammation in histopathologic evaluation [230]. Moreover, nicotine suppresses M1 microglia cell differentiation and decreases TNF-α release from these cells, impairs Th1 and th17 cells differentiation which consequently resulted to the protection of neurons [231]. The ability of nicotine to lessen the infiltration of CCR2+ Ly6Chigh monocyte and neutrophil into the CNS of nicotine-treated EAE mice is mediated by both α7- and α9nAChRs. Notably, nicotine reduced the mRNA transcript levels of CCL2 and CXCL2 chemokines involved in the chemotaxis of pro-inflammatory monocytes and neutrophils, respectively, in the brain of EAE mice [232]. Recently, it has been explored that the therapeutic effect of nicotine in CNS inflammation requires the presence of B1a lymphocytes. Moreover, binding of nicotine to α7nAChR reduces the secretion of proinflammatory cytokines (TNF-α, IL-1β, IL6, and IL18) from LPS-stimulated human monocytic cell line and diminishes paralysis in EAE [233].
Nicotine in Diseases of Metabolic System
Obesity
Obesity is the extra or abnormal accumulation of fat in the body and is an essential health-threatening factor [234]. Fatness is usually calculated as body mass index (BMI; body weight in kg divided by the square of the height in meters). Accordingly, the worldwide prevalence of overweight (BMI ≥ 25 kg/m2), obese (BMI ≥ 30 kg/m2), and severely obese individuals (BMI ≥ 40 kg/m2) are increasing [234]. Notably, obesity is associated with coronary heart disease, end-stage renal diseases, hypertension, type 2 diabetes mellitus (T2DM), osteoarthritis, and fatty liver disease [235].
Nicotine plays a dual role in obesity as a low-grade inflammatory disease. Maternal nicotine consumption not only dysregulates energy metabolism, including hormonal imbalance (leptin, insulin), even it increases the risk of metabolic syndromes such as obesity and hyperleptinemia and larger adipocytes in the offspring during adulthood [236, 237]. In obese patients, nicotine’s positive effect on energy metabolism, including glucose and insulin, is associated with the suppression of inflammatory signaling pathways [238]. There are also reports indicating nicotine involvement in weight loss via induction of thermogenesis in brown adipose tissue and decreasing food intake [239]. Seemingly, nicotine suppresses weight gain and obesity independent from food uptake [240]. Smokers and COPD patients, who suffer from weight loss, are less prone to obesity, and smoking cessation is associated with weight gain in active smokers [241, 242]. Nicotine’s negative energy regulation is mediated via AMP-activated protein kinase (AMPK) [243, 244], that plays a central role in the regulation of feeding and energy metabolism. Nicotine increases energy metabolism and decreases the efficacy of energy conversion [245]. As well, nicotine downregulates the expression of CCL2 and F4/80 in adipose tissue, which diminishes the infiltration of M1 macrophages [246].
Additionally, nicotine induces insulin resistance via the MKP1-P38MAPK-cJun-IRS1 signaling axis and mTOR activation in skeletal muscle cells, which intensifies lipolysis and weight loss [247]. Nicotine can regulate appetite and food intake genes. For example, nicotine induces uncoupling protein1 (UCP1), a protein involved in thermogenesis in BAT, in white and brown adipose tissues [248, 249]. Also, nicotine seems to up-regulate the neuropeptide Y (NPY) and orexins, molecules for feeding, while down-regulate their receptors [249].
Diabetes
Diabetes affects many organs, including the heart, kidney, eye, and nerve system, and induces foot pain and ulcers. Smoking increases the risk of T2DM by about 30–40% (women > men), and after secession, weight-gain is more in women [250]. Smoking intensifies the complications of diabetes, and the combination of smoking and diabetes increases the mortality and mortality rates. Diabetic smokers also have a higher level of cholesterol level and blood pressure. Besides, diabetic smokers control their blood sugar with larger insulin doses and cause resistance to insulin. Animal studies demonstrated adiposity and metabolic changes in fetal and neonates after nicotine exposure [251]. Remarkably, smoke during pregnancy has been shown to increase the risk of T2DM in the offspring. Nicotine also intensifies the renal injury in diabetic mice [252]. Moreover, nicotine uptake increases the blood sugar level due to the alteration of energy metabolism [253]. This regulation is mediated by stimulation of nAChRs in the hippocampus and inactivation of AMPK, and activation of TCF7L26, a diabetes-associated transcription factor. TCF7L2 activates neuronal signaling to the pancreas to release glucagon and less insulin and induction of hyperglycemia (blood glucose level) [254]. Significantly, hyperglycemia inhibits nAChR activity via a negative feedback loop involved in nicotine dependence. Despite all the above, nicotine also has promising effects on diabetic wound healing and angiogenesis in genetically diabetic mice. The distribution of nAChRs in endothelial cells of the mice was stimulated with nicotine and promoted angiogenesis. This function of nicotine-nAChR-angiogenesis was similar to the basic fibroblast growth factor (bFGF) [255].
Nicotine in Diseases of Digestive System
IBD
Inflammatory bowel disease (IBD) is a group of chronic complex inflammatory conditions of the gastrointestinal tract, which is identified as dysfunction and disruption of gastrointestinal epithelial barrier function and increased permeability of the basal membrane. IBD is a life-threatening disease with diverse symptoms ranging from mild to severe, including persistent diarrhea, abdominal pain, rectal bleeding/bloody stools, fatigue, reduced appetite, and weight loss. Ulcerative colitis (UC) and Crohn’s Disease (CD) are two main examples of IBD. Ulcerative Colitis (Th2-mediated) usually occurs in the large intestine and the rectum in which the inflamed areas spread continuously from the rectum toward the colon. However, CD (Th1-mediated) can spread in any area, from mouth to the anus, as patchy inflammatory areas within the healthy tissue [256]. Smoking has different effects on UC and CD; ameliorates UC’s severity but exacerbates CD [257]. While passive smoking is associated with an increased risk of intestinal surgeries in patients with CD [258], UC patients with a smoking history show improvements in their gut function after smoking, and tobacco cessation worsens their gut activity [170, 171, 259]. Remarkably, the mucosal epithelium of UC patients expresses less α3 subunit of nAChRs than healthy controls, possibly because of higher cell renewal [260].
Furthermore, surgical resection is more common in smokers with CD, and their gut lavage fluid contains less IgG, IL1b. This is a contradictory result since it has been demonstrating that smoking worsens CD and nicotine stimulates more inflammatory responses in CD patients [261]. Then, Bergeron, V. et al. demonstrated that there is a dysfunction in mononuclear cells from smokers with CD, and they produce a lower level of chemokines and cytokines in comparison to the non-smokers with CD and smokers with UC. There are also defective anti-oxidative and anti-inflammatory responses in cells from CD patients. Therefore, there is a different antioxidant potential in cells from CD and UC against smoke [262]. A transcriptomics study from colonoscopy samples of CD patients (smokers vs. non-smokers) showed differential upregulation of RNF138, MT2A, and STEAP3 genes in smokers [263].
Besides smoking, transdermal nicotine delivery in UC patients has shown protective effects and improves the symptoms [173, 264]. Some studies in IBD diseases have proposed the immunomodulatory function of nicotine on Th1/Th2 toward Th1, which is perhaps mediated via mir-124-IL6R signaling. The amount of miR-124 expression at the time of nicotine treatment switches the balance toward Th1 or Th2. Nicotine, along with higher miR-124 expression, stimulates Th1 polarization, and lower miR124 expression stimulates Th2 polarization. MiR-124 is downregulated in Crohn’s disease, in which nicotine treatment aggravates the condition toward intensified Th2 responses, whereas miR-124 is upregulated in UC, and nicotine treatment ameliorates the severity of the disease toward Th1 response [6]. Nicotine treatment of LPS-treated PBMC from both UC and CD patients downregulated production of IL1β, IL10, TGF-β, and TNFα [265]. As well, nicotine treated non-adherent mononuclear cells of CD patients, and in vivo nicotine patch showed downregulation of IL10 (Th2 inhibitory function) but did not affect IL2 and TNFα (Th1 responses) [266]. Also, the treatment of DC separated from CD and UC with cigarette smoke extract (CSE) upregulated MHC-II and costimulatory molecules and downregulated the expression of CXCL10 and CCL3 in UC than CD. Whereas CSE in CD increased Th1 polarization, it increased Foxp3 + CD4 + T cells and decreased the Th1 subset in UC samples [267].
Non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is the accumulation of fatty acid in non-alcoholic individuals and is mostly observed in obese and extremely obese people. Diabetes, overweight, and metabolic syndrome are other risk factors for NAFLD. The disease has two forms, including fatty liver (NAFL) and steatohepatitis (NASH) [268]. Nicotine is genotoxic and can play oncogenic and carcinogenic effects in the liver. By inducing direct and indirect damages in liver cells, nicotine is recognized as a risk factor for several liver diseases such as obesity-induced hepatic steatosis and NAFLD; it can exaggerate them [269].
The adverse effects of nicotine in the hepatic system include the upregulation of inflammatory cytokines, the development of secondary polycythemia, and blockage of proliferation and activation of apoptosis in lymphocytes. An animal study by Willis et al. showed a significant reduction in liver weight and enlargement of hepatocyte diameter and central hepatic vein due to nicotine consumption [270]. Biochemically, rats exposed to nicotine (2.5 mL/kg) showed a significant increase of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) in serum due to hepatocyte cell death/damage. Histologically, nicotine increased the inflammation and lymphocyte infiltration in the liver [66]. If combined with a high-fat diet (HFD), nicotine triggers and worsens hepatic steatosis. Besides, hepatic steatosis is associated with higher hepatocellular apoptosis, inactivation of AMPA, activation of acetyl-coenzyme A-carboxylase (ACC), and increases oxidative stress [271]. In HFD, nicotine elevates lipogenesis, lipid mobilization, and distribution and diminishes HFD-induced adiposity, contributing to hepatic steatosis [247, 271]. However, nicotine alone does not trigger hepatic steatosis [271]. Besides toxic effects, nicotine has immunomodulatory effects in the liver [272]. It downregulates and decreases the activity of CYP2A6 in the liver cells of monkeys [273, 274]. Additionally, CYP2A and CYP3A5 are downregulated in the airway of smokers [272, 275].
Nicotine in Diseases of Cardiovascular System
Cardiovascular diseases (CVDs) are the first cause of death worldwide. As a coronary artery disease, atherosclerosis is a major cause of heart attack, stroke, and peripheral arteries. Generally, chronic nicotine exposure enhances atherosclerosis in animal and human studies. After nicotine administration, impaired and diminished clearance of LDL and accelerated lipid transfer from HDL increases their deposition in the arterial wall, which result in the atherogenic lipid profile (increased LDL, decreased HDL/total cholesterol ratio) [276, 277]. Nicotine also raises heart rate and blood pressure, and platelet aggregation. The latter contributes to plaque growth and thrombosis [278]. Also, nicotine significantly induces vascular smooth muscle cells’ proliferation via the upregulation of bFGF mitogen and several MMPs, which leads to an augmentation of lesions and development of intimal hyperplasia, atherosclerosis, and aneurysm. Chronic nicotine consumption profoundly activates macrophages via nAChRs to express pro-inflammatory cytokines, aggravating atherosclerosis and aortic lesions [279]. Distribution of α7nAChRs on endothelial cells pointedly indicates nicotine-induced angiogenesis in the endothelium [278]. Nicotine induces proliferation, migration, and tube formation in endothelial cells. There is a close association between neovascularization and atherosclerotic plaque progression, which directly increased by nicotine [280]. However, chronic exposure of endothelial cells to nicotine impairs angiogenesis and decreased cell migration and tabular structure formation [281, 282]. In addition, nicotine stimulates the migration of vascular smooth muscle cell and structural changes in vascular endothelium via p38 and p44/42 signaling axis [283]. Treatment with nicotine also activates NF-κB signaling and increases proliferation of endothelial and smooth muscle cells [284].
Additionally, nicotine exposed mice show upregulated expression of VCAM-1, COX-2, platelet-derived growth factor β (PDGFβ), and molecules involved in downstream of NF-κB signaling pathway in aorta lesions [279]. The upregulation of PDGF promotes a phenotypic switching from vascular smooth muscle cells into myofibroblasts and osteoblast-like cells that secrete and modify the endothelium and cardia ECM by collagen and osteopontin [285]. Inhibition of α1nAChR reduced myofibroblasts in the aortic wall by 80% resulting in attenuated calcification and reduced immune cells in the lesions [286]. Treatment of endothelial cells by nicotine induces calcium internalization and accumulation via α7nAChR activation and induction of angiogenesis [287]. Furthermore, via the upregulation of VEGF and FGF, nicotine activates other angiogenic signaling pathways in endothelial cells [288,289,290]. Therefore, nicotine induces the proliferation of smooth muscle cells, endothelial cells, and accumulation of macrophages in plaques and lesions, promoting atherosclerosis and vessel hardening via calcification.
Nicotine in Diseases of Urogenital System
Kidneys are essential organ for filtering waste and toxic materials out of the blood. Diseases related to impaired kidney function is increasing with global prevalence of 9·1% (2019) [291]. Nicotine mainly affects kidneys via α7-nAChR signaling and worsens renal injury in chronic kidney disease (CKD), acute nephritis, and subtotal nephrectomy [292]. Different subtypes of nAChRs are distributed in renal cells, especially mesangial cells. Nicotine stimulates differentiation and specialization of cells in kidneys, the proliferation of mesangial cells, and fibronectin production, mediated through PKC activation, ERK1/2 phosphorylation, and NADPH oxidase and ROS generation [252, 292,293,294]. Also, COX-2 and prostaglandin generation are involved in nicotine-mediated renal injury [295]. Besides, nicotine mediates inflammation and fibrosis by TGF-β production (by activation of STAT3) and upregulation of vimentin, α-SMA, and fibronectin [296].
Several studies have also reported anti-inflammatory responses and attenuation of proteinuric renal injury due to nicotine treatment [297, 298]. Nicotine has renoprotective effects in kidney diseases. Oral nicotine delivery in rats with proteinuria-induced renal inflammation showed dose-dependent reduces proteinuria, glomerular desmin deposition, decreased glomerular podocin, decreased focal glomerulosclerosis (FGS) score, and reduced infiltration of macrophages and myofibroblasts. Renal inflammatory cytokines were also downregulated in monocytes of nicotine treated rats [299]. Furthermore, long-term nicotine delivery has been shown to slow-down proteinuria by reducing glomerulosclerosis and interstitial fibrosis [297, 300,301,302].
Impact of nicotine on Coronaviruses
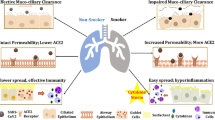
SARS, MERS, and CODIV-19 show similar aggressive pathology. They extensively affect multiple tissues and organs accompanied by uncontrolled cytokine storm and delayed interferon-gamma response, multi-organ fibrosis, impaired tissue remodeling, and organ failure [303, 304]. As mentioned in the former section, nAChRs are expressed in different lung cells, including epithelial cells, fibroblast cells, and RAS components [16]. The effect of nicotine in the renin-angiotensin system is also well studied. Overall, tobacco smoke and nicotine play complicated dynamic functions against coronavirus infections, which are mediated through different mechanisms such as overexpression of ACE2 and inhibition/dysregulation of inflammatory processes [305, 306].
Limited data are indicating the involvement of smoking at an increased risk of MERS infection. Infected smokers during the MERS-CoV epidemic of 2003 showed more mortality rate than infected non-smokers [307, 308]. Alraddadi et al. investigated the smoking history in a population of MERS infected cases and asserted that active smokers are more likely among patients than controls [309]. After this report, Seys et al. reported the upregulation (mRNA and protein level) of dipeptidyl peptidase-4 (DPP4/CD26), the receptor involved in MERS infection lung tissue of smokers and COPD patients. DPP4 is mostly expressed in various alveolar cells, including epithelial, endothelial, macrophages, and other immune cells in the submucosal region of airway epithelium and lymphoid aggregates, but not in bronchial epithelial cells [310]. Also, binding of the spike protein of SARS–CoV-2 to DPP4 is a critical factor for infection [311]. Interestingly, a positive correlation between ACE2 and DPP4 is reported recently in several normal tissues [312]. DPP4 is a natural surface marker for T cells and is involved in the immune regulation of several biological processes. DPP4, the same as ACE2, is much expressed in lower airways, lung parenchyma, interstitium, and pleural mesothelia, which are the most infected areas in infected patients [313,314,315]. Like DPP4, the upregulated expression of ACE2 has been shown in smokers and COPD patients [316,317,318]. However, it is downregulated after infection, but ACE remains unchanged, which results in severe acute respiratory failure [319,320,321]. Imai et al. demonstrated that loss of ACE2 expression in ace2 knockout mice causes very severe ARDS complications, including increased immune cell infiltration, lung edema, and vascular permeability [319]. Therefore, SARS infection downregulates the expression of ACE2 as a post-infection regulatory mechanism and ARDS [321]. Therefore, the upregulation of ACE2 via nicotine may play a protective role after SARS infections. We could not find any research regarding the direct effect of nicotine in MERS and SARS infections.
Nicotine has a dual effect on patients with COVID-19. On the one hand, nicotine has anti-inflammatory properties, and on the other hand, it would allow more viral entry. Since cytokine storm is the hallmark of SARS-CoV-2 infections, nicotine could diminish it through alpha7-contained nAChRs [322]. Furthermore, agonists of alpha7-nAChRs, nicotine or GTS-21, reduce cytokine storm mediators such as high mobility group box-1 (HMGB1) [323]. HMGB1 is a nuclear protein that is released into the extracellular spaces, including the airways and the blood circulation to promote inflammation [323]. High levels of HMGB1 have been shown in acute lung injury caused by infectious and severe COVID-19 patients, therefore, it could be a therapeutic target for patients with severe COVID-19 [324]. There are controversial reports regarding viral entry by smoking and nicotine use. Some studies showed that ACE2 increased by using nicotine and/or smoking inhabits [325,326,327,328]. While Caruso et al. demonstrated that the protein expression of ACE2 diminished in bronchial epithelial cells after smoking exposure [329]. another study reported ACE2 mRNA levels in bronchial epithelial cells from current smokers were similar to never smokers [330]. As mentioned above, in addition to receptors, proteases are needed for virus entrance into host cells. The effect of nicotine/smoking on these proteases is different. The cellular furin levels were reduced by smoking [331], whilst TMPRSS4 and TMPRSS2 increased and unchanged in bronchial epithelial cells from current smokers in comparison with never smokers [330].
Additionally, data mining and in silico studies for evaluating the effects of smoking/nicotine on COVID-19 patients were performed. For instance, to determine the relationship between vaping/smoking and the expression of inflammasomes and inflammatory cytokines, Lee et al. used transcriptome datasets (GSE138326 and GSE112073). They found upregulated pro-inflammatory markers and inflammasome genes in smoking and nicotine use as well as e-cigarettes containing flavor. The upregulation of CCL5 and CCR1 in cigarette smoking and CCL5 and CCR1 in vaping e-cigarettes containing flavor/nicotine have been reported. Also, the upregulation inflammasome-related genes (CXCL1, CXCL2, NOD2, and ASC) in smoking and vaping have been observed, revealing the negative effect of vaping/smoking on the inflammation and susceptibility to SARS-CoV-2 infection [328]. Furthermore, in silico studies showed interaction of α7 nAChRs with RBDs of SARS-CoV-2 spike glycoproteins, indicating dysregulation of the nicotinic cholinergic system and leading to cytokine storm in COVID-19 patients [332, 333]. Mohammadi et al. demonstrated the interaction between ACE2 human receptor and nicotine by molecular dynamic simulations. They also revealed the combination of favipiravir (antiviral therapeutic for SARS-CoV-2) with nicotine could successfully block 6LZG, a main active site of ACE2-S protein, suggesting the potential influence of this combination for blocking ACE2 versus SARS-CoV-2 [334].
Several systematic reviews and meta-analyses evaluated the association between smoking and COVID-19 hospitalization. Some of these studies showed low smoking prevalence among hospitalized patients with COVID-19, while others demonstrated the inverse trend [335,336,337,338,339,340,341,342,343,344,345,346]. The latest meta-analysis that included 109 studies with 517,020 patients showed that smoking was related to COVID-19 severity. Also smoking elevated the risk of ICU admission and death in patients with COVID-19, but was not relevant to mechanical ventilation. This study reported former smokers had a risk of progressing COVID-19 severity compared with current smokers. Current smokers were significantly associated with the severity of COVID-19 compared with non-smokers [344]. According to this meta-analysis that has been shown a positive correlation between smoking and COVID-19 progression, it is not clear whether nicotine would prevent negative outcomes among hospitalized patients with COVID-19.
Unfortunately, we were unable to find a publication that looked into the influence of nicotine dosage, age, and COVID disease. However, the findings of other research demonstrate that increasing age is a risk factor for mortality caused by COVID. COPD patients, smokers, quitters, and persons, who have never smoked, on the other hand, have various reactions to this virus [347].
To the best of our knowledge, there is no clinical trial to demonstrate the effects of smoking/nicotine on outcomes in COVID-19 patients. To clarify the effect of nicotine well-designed clinical trials are required for the assessment of its therapeutic agents.
Conclusion
SARS-CoV-2 infects and affects several body organs, including respiratory, digestive, nervous, cardiovascular, and urogenital systems. The early epidemiological reports indicated the lower incidence of smokers among SARS-CoV-2 infected and hospitalized patients. Then, several letters were published criticizing the higher risk of smoking for the infection. Currently, there is a discrepancy between the published multi-national epidemiological data and current basic data (higher expression of ACE2 in smokers and COPD patients). Of note, the latest meta-analysis that included 109 studies reported that smoking was associated to COVID-19 severity.
As shown in hyper-pneumonia sensitivity, generally similar to SARS-CoV-2 infection, the APC capacity and functioning are reduced in the airways, which attenuates the marshal of adaptive immunity and stimulation of uncontrolled cytokine storm and fibrotic responses. Besides, reports on SARS infections indicate the after-infection downregulation of ACE2 is followed by more severe pathogenesis that produces pro-inflammatory and pro-thrombotic cytokines.
In a brief conclusion, nicotine shows dual effects based on dose-dependent properties. Moreover, in some diseases, although nicotine has a worsening effect (e.g., diabetes) in the pathogenesis of the disease, but has a healing role in the treatment of some other diseases (e.g., multiple sclerosis). Besides, nicotine revealed a dual effect on the severity of COVID-19. Several publications are indicating the overexpression of ACE2 in smokers that cause further viral entry. We conclude that the overexpression of ACE2 in smokers and COPD patients is not enough to claim a higher risk for SARS-CoV-2 infections. The virus’s binding and entrance is the first step of the complex systemic immunological responses in SARS-CoV-2 infections and the activation of the innate immunity through macrophages and DCs, and consequently, T and B cells are also required. The useful effect of nicotine is to reduce cytokine storm mediators via alpha7-nAChRs. nAChrs are involved in many diseases including diseases (diabetes, obesity, cardiovascular, gastrointestinal etc.) which each show similarities with COVID-19 pathogenesis.
Therefore, clever studies are needed to check different doses of nicotine and monitor the background immunological characteristics of COVID-19 patients. Furthermore, all patients should be checked for predisposing factors and possible comorbidities, including cardiovascular diseases, IBD, diabetes, and NAFLD. Therefore, it seems that host factors, the dose of nicotine, and comorbidities determine the harmful or the protective effect of nicotine.
As a fundamental approach, we recommend assessing different doses of nicotine on host factors (including inflammatory pathways) to determine what dose, for who and when can be prescribed without any serious harmful effects. The status of macrophages, dendritic, and T and B cells should be also investigated in clinical studies of nicotine’s effect, especially in saliva and sputum samples in COVID-19 patients. The immunopathogenic similarity of COVID-19 and other inflammatory diseases and the effect of nicotine seems to be promising. To prevent the production of erroneous results and its generalization, the effect of smoking should be examined separately from the effect of nicotine. In other words, the results of these two types of studies cannot be easily generalized. Finally, we recommend a simultaneous check-up of RAS and cholinergic systems in nicotine related studies in COVID-19 patients (Fig. 5).

A puzzle diagram indicating the major questions and challenges to conclude the efficacy of nicotine in COVID-19.
Data Availability
All data and materials are available thought the corresponding authors upon request.
References
Centers for Disease Control and Prevention: SARS Basics Fact Sheet. 2020; Available from: https://www.cdc.gov/sars/about/fs-sars.html.
World Health Organization. : Epidemic and pandemic-prone diseases: MERS situation update, January 2020. 2020.
https://covid19.who.int/.
U.S. Department of Health and Human Services. The Health Consequences of Smoking-50 years of progress: a report of the Surgeon General. Atlanta (GA); 2014.
WHO global report on trends in prevalence of tobacco use 2000–2025 (Licence: CC BY-NC-SA 3.0 IGO). Third edition ed. 2019, Geneva: World Health Organization.
Qin Z, et al. MicroRNA124-IL6R mediates the Effect of Nicotine in Inflammatory Bowel Disease by shifting Th1/Th2 balance toward Th1. Front Immunol. 2020;11:235.
Guan WJ, et al. Clinical characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382(18):1708–20.
Vardavas CI, Nikitara K. COVID-19 and smoking: a systematic review of the evidence. Tob Induc Dis. 2020;18:20.
Cattaruzza MS, et al. Tobacco smoking and COVID-19 pandemic: old and new issues. A summary of the evidence from the scientific literature. Acta Biomed. 2020;91(2):106–12.
Yu T, et al. Association between Clinical Manifestations and Prognosis in patients with COVID-19. Clin Ther. 2020;42(6):964–72.
Ojo AS et al. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies Pulm Med, 2020. 2020: p. 6175964.
Nam HS, et al. High fatality rates and associated factors in two hospital outbreaks of MERS in Daejeon, the Republic of Korea. Int J Infect Dis. 2017;58:37–42.
Cai G. Bulk and single-cell transcriptomics identify tobacco-use disparity in lung gene expression of ACE2, the receptor of 2019-nCov. MedRxiv, 2020.
Smith JC, Sheltzer JM. Cigarette smoke triggers the expansion of a subpopulation of respiratory epithelial cells that express the SARS-CoV-2 receptor ACE2. bioRxiv, 2020.
Wang J, et al. Susceptibility analysis of COVID-19 in smokers based on ACE2. Preprints.org; 2020.
Oakes JM, et al. Nicotine and the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2018;315(5):R895–R906.
Propper RE. Does cigarette smoking protect against SARS-CoV-2 infection? Nicotine Tob Res; 2020.
Huang C, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Colombi D, et al. Well-aerated lung on admitting chest CT to predict adverse outcome in COVID-19 Pneumonia. Radiology. 2020;296(2):E86–E96.
Gonzalez-Rubio J, et al. Cytokine release syndrome (CRS) and nicotine in COVID-19 patients: trying to calm the storm. Front Immunol. 2020;11:1359.
Tajlil A, et al. Nicotine and smoking in the COVID-19 era. J Cardiovasc Thorac Res. 2020;12(2):136–9.
Williamson EJ, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584(7821):430–6.
Miyara M, Tubach F, Pourcher V. Low rate of daily active tobacco smoking in patients with symptomatic COVID-19. Qeios. 2020.
Rentsch CT, et al. Covid-19 Testing, Hospital Admission, and Intensive Care among 2,026,227 United States Veterans aged 54–75 years. medRxiv; 2020.
Almazeedi S, et al. Characteristics, risk factors and outcomes among the first consecutive 1096 patients diagnosed with COVID-19 in Kuwait. EClinicalMedicine. 2020;24:100448.
Jackson SE, et al. Association of the COVID-19 lockdown with smoking, drinking and attempts to quit in England: an analysis of 2019–20 data. Addiction. 2021;116(5):1233–44.
Theoharides TC. Potential association of mast cells with coronavirus disease 2019 Annals of Allergy, Asthma Immunology, 2021. 126(3): p. 217.
Polverino F, Kheradmand FJFiM et al. COVID-19, COPD, and AECOPD: immunologicpidemiological, and clinical aspects. 2021: p. 1121.
Zu ZY, et al. Coronavirus Disease 2019 (COVID-19): a perspective from China. Radiology. 2020;296(2):E15–e25.
Renu K, Prasanna PL, Valsala A, Gopalakrishnan. Coronaviruses pathogenesis, comorbidities and multi-organ damage - A review. Life Sci. 2020;255:117839.
Atri D, et al. COVID-19 for the cardiologist: a current review of the Virology, Clinical Epidemiology, Cardiac and other clinical manifestations and potential therapeutic strategies. JACC Basic Transl Sci; 2020.
Wang T, et al. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet. 2020;395(10228):e52.
Wu K, et al. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc Natl Acad Sci U S A. 2009;106(47):19970–4.
Olds JL, Kabbani N. Is nicotine exposure linked to cardiopulmonary vulnerability to COVID-19 in the general population? FEBS J, 2020.
Cai G. Tobacco-Use Disparity in Gene Expression of ACE2, the Receptor of 2019-nCov. Preprints, 2020.
Guzzi PH, et al. Master regulator analysis of the SARS-CoV-2/human interactome. J Clin Med. 2020;9(4):982.
Hoffmann M et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 2020.
Canrong W et al. Furin, a potential therapeutic target for COVID-19. ChinArxiv [Internet], 2020.
Feliciangeli SF, et al. Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J Biol Chem. 2006;281(23):16108–16.
Kuba K, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11(8):875–9.
Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93(5):543–8.
Bosnyak S, et al. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci. 2011;121(7):297–303.
Imai Y, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–6.
De Gasparo M et al. International union of pharmacology. XXIII. The angiotensin II receptors 2000. 52(3): p. 415–472.
Simmons G, et al. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 2013;100(3):605–14.
Palau V, Riera M, Soler MJ. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrology Dialysis Transplantation; 2020.
Althaus M, Clauss WG, Fronius M. Amiloride-sensitive sodium channels and pulmonary edema Pulmonary medicine, 2011. 2011.
Ji H-L, et al. SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am J Physiology-Lung Cell Mol Physiol. 2009;296(3):L372–83.
Anand P et al. SARS-CoV-2 selectively mimics a cleavable peptide of human ENaC in a strategic hijack of host proteolytic machinery. bioRxiv, 2020.
Kijima K, et al. Regulation of angiotensin II type 2 receptor gene by the protein kinase C–calcium pathway. Hypertension. 1996;27(3):529–34.
Bermejo-Martin JF, et al. Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J Infect. 2020;80(5):e23–4.
Chen G, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–9.
Liao M, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26(6):842–4.
Zhang X, et al. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-kappaB. Virology. 2007;365(2):324–35.
Chen IY, et al. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50.
Fan BE, et al. Hematologic parameters in patients with COVID-19 infection. Am J Hematol. 2020;95(6):E131–4.
Du F, Liu B, Zhang S. COVID-19: the role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J Thromb thrombolysis. 2021;51(2):313–29.
Fraga-Silva RA, et al. ACE2 activation promotes antithrombotic activity. Mol Med. 2010;16(5):210–5.
Gallagher PE, Ferrario CM, Tallant EA. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiology-Cell Physiol. 2008;295(5):C1169–74.
Talhout R, et al. Hazardous compounds in tobacco smoke. Int J Environ Res Public Health. 2011;8(2):613–28.
Bhalla DK, et al. Cigarette smoke, inflammation, and lung injury: a mechanistic perspective. J Toxicol Environ Health Part B. 2009;12(1):45–64.
Glantz SA, Parmley WW. Passive smoking and heart disease: mechanisms and risk. JAMA. 1995;273(13):1047–53.
Benowitz NL, Hukkanen J, Jacob P. Nicotine chemistry, metabolism, kinetics and biomarkers, in Nicotine psychopharmacology. Springer; 2009. pp. 29–60.
Zevin S, Gourlay SG, Benowitz NL. Clinical pharmacology of nicotine. Clin Dermatol. 1998;16(5):557–64.
Jensen K, et al. General mechanisms of nicotine-induced fibrogenesis. FASEB J. 2012;26(12):4778–87.
Benowitz NL, Hukkanen J, Jacob P 3. rd, Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol, 2009(192): p. 29–60.
Benowitz NL, et al. Nicotine metabolic profile in man: comparison of cigarette smoking and transdermal nicotine. J Pharmacol Exp Ther. 1994;268(1):296–303.
Costello MR, et al. Comparison of the reinforcing properties of nicotine and cigarette smoke extract in rats. 2014;39(8):1843–51.
Ji M et al. Nicotine component of cigarette smoke extract (CSE) decreases the cytotoxicity of CSE in BEAS-2B cells stably expressing human cytochrome P450 2A13 2017. 14(10): p. 1221.
Pomerleau OF. Nicotine and the central nervous system: biobehavioral effects of cigarette smoking. Am J Med. 1992;93(1A):2S–7S.
Carlisle DL, et al. Nicotine signals through muscle-type and neuronal nicotinic acetylcholine receptors in both human bronchial epithelial cells and airway fibroblasts. Respir Res. 2004;5(1):27.
Collins AC, Romm E, Wehner JM. Dissociation of the apparent relationship between nicotine tolerance and up-regulation of nicotinic receptors. Brain Res Bull. 1990;25(3):373–9.
Benowitz NL. Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Annu Rev Pharmacol Toxicol. 2009;49:57–71.
Conti-Tronconi BM, et al. The nicotinic acetylcholine receptor: structure and autoimmune pathology. Crit Rev Biochem Mol Biol. 1994;29(2):69–123.
Lewis AS, van Schalkwyk GI, Bloch MH. Alpha-7 nicotinic agonists for cognitive deficits in neuropsychiatric disorders: a translational meta-analysis of rodent and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2017;75:45–53.
Wang Y, et al. Human bronchial epithelial and endothelial cells express alpha7 nicotinic acetylcholine receptors. Mol Pharmacol. 2001;60(6):1201–9.
Maus AD, et al. Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol Pharmacol. 1998;54(5):779–88.
C SK, Kumar SA, Wei H. Comparative docking studies to understand the binding affinity of nicotine with soluble ACE2 (sACE2)-SARS-CoV-2 complex over sACE2. Toxicol Rep. 2020;7:1366–72.
Cuevas-Olguin R, et al. Nicotine smoking concentrations modulate GABAergic synaptic transmission in murine medial prefrontal cortex by activation of alpha7* and beta2* nicotinic receptors. Eur J Neurosci. 2020;51(3):781–92.
West KA, et al. Rapid akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111(1):81–90.
Klapproth H, Racke K, Wessler I. Acetylcholine and nicotine stimulate the release of granulocyte-macrophage colony stimulating factor from cultured human bronchial epithelial cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357(4):472–5.
Zia S, et al. Nicotine enhances expression of the alpha 3, alpha 4, alpha 5, and alpha 7 nicotinic receptors modulating calcium metabolism and regulating adhesion and motility of respiratory epithelial cells. Res Commun Mol Pathol Pharmacol. 1997;97(3):243–62.
Carlisle DL, et al. Nicotine signals through muscle-type and neuronal nicotinic acetylcholine receptors in both human bronchial epithelial cells and airway fibroblasts. Respir Res. 2004;5:27.
Merecz-Sadowska A et al. A Summary of In Vitro and In Vivo Studies Evaluating the Impact of E-Cigarette Exposure on Living Organisms and the Environment International Journal of Molecular Sciences, 2020. 21(2): p. 652.
Qiu F, et al. Impacts of cigarette smoking on immune responsiveness: up and down or upside down? Oncotarget. 2017;8(1):268.
Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. Elsevier Health Sciences; 2017.
Zia S, et al. Nicotine enhances expression of the alpha 3, alpha 4, alpha 5, and alpha 7 nicotinic receptors modulating calcium metabolism and regulating adhesion and motility of respiratory epithelial cells. Res Commun Mol Pathol Pharmacol. 1997;97(3):243–62.
Caldeira EJ, et al. Morphological alterations in the epithelium of the oral mucosa of rats (Rattus norvegicus) submitted to long-term systemic nicotine treatment. Arch Oral Biol. 2007;52(1):83–9.
Agius AM, et al. Smoking and middle ear ciliary beat frequency in otitis media with effusion. Acta Otolaryngol. 1995;115(1):44–9.
Arany I, et al. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiology-Renal Physiol. 2012;302(6):F722–9.
Arany I, et al. Chronic nicotine exposure exacerbates acute renal ischemic injury. Am J Physiology-Renal Physiol. 2011;301(1):F125–33.
Roomans G, et al. Effects of nicotine on intestinal and respiratory epithelium. J Submicrosc Cytol Pathol. 2002;34(4):381–8.
Klapproth H, Racké K, Wessler I. Acetylcholine and nicotine stimulate the release of granulocyte-macrophage colony stimulating factor from cultured human bronchial epithelial cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357(4):472–5.
West KA, et al. Rapid akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Investig. 2003;111(1):81–90.
Li Q, et al. Nicotine suppresses inflammatory factors in HBE16 airway epithelial cells after exposure to cigarette smoke extract and lipopolysaccharide. Translational Res. 2010;156(6):326–34.
Bodas M, et al. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic Biol Med. 2016;97:441–53.
Valdez-Miramontes C, et al. Nicotine modulates molecules of the innate immune response in epithelial cells and macrophages during infection with M. tuberculosis. Clin Experimental Immunol. 2020;199(2):230–43.
Alcorn JF. IL-22 plays a critical role in maintaining Epithelial Integrity during Pulmonary infection. Front Immunol. 2020;11:1160.
Nguyen HM-H et al. Nicotine Impairs the Response of Lung Epithelial Cells to IL-22 Mediators of Inflammation, 2020. 2020.
Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care, 2013. 22(8).
Crotty Alexander LE, et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am J Physiology-Regulatory Integr Comp Physiol. 2018;314(6):R834–47.
Aschner Y, Downey GP. Transforming growth factor-β: Master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol. 2016;54(5):647–55.
Rezonzew G, et al. Nicotine exposure and the progression of chronic kidney disease: role of the α7-nicotinic acetylcholine receptor. Am J Physiology-Renal Physiol. 2012;303(2):F304–12.
Soeda J, et al. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun. 2012;417(1):17–22.
Ramalingam A, et al. Angiotensin II type I receptor antagonism attenuates Nicotine-Induced Cardiac Remodeling, Dysfunction, and aggravation of myocardial ischemia-reperfusion Injury in rats. Front Pharmacol. 2019;10:1493.
Jensen K, et al. General mechanisms of nicotine-induced fibrogenesis. FASEB J. 2012;26(12):4778–87.
Wongtrakool C, et al. Nicotine stimulates nerve growth factor in lung fibroblasts through an NFκB-dependent mechanism. PLoS ONE. 2014;9(10):e109602.
Vicary GW, et al. Nicotine stimulates collagen type I expression in lung via α7 nicotinic acetylcholine receptors. Respir Res. 2017;18(1):1–12.
Ebrahimpour A, et al. Nicotine modulates growth factors and microRNA to promote inflammatory and fibrotic processes. J Pharmacol Exp Ther. 2019;368(2):169–78.
Félétou M. The endothelium, Part I: Multiple functions of the endothelial cells–focus on endothelium-derived vasoactive mediators. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. 2011.Morgan & Claypool Life Sciences.
Mercado C, Jaimes EA. Cigarette smoking as a risk factor for atherosclerosis and renal disease: novel pathogenic insights. Curr Hypertens Rep. 2007;9(1):66–72.
Heeschen C, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7(7):833–9.
Zhang S, Day I, Ye S. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis. 2001;154(2):277–83.
Farsalinos K et al. Nicotinic Cholinergic System and COVID-19: In Silico Identification of an Interaction between SARS-CoV-2 and Nicotinic Receptors with Potential Therapeutic Targeting Implications. Int J Mol Sci, 2020. 21(16).
Wang Z et al. Innate Immune Cells: A Potential and Promising Cell Population for Treating Osteosarcoma. Front Immunol, 2019. 10(1114).
Hossain MK, Wall KA. Use of dendritic cell receptors as targets for enhancing anti-cancer immune responses. Cancers. 2019;11(3):418.
Patente TA et al. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front Immunol, 2019. 9(3176).
Aicher A, et al. Nicotine strongly activates dendritic cell–mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003;107(4):604–11.
Guinet E, Yoshida K, Nouri-Shirazi M. Nicotinic environment affects the differentiation and functional maturation of monocytes derived dendritic cells (DCs). Immunol Lett. 2004;95(1):45–55.
Nouri-Shirazi M, Guinet E. Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology. 2003;109(3):365–73.
Nouri-Shirazi M, Tinajero R, Guinet E. Nicotine alters the biological activities of developing mouse bone marrow-derived dendritic cells (DCs). Immunol Lett. 2007;109(2):155–64.
Hu SX, et al. Lipopolysaccharide and dose of nicotine determine the effects of nicotine on murine bone marrow-derived dendritic cells. Mol Med Rep. 2012;5(4):1005–10.
Tao X, et al. Nicotine protects dendritic cells from apoptosis and support DCs-dependent CD4 + T-cell priming in vitro. Indian J Pharm Sci. 2019;81(6):1000–10.
Shapouri-Moghaddam A, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–40.
Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37.
Ko HK, et al. Regulation of cigarette smoke induction of IL-8 in macrophages by AMP‐activated protein kinase signaling. J Cell Physiol. 2015;230(8):1781–93.
Ween MP, et al. Phagocytosis and inflammation: exploring the effects of the components of E-cigarette vapor on macrophages. Physiological Rep. 2017;5(16):e13370.
Zhou M-S, et al. Nicotine potentiates proatherogenic effects of oxLDL by stimulating and upregulating macrophage CD36 signaling. Am J Physiol Heart Circ Physiol. 2013;305(4):H563–74.
Glynos C, et al. Comparison of the effects of e-cigarette vapor with cigarette smoke on lung function and inflammation in mice. Am J Physiology-Lung Cell Mol Physiol. 2018;315(5):L662–72.
Yanagita M, Kobayashi R, Murakami S. Nicotine can skew the characterization of the macrophage type-1 (MΦ1) phenotype differentiated with granulocyte-macrophage colony-stimulating factor to the MΦ2 phenotype. Biochem Biophys Res Commun. 2009;388(1):91–5.
Varone F, et al. Fibrotic hypersensitivity pneumonitis: diagnosis and management. Lung. 2020;198:429–40.
Yoshikawa H, et al. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-κB phosphorylation and nuclear factor‐κB transcriptional activity through nicotinic acetylcholine receptor α7. Clin Experimental Immunol. 2006;146(1):116–23.
de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–51.
Maldifassi MC, et al. A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PLoS ONE. 2014;9(9):e108397.
AlQasrawi D, Abdelli LS, Naser SA. Mystery solved: why smoke extract worsens disease in smokers with Crohn’s Disease and not ulcerative colitis? Gut MAP! Microorganisms. 2020;8(5):666.
Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers. 2016;8(3):36.
Vardavas CI, et al. Passive smoking alters circulating naïve/memory lymphocyte T-cell subpopulations in children. Pediatr Allergy Immunol. 2010;21(8):1171–8.
Nordman JC, et al. The α4 nicotinic receptor promotes CD4 + T-cell proliferation and a helper T-cell immune response. Mol Pharmacol. 2014;85(1):50–61.
Wasén C, et al. Smoking activates cytotoxic CD8 + T cells and causes survivin release in rheumatoid arthritis. J Autoimmun. 2017;78:101–10.
Nizri E, et al. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol. 2009;183(10):6681–8.
Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–41.
Tung JW, et al. Identification of B-cell subsets, in B Cell Protocols. Springer; 2004. pp. 37–58.
Cyster JG, Allen CD. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–40.
Maslinski W, Laskowska-Bozek H, Ryzewski J. Nicotinic receptors of rat lymphocytes during adjuvant polyarthritis. J Neurosci Res. 1992;31(2):336–40.
Skok MV, et al. Functional nicotinic acetylcholine receptors are expressed in B lymphocyte-derived cell lines. Mol Pharmacol. 2003;64(4):885–9.
Skok M, Grailhe R, Changeux JP. Nicotinic receptors regulate B lymphocyte activation and immune response. Eur J Pharmacol. 2005;517(3):246–51.
Skok M, et al. The role of nicotinic acetylcholine receptors in lymphocyte development. J Neuroimmunol. 2006;171(1–2):86–98.
Koval L, et al. α7 nicotinic acetylcholine receptors are involved in suppression of the antibody immune response. J Neuroimmunol. 2018;318:8–14.
Petri B, Sanz M-J. Neutrophil chemotaxis. Cell tissue research. 2018;371(3):425–36.
Iho S, et al. Nicotine induces human neutrophils to produce IL-8 through the generation of peroxynitrite and subsequent activation of NF‐κB. J Leukoc Biol. 2003;74(5):942–51.
Hosseinzadeh A, et al. Nicotine induces neutrophil extracellular traps. J Leukoc Biol. 2016;100(5):1105–12.
Schwartz LB. Mast cells: function and contents. Curr Opin Immunol. 1994;6(1):91–7.
Parameswaran K, et al. Cysteinyl leukotrienes promote human airway smooth muscle migration. Am J respiratory Crit care Med. 2002;166(5):738–42.
Henderson WR. The role of leukotrienes in inflammation Annals of internal medicine 1994. 121(9): p. 684–697.
Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunol Rev. 2000;173(1):131–40.
Turner H, Kinet J-P. Signalling through the high-affinity IgE receptor FcεRI. Nature. 1999;402(6760):24–30.
Kulka M, Befus AD. The dynamic and complex role of mast cells in allergic disease. Arch Immunologiae et Ther Exp. 2003;51(2):111–20.
Marone G, Lichtenstein LM, Galli FJ. Mast cells and basophils. 2000.
Church MK, Okayama Y, El-Lati S. Mediator secretion from human skin mast cells provoked by immunological and non-immunological stimulation. Skin Pharmacol Physiol. 1991;4(Suppl 1):15–24.
Mishra NC, et al. Nicotine inhibits FcεRI-induced cysteinyl leukotrienes and cytokine production without affecting mast cell degranulation through α7/α9/α10-nicotinic receptors. J Immunol. 2010;185(1):588–96.
Givi M et al. Cigarette smoke suppresses the surface expression of c-kit and FcεRI on mast cells Mediators of inflammation, 2013. 2013.
Hayashi EA, Akira S, Nobrega A. Role of TLR in B cell development: signaling through TLR4 promotes B cell maturation and is inhibited by TLR2. J Immunol. 2005;174(11):6639–47.
Inaba K, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–702.
Small-Howard A, Turner H. Exposure to tobacco-derived materials induces overproduction of secreted proteinases in mast cells. Toxicol Appl Pharmacol. 2005;204(2):152–63.
Yamamoto T, et al. Therapeutic effect of kakkonto in a mouse model of food allergy with gastrointestinal symptoms. Int Archives Allergy Immunol. 2009;148(3):175–85.
Yamamoto T, et al. Anti-allergic role of cholinergic neuronal pathway via α7 nicotinic ACh receptors on mucosal mast cells in a murine food allergy model. PLoS ONE. 2014;9(1):e85888.
Alberg A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology. 2002;180(2):121–37.
Vassallo R, et al. Nicotine and oxidative cigarette smoke constituents induce immune-modulatory and pro-inflammatory dendritic cell responses. Mol Immunol. 2008;45(12):3321–9.
McGrath J, McDonald JW, MacDonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database of Systematic Reviews, 2004(4).
Pullan RD, et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med. 1994;330(12):811–5.
Ingram JR, et al. Nicotine enemas for treatment of ulcerative colitis: a study of the pharmacokinetics and adverse events associated with three doses of nicotine. Aliment Pharmacol Ther. 2004;20(8):859–65.
Thomas GA, Rhodes J, Ingram JR. Mechanisms of disease: nicotine–a review of its actions in the context of gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2(11):536–44.
Lakhan SE, Kirchgessner A. Anti-inflammatory effects of nicotine in obesity and ulcerative colitis. J Transl Med. 2011;9:129.
de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–51.
Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–8.
Metz CN, Tracey KJ. It takes nerve to dampen inflammation. Nat Immunol. 2005;6(8):756–7.
Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–62.
Barthélémy H et al. Smoking increases the risk of post-acute COVID-19 syndrome: Results from a French community-based survey 2022. 20.
Oronsky B et al. A review of persistent post-COVID syndrome (PPCS). 2021: p. 1–9.
Crook H et al. Long covid—mechanisms, risk factors, and management 2021. 374.
Hanson SW et al. Estimated global proportions of individuals with persistent fatigue, cognitive, and respiratory symptom clusters following symptomatic COVID-19 in 2020 and 2021. 2022. 328(16): p. 1604–15.
Hussein AAM et al. Post-COVID-19 functional status: Relation to age, smoking, hospitalization, and previous comorbidities 2021. 16(3): p. 260.
de Granda-Orive JI, Solano-Reina S, C.A.J.O.. R.A. Jiménez-Ruiz, are smoking and vaping risk factors. of Developing Long and Persistent Post-COVID-19?; 2022.
Greenberger PA. Hypersensitivity pneumonitis: a fibrosing alveolitis produced by inhalation of diverse antigens. J Allergy Clin Immunol. 2019;143(4):1295–301.
Barrera L, et al. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177(1):44–55.
Vasakova M, et al. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Med. 2019;200(3):301–8.
Israel-Assayag E, et al. Expression of costimulatory molecules on alveolar macrophages in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1999;159(6):1830–4.
Selman M, Buendia-Roldan I. Immunopathology, diagnosis, and management of hypersensitivity pneumonitis. Semin Respir Crit Care Med. 2012;33(5):543–54.
Costabel U, Bonella F, Guzman J. Chronic hypersensitivity pneumonitis. Clin Chest Med. 2012;33(1):151–63.
Varone F, et al. Fibrotic hypersensitivity pneumonitis: diagnosis and management. Lung. 2020;198(3):429–40.
Morens D, et al. Cigarette smoking and protection from Parkinson’s disease: false association or etiologic clue? Neurology. 1995;45(6):1041–51.
Grandinetti A, et al. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson’s disease. Am J Epidemiol. 1994;139(12):1129–38.
Pavlov VA, Tracey KJ. Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci. 2017;20(2):156.
Posadas I, López-Hernández B, Ceña V. Nicotinic receptors in neurodegeneration. Curr Neuropharmacol. 2013;11(3):298–314.
Papke RL. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem Pharmacol. 2014;89(1):1–11.
Feigin VL, et al. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the global burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–80.
Nichols E, et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the global burden of Disease Study 2016. Lancet Neurol. 2019;18(1):88–106.
Kumar A, Tsao JW. Alzheimer disease. 2019.
Guo T, et al. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegeneration. 2020;15(1):1–37.
Durazzo TC, et al. Smoking and increased Alzheimer’s disease risk: a review of potential mechanisms. Alzheimer’s Dement. 2014;10:S122–45.
Sahakian B, et al. The effects of nicotine on attention, information processing, and short-term memory in patients with dementia of the Alzheimer type. Br J Psychiatry. 1989;154(6):797–800.
Newhouse P, et al. Nicotine treatment of mild cognitive impairment: a 6-month double-blind pilot clinical trial. Neurology. 2012;78(2):91–101.
Newhouse PA, et al. Intravenous nicotine in Alzheimer’s disease: a pilot study. Psychopharmacology. 1988;95(2):171–5.
Zhang J, et al. Nicotine attenuates the β-amyloid neurotoxicity through regulating metal homeostasis. FASEB J. 2006;20(8):1212–4.
Wallin C, et al. Alzheimer’s disease and cigarette smoke components: effects of nicotine, PAHs, and cd (II), cr (III), pb (II), pb (IV) ions on amyloid-β peptide aggregation. Sci Rep. 2017;7(1):1–14.
de Oliveira ASA, et al. BAG2 expression dictates a functional intracellular switch between the p38-dependent effects of nicotine on tau phosphorylation levels via the α7 nicotinic receptor. Exp Neurol. 2016;275:69–77.
Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. 2020;323(6):548–60.
Dorsey ER, et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the global burden of Disease Study 2016. Lancet Neurol. 2018;17(11):939–53.
Maiti P, Manna J, Dunbar GL. Current understanding of the molecular mechanisms in Parkinson’s disease: targets for potential treatments. Translational neurodegeneration. 2017;6(1):28.
Garretti F, et al. Autoimmunity in Parkinson’s Disease: the role of α-synuclein-specific T cells. Front Immunol. 2019;10:303.
Jiang T et al. The Challenge of the Pathogenesis of Parkinson’s Disease: Is Autoimmunity the Culprit? Frontiers in immunology, 2018. 9: p. 2047.
Mappin-Kasirer B, et al. Tobacco smoking and the risk of Parkinson disease: a 65-year follow-up of 30,000 male british doctors. Neurology. 2020;94(20):e2132–8.
Gallo V, et al. Exploring causality of the association between smoking and Parkinson’s disease. Int J Epidemiol. 2019;48(3):912–25.
Fagerström KO, et al. Nicotine may relieve symptoms of Parkinson’s disease. Psychopharmacology. 1994;116(1):117–9.
Kelton M, et al. The effects of nicotine on Parkinson’s disease. Brain and cognition; 2000.
Vieregge A, et al. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology. 2001;57(6):1032–5.
Lieberman A, et al. Nicotine bitartrate reduces falls and freezing of Gait in Parkinson disease: a reanalysis. Front Neurol. 2019;10:424.
Cormier A, et al. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology. 2003;44(5):642–52.
Miksys S, Tyndale R. Nicotine induces brain CYP enzymes: relevance to Parkinson’s disease, Parkinson’s Disease and Related Disorders. 2006,Springer. 177–80.
Hong D-P, Fink AL, Uversky VN. Smoking and Parkinson’s disease: does nicotine affect α-synuclein fibrillation? Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 2009. 1794(2): p. 282–90.
Mudo G, et al. Acute intermittent nicotine treatment induces fibroblast growth factor-2 in the subventricular zone of the adult rat brain and enhances neuronal precursor cell proliferation. Neuroscience. 2007;145(2):470–83.
Getachew B, et al. Nicotine protects against manganese and iron-induced toxicity in SH-SY5Y cells: implication for Parkinson’s disease. Neurochem Int. 2019;124:19–24.
Nicholatos JW, et al. Nicotine promotes neuron survival and partially protects from Parkinson’s disease by suppressing SIRT6. Acta Neuropathol Commun. 2018;6(1):1–18.
Wallin MT, et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the global burden of Disease Study 2016. Lancet Neurol. 2019;18(3):269–85.
Goldenberg MM. Multiple sclerosis review. Pharm Ther. 2012;37(3):175.
Dobson R, Giovannoni G. Multiple sclerosis–a review. Eur J Neurol. 2019;26(1):27–40.
Hedström AK, et al. Tobacco smoking, but not swedish snuff use, increases the risk of multiple sclerosis. Neurology. 2009;73(9):696–701.
Hedström A, et al. Nicotine might have a protective effect in the etiology of multiple sclerosis. Multiple Scler J. 2013;19(8):1009–13.
Shi F-D, et al. Nicotinic attenuation of central nervous system inflammation and autoimmunity. J Immunol. 2009;182(3):1730–9.
Naddafi F, et al. Novel therapeutic approach by nicotine in experimental model of multiple sclerosis. Innovations in clinical neuroscience. 2013;10(4):20.
Gao Z, et al. The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS ONE. 2014;9(9):e107979.
Jiang W, et al. Infiltration of CCR2 + Ly6Chigh proinflammatory monocytes and neutrophils into the central nervous system is modulated by nicotinic acetylcholine receptors in a model of multiple sclerosis. J Immunol. 2016;196(5):2095–108.
Rothbard JB et al. Identification of a common immune regulatory pathway induced by small heat shock proteins, amyloid fibrils, and nicotine Proceedings of the National Academy of Sciences, 2018. 115(27): p. 7081–7086.
Tomiyama AJ. Stress and obesity. Ann Rev Psychol. 2019;70(1):703–18.
Craig M, Hales, et al. In: Surveys, editor. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. Editor: Hyattsville, MD; 2020. f.H.S.D.o.H.N.E.
Zhang WX, et al. Perinatal nicotine exposure increases obesity susceptibility by peripheral leptin resistance in adult female rat offspring. Toxicol Lett. 2018;283:91–9.
Costa SO, et al. Maternal high fat diet consumption reduces liver alpha7 nicotinic cholinergic receptor expression and impairs insulin signalling in the offspring. Sci Rep. 2020;10(1):48.
Stojakovic A, et al. Effects of nicotine on homeostatic and hedonic components of food intake. J Endocrinol. 2017;235(1):R13–R31.
Seoane-Collazo P, et al. Nicotine improves obesity and hepatic steatosis and ER stress in diet-induced obese male rats. Endocrinology. 2014;155(5):1679–89.
Rupprecht LE, et al. Self-administered nicotine differentially impacts body weight gain in obesity-prone and obesity-resistant rats. Physiol Behav. 2017;176:71–5.
Cropsey KL, et al. The impact of quitting smoking on weight among women prisoners participating in a smoking cessation intervention. Am J Public Health. 2010;100(8):1442–8.
Williamson DF, et al. Smoking cessation and severity of weight gain in a national cohort. N Engl J Med. 1991;324(11):739–45.
Nogueiras R, Dieguez C, Lopez M. Come to where insulin resistance is, come to AMPK Country. Cell Metab. 2015;21(5):663–5.
Martinez de Morentin PB, et al. Nicotine induces negative energy balance through hypothalamic AMP-activated protein kinase. Diabetes. 2012;61(4):807–17.
Tucci SA. Phytochemicals in the control of human appetite and body weight. Pharmaceuticals (Basel). 2010;3(3):748–63.
Wang X, et al. Activation of the cholinergic antiinflammatory pathway ameliorates obesity-induced inflammation and insulin resistance. Endocrinology. 2011;152(3):836–46.
Wu Y, et al. Activation of AMPKalpha2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. 2015;21(4):373–82.
Yoshida T, et al. Nicotine induces uncoupling protein 1 in white adipose tissue of obese mice. Int J Obes Relat Metab Disord. 1999;23(6):570–5.
Li MD, Kane JK, Konu O. Nicotine, body weight and potential implications in the treatment of obesity. Curr Top Med Chem. 2003;3(8):899–919.
Pirie PL, Murray DM, Luepker RV. Gender differences in cigarette smoking and quitting in a cohort of young adults. Am J Public Health. 1991;81(3):324–7.
Holloway AC, et al. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia. 2005;48(12):2661–6.
Hua P, et al. Nicotine worsens the severity of nephropathy in diabetic mice: implications for the progression of kidney disease in smokers. Am J Physiol Renal Physiol. 2010;299(4):F732–9.
Chiolero A, et al. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008;87(4):801–9.
Duncan A, et al. Habenular TCF7L2 links nicotine addiction to diabetes. Nature. 2019;574(7778):372–7.
Jacobi J, et al. Nicotine accelerates angiogenesis and wound healing in genetically diabetic mice. Am J Pathol. 2002;161(1):97–104.
Katz JA, Melmed G, Sands BE. The FACTS ABOUT inflammatory Bowel Diseases. New York: Crohn’s & Colitis Foundation of America; 2011.
AlQasrawi D, Abdelli LS, Naser SA. Mystery Solved: Why Smoke Extract Worsens Disease in Smokers with Crohn’s Disease and Not Ulcerative Colitis? Gut MAP! Microorganisms, 2020. 8(5).
Scharrer S, et al. Passive Smoking increases the risk for intestinal Surgeries in patients with Crohn’s Disease. Inflamm Bowel Dis; 2020.
Sandborn WJ. Nicotine therapy for ulcerative colitis: a review of rationale, mechanisms, pharmacology, and clinical results. Am J Gastroenterol. 1999;94(5):1161–71.
Richardson CE, et al. Effect of smoking and transdermal nicotine on colonic nicotinic acetylcholine receptors in ulcerative colitis. QJM. 2003;96(1):57–65.
Arnott ID, et al. Whole gut lavage fluid interleukin-1beta and interleukin-8 in smokers and non-smokers with Crohn’s disease in clinical remission. Dig Liver Dis. 2002;34(6):424–9.
Bergeron V, et al. Current smoking differentially affects blood mononuclear cells from patients with Crohn’s disease and ulcerative colitis: relevance to its adverse role in the disease. Inflamm Bowel Dis. 2012;18(6):1101–11.
Nielsen OH, et al. Influence of smoking on colonic gene expression profile in Crohn’s disease. PLoS ONE. 2009;4(7):e6210.
Ingram JR, et al. A randomized trial of nicotine enemas for active ulcerative colitis. Clin Gastroenterol Hepatol. 2005;3(11):1107–14.
Aldhous MC, et al. Does nicotine influence cytokine profile and subsequent cell cycling/apoptotic responses in inflammatory bowel disease? Inflamm Bowel Dis. 2008;14(11):1469–82.
Madretsma S, et al. In-vivo effect of nicotine on cytokine production by human non-adherent mononuclear cells. Eur J Gastroenterol Hepatol. 1996;8(10):1017–20.
Ueno A, et al. Opposing effects of smoking in ulcerative colitis and Crohn’s disease may be explained by differential effects on dendritic cells. Inflamm Bowel Dis. 2014;20(5):800–10.
Wong VW, et al. Asia-Pacific Working Party on non-alcoholic fatty liver Disease guidelines 2017-Part 1: definition, risk factors and assessment. J Gastroenterol Hepatol. 2018;33(1):70–85.
Sinha-Hikim AP, Sinha-Hikim I, Friedman TC. Connection of nicotine to Diet-Induced obesity and non-alcoholic fatty liver disease: Cellular and mechanistic insights. Front Endocrinol (Lausanne). 2017;8:23.
Willis DN, et al. Toxicity of gutkha, a smokeless tobacco product gone global: is there more to the toxicity than nicotine? Int J Environ Res Public Health. 2014;11(1):919–33.
Friedman TC, et al. Additive effects of nicotine and high-fat diet on hepatic steatosis in male mice. Endocrinology. 2012;153(12):5809–20.
Hukkanen J, Jacob P 3rd, and, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57(1):79–115.
Denton TT, Zhang X, Cashman JR. Nicotine-related alkaloids and metabolites as inhibitors of human cytochrome P-450 2A6. Biochem Pharmacol. 2004;67(4):751–6.
Schoedel KA, et al. Down-regulation of hepatic nicotine metabolism and a CYP2A6-like enzyme in african green monkeys after long-term nicotine administration. Mol Pharmacol. 2003;63(1):96–104.
Crawford EL, et al. Measurement of cytochrome P450 2A6 and 2E1 gene expression in primary human bronchial epithelial cells. Carcinogenesis. 1998;19(10):1867–71.
Cluette-Brown J, et al. Oral nicotine induces an atherogenic lipoprotein profile. Proc Soc Exp Biol Med. 1986;182(3):409–13.
Hojnacki J, et al. Oral nicotine impairs clearance of plasma low density lipoproteins. Proc Soc Exp Biol Med. 1986;182(3):414–8.
Lee J, Cooke JP. Nicotine and pathological angiogenesis. Life Sci. 2012;91(21–22):1058–64.
Lau PP, et al. Nicotine induces proinflammatory responses in macrophages and the aorta leading to acceleration of atherosclerosis in low-density lipoprotein receptor(-/-) mice. Arterioscler Thromb Vasc Biol. 2006;26(1):143–9.
Heeschen C, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7(7):833–9.
Konishi H, Wu J, Cooke JP. Chronic exposure to nicotine impairs cholinergic angiogenesis. Vasc Med. 2010;15(1):47–54.
Park HS, et al. Chronic nicotine exposure attenuates proangiogenic activity on human umbilical vein endothelial cells. J Cardiovasc Pharmacol. 2011;57(3):287–93.
Di Luozzo G, et al. Nicotine induces mitogen-activated protein kinase dependent vascular smooth muscle cell migration. Atherosclerosis. 2005;178(2):271–7.
Villablanca AC. Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J Appl Physiol (1985). 1998;84(6):2089–98.
Carty CS et al. Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cells. J Vasc Surg, 1996. 24(6): p. 927 – 34; discussion 934-5.
Zhang Q, et al. Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin Cancer Res. 2007;13(16):4686–94.
Wu JC, et al. Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor. J Cell Biochem. 2009;108(2):433–46.
Heeschen C, et al. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest. 2002;110(4):527–36.
Ng MK, et al. A central role for nicotinic cholinergic regulation of growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol. 2007;27(1):106–12.
Conklin BS, et al. Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. Am J Pathol. 2002;160(2):413–8.
Bikbov B, et al. Global, regional, and national burden of chronic kidney disease, 1990&-2017: a systematic analysis for the global burden of Disease Study 2017. The Lancet. 2020;395(10225):709–33.
Jaimes EA, Tian RX, Raij L. Nicotine: the link between cigarette smoking and the progression of renal injury? Am J Physiol Heart Circ Physiol. 2007;292(1):H76–82.
Jaimes EA, Galceran JM, Raij L. Angiotensin II induces superoxide anion production by mesangial cells. Kidney Int. 1998;54(3):775–84.
Sharma P, et al. NADPH-oxidase activation by protein kinase C-isotypes. Biochem Biophys Res Commun. 1991;177(3):1033–40.
Jaimes EA, et al. Nicotine augments glomerular injury in a rat model of acute nephritis. Am J Nephrol. 2009;29(4):319–26.
Arany I, et al. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol. 2012;302(6):F722–9.
Yeboah MM, et al. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int. 2008;74(1):62–9.
Yeboah MM, et al. Nicotinic acetylcholine receptor expression and regulation in the rat kidney after ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2008;295(3):F654–61.
Agarwal PK, et al. Renoprotective effects of long-term oral nicotine in a rat model of spontaneous proteinuria. Am J Physiol Renal Physiol. 2012;302(7):F895–904.
Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30(3):234–54.
Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79(2):319–26.
Sadis C, et al. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS ONE. 2007;2(5):e469.
Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–39.
Channappanavar R, et al. Dysregulated type I Interferon and inflammatory monocyte-macrophage responses cause Lethal Pneumonia in SARS-CoV-Infected mice. Cell Host Microbe. 2016;19(2):181–93.
Hirano T, Murakami M. COVID-19: a New Virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52(5):731–3.
Birrell MA, et al. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J Cell Physiol. 2008;214(1):27–37.
Park JE, et al. MERS transmission and risk factors: a systematic review. BMC Public Health. 2018;18(1):574.
Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206–16.
Alraddadi BM, et al. Risk factors for primary Middle East Respiratory Syndrome Coronavirus illness in humans, Saudi Arabia, 2014. Emerg Infect Dis. 2016;22(1):49–55.
Seys LJM, et al. DPP4, the Middle East respiratory syndrome coronavirus receptor, is upregulated in lungs of smokers and chronic obstructive Pulmonary Disease Patients. Clin Infect Dis. 2018;66(1):45–53.
Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9(1):601–4.
Qi F, et al. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. 2020;526(1):135–40.
Meyerholz DK, Lambertz AM, McCray PB. Jr., Dipeptidyl peptidase 4 distribution in the human respiratory tract: implications for the Middle East Respiratory Syndrome. Am J Pathol. 2016;186(1):78–86.
Drosten C, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–76.
Solerte SB, et al. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020;57(7):779–83.
Cai G, et al. Tobacco Smoking increases the lung gene expression of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;201(12):1557–9.
Cai G, et al. Reply to Polverino: cigarette smoking and COVID-19: a Complex Interaction. Am J Respir Crit Care Med. 2020;202(3):472–4.
Li G, et al. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun. 2020;112:102463.
Imai Y, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–6.
Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93(5):543–8.
Kuba K, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–9.
Andersson U. The cholinergic anti-inflammatory pathway alleviates acute lung injury. Mol Med. 2020;26(1):1–4.
Gauthier AG, et al. From nicotine to the cholinergic anti-inflammatory reflex–can nicotine alleviate the dysregulated inflammation in COVID-19? J Immunotoxicol. 2021;18(1):23–9.
Chen R, et al. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon. 2020;6(12):e05672.
Naidu V, Zeki AA, Sharma P. Sex differences in the induction of angiotensin converting enzyme 2 (ACE-2) in mouse lungs after e-cigarette vapor exposure and its relevance to COVID-19. J Investig Med. 2021;69(5):954–61.
McAlinden KD, et al. Electronic cigarette aerosol is cytotoxic and increases ACE2 expression on human airway epithelial cells: implications for SARS-CoV-2 (COVID-19). J Clin Med. 2021;10(5):1028.
Maggi F et al. Nicotine upregulates ACE2 expression and increases competence for SARS-CoV-2 in human pneumocytes. ERJ Open Research, 2021. 7(2).
Lee AC, et al. Tobacco, but not nicotine and flavor-less electronic cigarettes, induces ACE2 and immune dysregulation. Int J Mol Sci. 2020;21(15):5513.
Caruso M, et al. Role of cigarette smoke on angiotensin-converting Enzyme-2 protein membrane expression in bronchial epithelial cells using an air-liquid interface model. Front Pharmacol. 2021;12:335.
Voinsky I, Gurwitz D. Smoking and COVID-19: similar bronchial ACE2 and TMPRSS2 expression and higher TMPRSS4 expression in current versus never smokers. Drug Dev Res. 2020;81(8):1073–80.
AbdelMassih AF et al. A multicenter consensus: A role of furin in the endothelial tropism in obese patients with COVID-19 infection. Obes Med, 2020: p. 100281.
Lagoumintzis G, et al. Nicotinic cholinergic system and COVID-19: in silico identification of interactions between α7 nicotinic acetylcholine receptor and the cryptic epitopes of SARS-Co-V and SARS-CoV-2 Spike glycoproteins. Food Chem Toxicol. 2021;149:112009.
Tanmay S, et al. Is SARS-CoV-2 spike glycoprotein impairing macrophage function via alpha7-nicotinic acetylcholine receptors? Food Chem Toxicol. 2021;152:112184.
Mohammadi S, et al. In silico investigation on the inhibiting role of Nicotine/Caffeine by blocking the S protein of SARS-CoV-2 Versus ACE2 receptor. Microorganisms. 2020;8(10):1600.
Patanavanich R, Glantz SA. Smoking is associated with COVID-19 progression: a meta-analysis.Nicotine Tobacco Research. 2020. 22(9): p. 1653–6.
Reddy RK, et al. The effect of smoking on COVID-19 severity: a systematic review and meta‐analysis. J Med Virol. 2021;93(2):1045–56.
Farsalinos K, et al. Current smoking, former smoking, and adverse outcome among hospitalized COVID-19 patients: a systematic review and meta-analysis. Therapeutic Adv chronic disease. 2020;11:2040622320935765.
González-Rubio J, et al. A systematic review and meta-analysis of hospitalised current smokers and COVID-19. Int J Environ Res. 2020;17(20):7394.
Zhao Q, et al. The impact of COPD and smoking history on the severity of COVID-19: a systemic review and meta‐analysis. J Med Virol. 2020;92(10):1915–21.
Guo FR. Smoking links to the severity of COVID-19: an update of a meta‐analysis. J Med Virol. 2020;92(11):2304–5.
Gülsen A et al. The effect of smoking on COVID-19 symptom severity: Systematic review and meta-analysis Pulmonary medicine, 2020. 2020.
Alqahtani JS, et al. Prevalence, severity and mortality associated with COPD and smoking in patients with COVID-19: a Rapid systematic review and Meta-analysis. PLoS ONE. 2020;15(5):e0233147.
Umnuaypornlert A, et al. Smoking and risk of negative outcomes among COVID-19 patients: a systematic review and meta-analysis. Tobacco induced diseases; 2021. p. 19.
Zhang H et al. Association of smoking history with severe and critical outcome in COVID-19 patients: A systemic review and meta-analysis. Eur J Integr Med, 2021: p. 101313.
Guo FR. Active smoking is associated with severity of coronavirus disease 2019 (COVID-19): an update of a meta-analysis. Tobacco induced diseases; 2020. p. 18.
Farsalinos K, et al. Smoking prevalence among hospitalized COVID-19 patients and its association with disease severity and mortality: an expanded re-analysis of a recent publication. Harm Reduct J. 2021;18(1):9.
Farsalinos K et al. Current smoking, former smoking, and adverse outcome among hospitalized COVID-19 patients: a systematic review and meta-analysis. 2020. 11: p. 2040622320935765.
Patanavanich R, Glantz SA. Smoking is Associated with COVID-19 progression: a Meta-analysis. Nicotine & Tobacco Research; 2020. pp. 1653–6.
Zhang H, et al. Association of smoking history with severe and critical outcomes in COVID-19 patients: a systemic review and meta-analysis. Eur J Integr Med. 2021;43:101313.
Umnuaypornlert A, et al. Smoking and risk of negative outcomes among COVID-19 patients: a systematic review and meta-analysis. Tob Induc Dis. 2021;19:09.
Reddy RK, et al. The effect of smoking on COVID-19 severity: a systematic review and meta-analysis. J Med Virol. 2021;93(2):1045–56.
Zhao Q, et al. The impact of COPD and smoking history on the severity of COVID-19: a systemic review and meta-analysis. J Med Virol. 2020;92(10):1915–21.
Zhang T, et al. Risk factors and predictors associated with the severity of COVID-19 in China: a systematic review, meta-analysis, and meta-regression. J Thorac Dis. 2020;12(12):7429–41.
Zeng L, et al. Clinical characteristics of COVID-19 with cardiac injury: a systematic review and meta-analysis. Epidemiol Infect. 2020;148:e266.
Xie J, et al. Clinical characteristics, laboratory abnormalities and CT findings of COVID-19 patients and risk factors of severe disease: a systematic review and meta-analysis. Ann Palliat Med. 2021;10(2):1928–49.
Xiang G, et al. Clinical risk factors for mortality of hospitalized patients with COVID-19: systematic review and meta-analysis. Ann Palliat Med. 2021;10(3):2723–35.
Taylor EH et al. Factors associated with mortality in patients with COVID-19 admitted to intensive care: a systematic review and meta-analysis. Anaesthesia, 2021.
Simons D, et al. The association of smoking status with SARS-CoV-2 infection, hospitalization and mortality from COVID-19: a living rapid evidence review with bayesian meta-analyses (version 7). Addiction. 2021;116(6):1319–68.
Silverio A, et al. Cardiovascular risk factors and mortality in hospitalized patients with COVID-19: systematic review and meta-analysis of 45 studies and 18,300 patients. BMC Cardiovasc Disord. 2021;21(1):23.
Shoar S, et al. Meta-analysis of Cardiovascular events and related biomarkers comparing Survivors Versus Non-survivors in patients with COVID-19. Am J Cardiol. 2020;135:50–61.
Shi C, et al. Predictors of mortality in patients with coronavirus disease 2019: a systematic review and meta-analysis. BMC Infect Dis. 2021;21(1):663.
Mesas AE, et al. Predictors of in-hospital COVID-19 mortality: a comprehensive systematic review and meta-analysis exploring differences by age, sex and health conditions. PLoS ONE. 2020;15(11):e0241742.
Li Y, et al. Risk factors for poor outcomes in hospitalised COVID-19 patients: a systematic review and meta-analysis. J Glob Health. 2021;11:10001.
Li X, et al. Clinical determinants of the severity of COVID-19: a systematic review and meta-analysis. PLoS ONE. 2021;16(5):e0250602.
Li J et al. Meta-analysis investigating the relationship between clinical features, outcomes, and severity of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pneumonia. Am J Infect Control, 2020.
Lassi ZS, et al. A systematic review and meta-analysis of data on pregnant women with confirmed COVID-19: clinical presentation, and pregnancy and perinatal outcomes based on COVID-19 severity. J Glob Health. 2021;11:05018.
Karanasos A, et al. Impact of Smoking Status on Disease Severity and Mortality of hospitalized patients with COVID-19 infection: a systematic review and Meta-analysis. Nicotine Tob Res. 2020;22(9):1657–9.
Kang S, Gong X, Yuan Y. Association of smoking and cardiovascular disease with disease progression in COVID-19: A systematic review and meta-analysis. Epidemiol Infect, 2021: p. 1–26.
Huang I, Pranata R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): systematic review and meta-analysis. J Intensive Care. 2020;8:36.
Hou H et al. Smoking is independently associated with an increased risk for COVID-19 mortality: A systematic review and meta-analysis based on adjusted effect estimates. Nicotine Tob Res, 2021.
Figliozzi S, et al. Predictors of adverse prognosis in COVID-19: a systematic review and meta-analysis. Eur J Clin Invest. 2020;50(10):e13362.
Zheng Z, et al. Risk factors of critical & mortal COVID-19 cases: a systematic literature review and meta-analysis. J Infect. 2020;81(2):e16–e25.
Sanchez-Ramirez DC, Mackey D. Underlying respiratory diseases, specifically COPD, and smoking are associated with severe COVID-19 outcomes: a systematic review and meta-analysis. Respir Med. 2020;171:106096.
Abate BB, et al. Sex difference in coronavirus disease (COVID-19): a systematic review and meta-analysis. BMJ Open. 2020;10(10):e040129.
Baradaran A, et al. Prevalence of Comorbidities in COVID-19 patients: a systematic review and Meta-analysis. Arch Bone Jt Surg. 2020;8(Suppl 1):247–55.
Gonzalez-Rubio J et al. A Systematic Review and Meta-Analysis of Hospitalised Current Smokers and COVID-19. Int J Environ Res Public Health, 2020. 17(20).
Farsalinos K, et al. Current smoking, former smoking, and adverse outcome among hospitalized COVID-19 patients: a systematic review and meta-analysis. Ther Adv Chronic Dis. 2020;11:2040622320935765.
Farsalinos K, Barbouni A, Niaura R. Systematic review of the prevalence of current smoking among hospitalized COVID-19 patients in China: could nicotine be a therapeutic option? Intern Emerg Med. 2020;15(5):845–52.
Dorjee K, et al. Prevalence and predictors of death and severe disease in patients hospitalized due to COVID-19: a comprehensive systematic review and meta-analysis of 77 studies and 38,000 patients. PLoS ONE. 2020;15(12):e0243191.
Del Sole F, et al. Features of severe COVID-19: a systematic review and meta-analysis. Eur J Clin Invest. 2020;50(10):e13378.
Acknowledgements
We would like to express our gratitude for assistance of Baqiyatallah University of Medical Sciences.
Funding
NA.
Author information
Authors and Affiliations
Contributions
ZS, SAJ, and MG have made substantial contributions to design the study. ZS, SAJ, and BFN have worked in writing the original draft. All authors approved the final version to be published; they all agreed to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
NA.
Consent for publication
NA.
Competing interests
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salehi, Z., Motlagh Ghoochani, B., Hasani Nourian, Y. et al. The controversial effect of smoking and nicotine in SARS-CoV-2 infection. Allergy Asthma Clin Immunol 19, 49 (2023). https://doi.org/10.1186/s13223-023-00797-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13223-023-00797-0