
Abstract
Purpose
To identify the differences in gut bacterial community of Parabramis pekinensis under different growth conditions, and the effect of the diet in a controlled habitat on the community structure, aiming to provide a comprehensive survey of how the gut microbiota in P. pekinensis varies depending on habitat.
Methods
A total of 73 P. pekinensis from Yangtze River (W), rivers in the outskirts of Jingjiang (Jiangsu province, China, R), and farms (C) were collected to analyze the intestinal microbiota using high-throughput sequencing of the V3-V4 16S ribosomal RNA gene. We also subdivided the gut into the foregut (F), midgut (M), and hindgut (B) to analyze the differences between them.
Results
The dominant bacterial phyla in P. pekinensis were Fusobacteria, Firmicutes, Proteobacteria, and Actinobacteria; meanwhile, Cyanobacteria, Bacteroidetes, Chloroflexi, and Verrucomicrobia were also highly abundant. It is worth noting that the abundance of Fusobacteria Cetobacterium was also very high. The abundance and diversity of the intestinal microbiota structure of fish taken from breeding farm were significantly lower than those taken from Yangtze river and Suburban river, and the abundance of Aeromonas in the gut of fish taken from Yangtze river was much higher than that of fish taken from Suburban river. Compared to midgut, foregut and hindgut have similar microbiota structures, but did not differ significantly in them.
Conclusions
The core intestinal microbiota of P. pekinensis is the same to other herbivorous and partially omnivorous fish. There were significant differences in the intestinal microbiota structure of P. pekinensis from different habitats, but no significant differences in the microbiota abundance and diversity between the different parts of the intestine.
Similar content being viewed by others
Background
Fish are the most diverse and abundant vertebrate groups in the world. They are of great importance in aquatic ecosystem health and human nutrition. The study of intestinal microbiota in fishes has become a leading research topic in recent years (Talwar et al. 2018). As several studies have shown, symbiotic micro-organisms govern most biological traits of fish such as immune response(Gomez and Balcazar 2008; Vasquez et al. 2019; Yuan et al. 2020; Yukgehnaish et al. 2020), nutrient absorption and energy regulation (Reda et al. 2018; Butt and Volkoff 2019), lipid metabolism (Semova et al. 2012; Yu et al. 2019), and reproduction (Carnevali et al. 2017; Aydın and Çek-Yalnız 2019). Many factors can affect the microbial composition in the gastrointestinal tract of fish such as gender, age, diet, environment, and behavior (Wong and Rawls 2012; Baldo et al. 2015; Li et al. 2016; Piazzon et al. 2019; Solovyev et al. 2019; Le and Wang 2020). Inside the gastrointestinal system of fish, the microbial community varies across parts (Clements et al. 2014; Banerjee and Ray 2017).
As one of the Cyprinidae fish in the Yangtze River, the wild resources of P. pekinensis are also declining rapidly (Yi et al. 2010). However, there is still very few research on the species, and there has not yet been a breakthrough in artificial breeding of P. pekinensis. In order to identify its microbiote composition under different growth conditions, high-throughput sequencing technology was used to study the bacterial community of P. pekinensis samples originated from different habitats in the eastern area of China.
Results
Operational taxonomic unit (OTU) partition and data analysis
A total of 5,044,643 valid reads (including 3,527,456 from the intestinal samples) were obtained from 39 samples. After quality control and filtration, 3,101,597 high-quality reads (including 2,415,872 from the intestinal samples) were obtained, with an average length of 407 bp. Merging and OTU partition were performed at a 97% similarity level, and a total of 615 OTUs were obtained. Among them, a total of 552 OTUs were from the intestinal samples. Most of the microbial diversity in these samples were successfully captured by the sequencing (Additional files 1 and 2), and the sampling quantity was sufficient for analysis (Additional file 3).
Bacterial communities in gastrointestinal tract, the water, and the compound feed samples
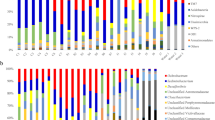
A total of 201 genera belonging to 14 phyla were identified in the intestinal samples (Fig. 1). On the whole, P. pekinensis was numerically dominated by phyla Fusobacteria (39%), Firmicutes (29.79%), Proteobacteria (13.68%), Actinobacteria (12.05%), and Cyanobacteria (3.13%).These five phyla represented 97.65% of the total bacterial sequences. At the genus level, the relative abundances of dominant intestinal microbiota were Cetobacterium (39%), uncultured (Propionibacteriaceae) (10.62%), Romboutsia (9.38%), Erysipelothrix (8.64%), and Cyanobium PCC-6307 (3.13%), which was 70.77% of the total.

Phylum-level gut microbiota composition of Parabramis pekinensis shows the overall abundance of bacteria detected in all samples. “CW1,” “CW2,” “CW3,” “RW1,” “RW2,” “RW3,” “WW1,” “WW2,” and “WW3”are water samples from three sampling sites. “F1,” “F2,” and “F3”are compound feed samples from the farm
In contrast, the water samples were numerically dominated by phyla Proteobacteria (35.86%), Firmicutes (21.83%), Fusobacteria (20.97%), Bacteroidetes (11.24%), and Actinobacteria (7.06%), which represented 96.96% of the total bacterial sequences. The compound feed samples were numerically dominated by phyla Proteobacteria (49.03%), Fusobacteria (25.03%), Firmicutes (18.53%), Actinobacteria (4.52%), and Bacteroidetes (1.92%), which represented 99.03% of the total bacterial sequences (Fig. 1).
The species of the dominant microbiota (Fusobacteria, Firmicutes, Proteobacteria, Actinobacteria) in the intestinal tract of each group was almost the same as the dominant microbiota in the water for each group, but they had different proportions, and the results were similar in the compound feed samples (Fig. 1) and the culture group.
The difference of intestinal microbiota structure between different habitats
The results showed that the dominant bacteria were the same in the intestines of the fish in three different habitats, mainly phyla Fusobacteria, Firmicutes, Actinobacteria, and Proteobacteria (Additional file 4). However, there were significant differences in abundance. Alpha diversity indices confirmed that the abundance (Additional file 5 & Additional file 6) of intestinal microbiota in C group were significantly lower than those in R and W groups. However, the same results indicate that there was no significant difference between the R and W groups. Principal component analysis (PCA) (Fig. 2a) and non-metric multi-dimensional scaling (NMDS) analysis (Fig. 2c, e) showed a significant difference in the microbiota structure between the three groups. A clear distinction in the intestinal microbiota of different groups was also revealed by ANOSIM (R = 0.4113, P = 0.001). UniFrac Heatmap analysis was performed on the samples using weighted and unweighted algorithms (weighted algorithms consider the abundance of the sequence, while unweighted algorithms neglect the abundance of the sequence). According to the results of the weighted UniFrac (Fig. 3a), the three groups tend to cluster separately, which illustrate the differences between them. Meanwhile, unweighted UniFrac (Fig. 3b) showed that the C and W groups tend to cluster individually, while the R group disperses between them, illustrating the apparent differences between the C group and W group, while the differences between them and the R group are not so distinct.

β-Diversity analysis of the gut microbiota of Parabramis pekinensis. The PCA, NMDS based on weighted and unweighted analysis for different habitats (a, c, e) and different sections of the intestine (b, d, f) are shown

UniFrac Heatmap shows the difference between every two samples. a Weighted UniFrac Heatmap of different habitats. b Unweighted UniFrac Heatmap of different habitats. c Weighted UniFrac Heatmap of different sections of the intestine. d Unweighted UniFrac Heatmap of different sections of the intestine
LEfSe (absolute linear discriminant analysis score log10 ≥ 2.0) analysis results identified the different main species among the groups. Taking the order level as an example (Additional file 7), order Fusobacteriales had significantly higher abundance in the C group (80.68%) than R (8.11%) and W groups (27.83%). Orders Erysipelotrichales, Propionibacteriales, Synechococcales, and Bacteroidales had significantly higher abundances in the R group (32.4%, 30.52%, 9.02%, 4.26%, respectively) than the C (2.32%, 0.16%, 0.08%, 0.43%, respectively) and W groups (3.65%, 1.14%, 0.33%, 0.38%, respectively). Orders Clostridiales and Aeromonadales had significantly higher abundances in the W group (41.17%, 7.65%, respectively) than the C (3.45%, 1.02%, respectively) and R groups (4.65%, 0.2%, respectively).
The difference of microbiota content in different parts of the intestine
Overall, all three parts of the intestine (F, M, and B) were numerically dominated by phyla Fusobacteria, Firmicutes, Actinobacteria, and Proteobacteria (Additional file 8), and there were no significant differences in the microbiota abundance (Additional file 9) or microbiota diversity (Additional file 10) between the three parts of the intestine. The used ordination techniques (PCA (Fig. 2b), NMDS (Fig. 2d, f) and ANOSIM (R = 0.0057, P = 0.383) analysis) showed no significant differences in the microbiota content between the three groups. Fusobacteria was the most abundant bacteria in F and M, while Firmicutes was the most abundant bacteria in B. The results of weighted UniFrac (Fig.. 3c) and unweighted UniFrac (Fig. 3d) analysis showed that the three groups were not clustered, indicating that the differences in the microbiota content in the three parts were not significant.
LEfSe (absolute linear discriminant analysis score log10 ≥ 2.0) analysis results showed that (Additional file 11) the significant differences in the microbiota content between the F, M, and B were due to the order Verrucomicrobiales (0.03%, 0.06%, 0.01%, respectively), Chthoniobacterales (0.04%, 0.23%, 0.01%, respectively), Caldilineales (0.04%, 0.1%, 0.02%, respectively), and uncultured α-proteobacteria (0.02%, 0.06%, 0.01%, respectively). These had a relatively higher abundance in M, but a very small ratio in the overall microbiota content. However, when the habitats were compared, there were still some differences in the dominant microbiota in different parts of the intestinal tract. The microbiota content of the C and W groups were similar; phyla Fusobacteria, Firmicutes and Proteobacteria had the highest abundance in F, M, and B of the two groups; and the abundance of the dominant microbiota in all three parts of the intestinal tract of the W group was evener. However, the R group was different. Phylum Actinobacteria had the highest abundance in their F and M, followed by Firmicutes, and Firmicutes having the highest abundance in B, followed by Fusobacteria.
Biological pathways and functional categories deduced from the 16S data
PICRUSt algorithm was used to predict microbial functions. In this study, 41KEGG pathways (level 2KEGG orthology groups) were identified (Fig. 4). The top five pathways for abundance were mainly related to membrane transport (12.33–13.64%), carbohydrate metabolism (10.15–11.86%), amino acid metabolism (9.81–10.23%), replication and repair (7.40–8.15%), and energy metabolism (5.57–6.70%). Their main biological functions include transporters, ABC transporters, DNA repair, and recombination, purine metabolism, and general function (Additional file 12).

Histogram showing the ten dominant gene families across three different habitats (level 2KEGG orthology groups)
Discussion
Bacterial community structure in P. pekinensis intestine
At present, the wild resources of P. pekinensis are declining day by day, and many efforts are deployed to preserving these fish stocks. Fish farming constitute a promising strategy to adjust the amount of this endangered species. However, artificial P. pekinensis breeding is far from being mastered, since the lack of information relating to its biological requirements. In this study, high-throughput sequencing technology was used to detect the intestinal microbiota content in different parts of the intestinal tract of P. pekinensis. Our results show that P. pekinensis share a core freshwater herbivorous fish microbiota (van Kessel et al. 2011; Wu et al. 2012; Ni et al. 2014). Phyla Fusobacteria, Firmicutes, Proteobacteria, and Actinobacteria were the dominant microbiota in the intestines of P. pekinensis, what appears to be the same bacterial community observed for the grass carp (Wu et al. 2012). Phyla Cyanobacteria, Bacteroidetes, Chloroflexi, and Verrucomicrobia are closely associated with omnivorous fish (van Kessel et al. 2011; Meng et al. 2018; Wang et al. 2018). In the intestine of P. pekinensis, Cyanobacteria is the fifth largest group of bacteria, and Bacteroidetes, Chloroflexi, and Verrucomicrobia have also a higher abundance, which may indicate that P. pekinensis is a typical herbivorous and partially omnivorous fish. It is worth noting that Pseudomonas, Aeromonas, and Allorhizobium were more abundant in the intestine of herbivorous fishes than other microbiotas since they are closely associated with cellulose degradation (He et al. 2010; Jiang et al. 2011; Wu et al. 2012; Liu et al. 2016; Talwar et al. 2018), Although the same is true for P. pekinensis, the highest abundance of intestinal microbiota in P. pekinensis was Cetobacerium (33.64%). Studies have shown that Cetobacterium is closely related to the generation of protease and lipase; it is commonly seen at a high abundance in the digestive tract of carnivorous fish (Kim et al. 2007; Merrifield et al. 2009; Liu et al. 2016; Zeng et al. 2020). In addition, our previous breeding experiments found that, during intensive parental cultivation of P. pekinensis, a low level of protein (crude protein content was 25%) in the compound feed would result in slow growth and difficulty in the maturation of the gonads, and the situation was improved by changing to feed with higher protein content(crude protein content was 30%)
The difference in the intestinal microbiota according to different habitats and different parts of the intestine
The collection of all samples was completed within 3 days; there was no direct communication between the three groups. The C group was collected in the runway of the industrial circulating water pond. All the fish collected own a good health, were at the same age (all 2 ages), and were the same gender (all females). The water of the three sampling sites had similar temperature (20.5 °C), oxygen (10.27 ± 0.35 mg/L), pH (7.7 ± 0.16), and ammonia concentration (0.28 ± 0.03 mg/L, Nassler colorimetric method). They all belong to the category of slow-moving fresh water, although their flow rate (0.2–0.5 m/s) and salinity (0.1–1‰) differ significantly in numerical values (Table 1). The results showed that there were significant differences in the abundance, diversity, and structural characteristics of the intestinal microbiota of P. pekinensis under different growth conditions. The diversity of intestinal microbiota of the cultured group was significantly lower than that of the R and W groups, and the Fusobacteria Cetobacterium got an absolute advantage, with the abundance accounting for 80.68%. We speculate that this may be due to the single composition of the diet. Fish taken from the breeding farm live in a narrow breeding track; the compound feed was their only food, which has a relatively high content of protein (Michl et al. 2017). In addition to their role in protein intake, Cetobacterium can provide protection to the host against pathogens (Tsuchiya et al. 2008; Silva et al. 2011; Larsen et al. 2014). So this may be another reason for the abundance of Cetobacterium in the intestines of the C group. In the intestines of the R and W groups, the distribution of microbiota was relatively even, and these are also the most common microbiota content in the intestines of herbivorous fish and omnivorous fish (van Kessel et al. 2011; Wu et al. 2012). In the intestinal tract of the R group, Firmicutes (Erysipelothrix), Actinobacteria (Propionibacteriaceae), and Cyanobacteria had the highest abundance. In the intestinal tract of the W group, Firmicutes (Peptostreptococcaceae), Fusobacteria (Cetobacterium), and Proteobacteria (Aeromonas) had the highest abundance. Phylum Firmicutes is one of the most important types of bacteria in the intestinal microbiota of freshwater fish, and they play a crucial role in promoting the absorption of fat in the intestinal epithelial, and liver tissues, as well as the formation of fat droplets (Semova et al. 2012). In our study, phylum Firmicutes was the most abundant in the intestinal tract of both R and W groups, which is the same as that of the wild Megalobrama amblycephala (Li et al. 2014; Liu et al. 2016; Wei et al. 2018). In the intestinal tract of the C group, the percentage of Firmicutes reach only 6.64%. This fully demonstrates that diet can significantly affect and determine the host’s intestinal microbiota content (Michl et al. 2017; Catalan et al. 2018). In the wild habitats without artificial feeding, their nutrition was understandably more complicated. They would choose foods that were rich in fiber and fatty acids; therefore, the abundance of Firmicutes was very high. Chitin is the main component of zooplankton biomass in the environment, second only to cellulose (Ashhurst 2001), as digesting chitin on zooplankton is very important for fish (Sakata 1980; Macdonald 1986; Ashhurst 2001; Sugita and Ito 2006; Askarian et al. 2012; Ray et al. 2012). Aeromonas, Bacillus, and Acinetobacter have been closely associated with chitin digestion (Sakata 1980; Macdonald 1986; Sugita and Ito 2006; Askarian et al. 2012). We found that the abundance of Aeromonas, Bacillus, and Acinetobacter in the intestinal tract of the W group was higher than that of R group, which may indicate that P. pekinensis has a different eating preference in these two different habitats, and this needs further research to confirm.
At present, studies on the differences between different parts of intestinal tract mainly focus on the comparative analysis of the whole digestive tract, including the stomach, pylorus, and intestines (McDonald et al. 2012), while there were few studies on the different parts of the intestinal tract. Similar studies in some higher vertebrates have shown that there are certain differences in the abundance and diversity of microbiota in different parts of the intestinal tract (Walker et al. 2011). However, our study showed that there were no significant differences between the dominant microbiota in different parts of the intestinal tract of P. pekinensis. Although the phyla Fusobacteria had the highest abundance in F and M, and the phyla Firmicutes had the highest abundance in B, but there was no significant difference in their abundance; our results are similar to that of Ringo and Strom (1994). Their study showed that there was no significant difference in the species of bacteria in the small and large intestines. However, some bacteria with a relatively low abundance, such as Verrucomicrobiales, Chthoniobacterales, Caldilineales, and α-proteobacteria were significantly higher in M than F and B, which suggests that F and B have similar structures.
The dominant microbiota detected in the water and feed samples were similar to that of the intestinal tract, and there was an absolute one-to-one correlation between the changes of microbial abundance. For example, Actinobacteria, Bacteroidetes, and Cyanobacteria had higher abundance in the intestinal tract of R group than the W group, and the abundance of these bacteria in the Suburban River was also higher than Yangtze River. The abundance of Fusobacteria and Proteobacteria in the intestinal tract of the R group was lower than that of the W group, and the abundance of these in the Suburban River was also lower than that of Yangtze River. This indicates that environmental factors can influence the colonization of fish intestinal microbiota to a certain extent, which was confirmed by many similar studies (Benson et al. 2010; Larsen et al. 2014; Li et al. 2014; Liu et al. 2016).
Functional analysis
The critical role of bacteria living in the intestines of the host has been widely studied. For example, sterile zebrafish cannot absorb macromolecular proteins (Bates et al. 2006), and intestinal microbiota can inhibit the expression of lipoprotein lipase circulating inhibitors (Angptl4/Fiaf) in the intestinal epithelium, thus promoting the accumulation of fat in the adipose tissue (Camp et al. 2012).In our study, the intestinal microbiota of P. pekinensis was mainly related to environmental information processing, genetic information processing, metabolism, and others. The main biological functions were transporters, ABC transporters, and DNA repair and recombination proteins. Transporters are mainly related to transport and metabolism of amino acids, carbohydrates, lipids, inorganic ions, and coenzymes. ABC transporter, first discovered in bacteria, is a transport ATPase on the bacterial plasma membrane and belongs to a large and diverse protein family. In this protein family, there are members that transport ions, amino acids, nucleotides, polysaccharides, peptides, and even proteins, and multiple studies have shown that they are also associated with drug resistance (Yang et al. 2020; Yin et al. 2020). This also confirmed that intestinal microbiota plays an essential role in the host’s nutrition metabolism, disease immunity, and other aspects.
Conclusions
Fusobacteria, Firmicutes, Proteobacteria, and Actinobacteria dominate the intestinal microbiota in P. pekinensis. According to the structure of the intestinal microbiota, P. pekinensis is a typical herbivorous and partially omnivorous fish. Cetobacterium also has a very important proportion in its gut. Water environment, habitat, and feeding source have a significant influence on the intestinal microbiota structure, but there is no significant difference in different parts of the intestinal tract, and the F and B have a relative more similar microbiota structure.
Methods
Sampling and experimental design
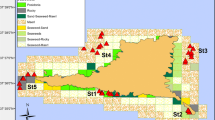
Between October 23 and 25, 2019, we collected a total of 73 P. pekinensis for this study. When sampling, we only collect fish from one habitat in 1 day for experiment, and the sampling sequence is Yangtze river population, outskirts river population, and breeding population. Among them (Fig. 5), 26 were from the Yangtze River (120° 12′ 53.31″ East, 31° 94′ 86.22″ North), 17 from a Suburban river (120° 15′ 19.25″ East, 32° 01′ 12.89″ North), and 30 2-year-old fish from a circulating aquaculture pond (120° 29′ 98.31″ East, 32° 12′ 92.06″ North). The breeding farm’s total breeding area is nearly 110,000 square meters, and the farmed P. pekinensis are raised on fry produced by wild parents. The three groups were situated far apart from each other, and there was no direct contact among the three groups in water. The fish taken from the breeding farm were sampled before feed to ensure that the intestines are not filled with feed, and they had neither been sick in the last 2 months nor treated with any drugs. During the sampling of each group, 15 water samples (3 for each location, at a depth of 1 m) were collected simultaneously from five locations within the range of 1000 square meters. For water samples, we first use a 2-L water sample collector to collect water at five different locations of the same sampling site, and fully mix the five collected water; then, a centrifuge tube is used to take 50 ml as a sample. The water quality of the three sampling sites were also detected (Table 1). The compound feed of 100 g, which was prepared by the farm (crude protein content was 30%) was also collected for the C group. For the feed samples, we randomly selected 5 bags of feed in the warehouse and weighed 1 kg respectively, and fully mix the five collected feeds, and then, 100 g was taken as a sample.

Map of the sampling sites
The experimental set up was designed in three parallel sets. Fish collected in the same habitat were maintained in three different groups, each containing 5–10 fish of similar size, and mixed as a whole sample to eliminate the differences between individuals (Table 2). The acquisition of intestinal microbiota was carried out on the workbench following aseptic technique throughout the procedure. First, the fish was placed on ice, and after abdominal disinfection (75% ethanol), the fish abdomen was cut open to straighten out the intestines. Then, the intestines were cut into three sections with a scalpel following the natural folding of the intestines, including the foregut, midgut, and hindgut (Wang et al. 2010; Oehlers et al. 2011). After the intestinal tract was dissected, the intestinal contents were rinsed with sterile normal saline and then placed in a sterile tube. Finally, samples were labeled and stored at − 80 °C until DNA extraction. In order to avoid cross-contamination between the samples, the dissection of the three groups was conducted by three different laboratory personnel on three different aseptic workbenches, using different dissecting scissors and scalpels for different intestinal sections. The water samples collected from five different locations of the same habitat were thoroughly mixed into a 50-ml aseptic centrifuge tube and then filtered with polyester cellulose filters (Millipore, Billerica, MA, USA); the size of pore was 0.2 μm. Filtrates from the same habitat were stored in the same tube, immediately filled with anhydrous ethanol, and stored at − 80 °C until DNA extraction. The feed samples were transferred into a sterile tube and stored at − 80 °C to be ground into powder in liquid nitrogen before DNA extraction.
DNA extraction, amplicon library preparation, and sequencing
DNA was extracted from the intestinal samples using the QIAamp DNA Stool Mini Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. Next, the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) was used to determine the concentration and purity of total DNA. PCR reactions were performed to amplify the V3-V4 region of bacterial 16S rRNA genes using primers: 341F,5′CCTAYGGGRBGCASCAG3′;806R, 5′GGACTACNNGGGTATCTAAT3′ (Hjelmso et al. 2014). Then the PCR products were visualized, purified (AxyPrep DNA Gel Extraction Kit, AxygenBiosciences, Union City, CA, USA), quantified (QuantiFluor™-ST, Promega, USA), and homogenized to form a DNA pool. Finally, the pooled products were sent to Novogene Co., Ltd. (Beijing, China) for paired-end sequencing with an Illumina HiSeq 2500 platform following the standard protocols. The sequencing data are deposited in the NCBI short read archive with the accession number PRJNA608608.
16S rRNA gene sequence and statistical analysis
After revealing the overlap relationship between paired-end reads (Caporaso et al. 2010), FLASH v1.2.7 software, Trimmomatic v0.33 software, and UCHIME v4.2 software were used to conduct quality filtering of the reads and the stitching effect, and finally, Effective Tags were obtained. UCLUST in QIIME (version 1.9.1) software was used to merge the Tags and classify OTU at a 97% similarity level (Edgar 2013). Then, the highest abundance sequence was selected as the representative sequence, which was used to annotate OTU based on Silva 132 (Quast et al. 2013; Yilmaz et al. 2014) taxonomy database. The Alpha diversity (Chao1 and Shannon), principal component analysis (PCA), and NMDS of the different groups were analyzed to evaluate the differences in the intestinal microbiota composition. Analysis of similarities (ANOSIM) was used to test the significance of the difference among defined categories. LEfSe (Segata et al. 2011) was used to analyze the significance of intra-group and inter-group differences. PICRUSt (Langille et al. 2013) was employed to predict the potential molecular functions of the 16S rRNA sequencing data based on the Kyoto Encyclopedia of Gene and Genomes (KEGG) database.
Availability of data and materials
The sequencing data are deposited in the NCBI Short Read Archive (SRA) with the accession numberPRJNA608608.
Abbreviations
- OTU:
-
Operational taxonomic unit
- NMDS:
-
Non-metricmulti-dimensional scaling
- W:
-
Fish from Yangtze River
- R:
-
Fish from Suburban River
- C:
-
Fish from aquaculture pond
- F:
-
Foregut
- M:
-
Midgut
- B:
-
Hindgut
References
Ashhurst DE (2001) Chitin and chitinases. Cell Biochem Funct 19:228–228
Askarian F, Zhou Z, Olsen RE, Sperstad S, Ringø E (2012) Culturable autochthonous gut bacteria in Atlantic salmon (Salmo salar L.) fed diets with or without chitin. Characterization by 16S rRNA gene sequencing, ability to produce enzymes and in vitro growth inhibition of four fish pathogens. Aquaculture 326-329:1–8
Aydın F, Çek-Yalnız Ş (2019) Effect of probiotics on reproductive performance of fish. Nat Eng Sci 4:153–162
Baldo L, Riera JL, Tooming-Klunderud A, Alba MM, Salzburger W (2015) Gut microbiota dynamics during dietary shift in Eastern African cichlid fishes. Plos One 10:e0127462
Banerjee G, Ray AK (2017) Bacterial symbiosis in the fish gut and its role in health and metabolism. Symbiosis 72:1–11
Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K (2006) Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297:374–386
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A 107:18933–18938
Butt RL, Volkoff H (2019) Gut microbiota and energy homeostasis in fish. Front Endocrinol (Lausanne) 10:9
Camp JG, Jazwa AL, Trent CM, Rawls JF (2012) Intronic cis-regulatory modules mediate tissue-specific and microbial control of angptl4/fiaf transcription. PLoS Genet 8:e1002585
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carnevali O, Maradonna F, Gioacchini G (2017) Integrated control of fish metabolism, wellbeing and reproduction: the role of probiotic. Aquaculture 472:144–155
Catalan N, Villasante A, Wacyk J, Ramirez C, Romero J (2018) Fermented soybean meal increases lactic acid bacteria in gut microbiota of Atlantic salmon (Salmo salar). Probiotics Antimicrob Proteins 10:566–576
Clements KD, Angert ER, Montgomery WL, Choat JH (2014) Intestinal microbiota in fishes: what's known and what's not. Mol Ecol 23:1891–1898
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Gomez GD, Balcazar JL (2008) A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol 52:145–154
He L, Hao B, Xie C, Luo X, Zhang Z, Zhu X (2010) Isolation and identification of major cellulase-producing fungi in intestine of grass carp*. Chinese J Apppl Environ Biol 2009:414–418
Hjelmso MH, Hansen LH, Baelum J, Feld L, Holben WE, Jacobsen CS (2014) High-resolution melt analysis for rapid comparison of bacterial community compositions. Appl Environ Microbiol 80:3568–3575
Jiang Y, Xie C, Yang G, Gong X, Chen X, Xu L, Bao B (2011) Cellulase-producing bacteria of Aeromonas are dominant and indigenous in the gut of Ctenopharyngodon idellus (Valenciennes). Aquaculture Res 42:499–505
Kim DH, Brunt J, Austin B (2007) Microbial diversity of intestinal contents and mucus in rainbow trout (Oncorhynchus mykiss). J Appl Microbiol 102:1654–1664
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821
Larsen AM, Mohammed HH, Arias CR (2014) Characterization of the gut microbiota of three commercially valuable warmwater fish species. J Appl Microbiol 116:1396–1404
Le, M.H., ; Wang, D. , 2020. Structure and membership of gut microbial communities in multiple fish cryptic species under potential migratory effects. Sci Rep 10, 1-12.
Li J, Ni J, Li J, Wang C, Li X, Wu S, Zhang T, Yu Y, Yan Q (2014) Comparative study on gastrointestinal microbiota of eight fish species with different feeding habits. J Appl Microbiol 117:1750–1760
Li X, Yan Q, Ringo E, Wu X, He Y, Yang D (2016) The influence of weight and gender on intestinal bacterial community of wild largemouth bronze gudgeon (Coreius guichenoti, 1874). BMC Microbiol 16:191
Liu H, Guo X, Gooneratne R, Lai R, Zeng C, Zhan F, Wang W (2016) The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci Rep 6:24340
Macdonald NL, SJ, Austin B, (1986) Bacterial microflora in the gastro-intestinal tract of dover sole (Solea solea L.), with emphasis on the possible role of bacteria in the nutrition of the host. FEMS Microbiol Lett 35:107–111
McDonald R, Schreier HJ, Watts JE (2012) Phylogenetic analysis of microbial communities in different regions of the gastrointestinal tract in Panaque nigrolineatus, a wood-eating fish. Plos One 7:e48018
Meng XL, Li S, Qin CB, Zhu ZX, Hu WP, Yang LP, Lu RH, Li WJ, Nie GX (2018) Intestinal microbiota and lipid metabolism responses in the common carp (Cyprinus carpio L.) following copper exposure. Ecotoxicol Environ Saf 160:257–264
Merrifield DL, Dimitroglou A, Bradley G, Baker RT, Davies SJ (2009) Soybean meal alters autochthonous microbial populations, microvilli morphology and compromises intestinal enterocyte integrity of rainbow trout, Oncorhynchus mykiss (Walbaum). J Fish Dis 32:755–766
Michl SC, Ratten JM, Beyer M, Hasler M, LaRoche J, Schulz C (2017) The malleable gut microbiome of juvenile rainbow trout (Oncorhynchus mykiss): Diet-dependent shifts of bacterial community structures. Plos One 12:e0177735
Ni J, Yan Q, Yu Y, Zhang T (2014) Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol Ecol 87:704–714
Oehlers SH, Flores MV, Chen T, Hall CJ, Crosier KE, Crosier PS (2011) Topographical distribution of antimicrobial genes in the zebrafish intestine. Dev Comp Immunol 35:385–391
Piazzon MC, Naya-Catala F, Simo-Mirabet P, Picard-Sanchez A, Roig FJ, Calduch-Giner JA, Sitja-Bobadilla A, Perez-Sanchez J (2019) Sex, age, and bacteria: how the intestinal microbiota is modulated in a protandrous hermaphrodite fish. Front Microbiol 10:2512
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Ray AK, Ghosh K, Ringø E (2012) Enzyme-producing bacteria isolated from fish gut: a review. Aquaculture Nutrition 18:465–492
Reda RM, Selim KM, El-Sayed HM, El-Hady MA (2018) In vitro selection and identification of potential probiotics isolated from the gastrointestinal tract of Nile tilapia, Oreochromis niloticus. Probiotics Antimicrob Proteins 10:692–703
Ringo E, Strom E (1994) Microflora of Arctic charr, salvelinus alpinus (L.): gastrointestinal microflora of free-living fish and effect of diet and salinity on intestinal microflora. Aquaculture Fisheries Manage 25:623–630
Sakata T, O.J, Kakimoto D. (1980) Variations in the intestinal microflora of Tilapia reared in fresh and sea water. Nihon-suisan-gakkai-shi 46:313–17
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60
Semova I, Carten JD, Stombaugh J, Mackey LC, Knight R, Farber SA, Rawls JF (2012) Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12:277–288
Silva FC, Nicoli JR, Zambonino-Infante JL, Kaushik S, Gatesoupe FJ (2011) Influence of the diet on the microbial diversity of faecal and gastrointestinal contents in gilthead sea bream (Sparus aurata) and intestinal contents in goldfish (Carassius auratus). FEMS Microbiol Ecol 78:285–296
Solovyev MM, Kashinskaya EN, Bochkarev NA, Andree KB, Simonov E (2019) The effect of diet on the structure of gut bacterial community of sympatric pair of whitefishes (Coregonus lavaretus): one story more. PeerJ 7:e8005
Sugita H, Ito Y (2006) Identification of intestinal bacteria from Japanese flounder (Paralichthys olivaceus) and their ability to digest chitin. Lett Appl Microbiol 43:336–342
Talwar C, Nagar S, Lal R, Negi RK (2018) Fish gut microbiome: current approaches and future perspectives. Indian J Microbiol 58:397–414
Tsuchiya C, Sakata T, Sugita H (2008) Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett Appl Microbiol 46:43–48
van Kessel MA, Dutilh BE, Neveling K, Kwint MP, Veltman JA, Flik G, Jetten MS, Klaren PH, Opden Camp HJ (2011) Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express 1:41. https://doi.org/10.1186/2191-0855-1-41
Vasquez EC, Pereira TMC, Campos-Toimil M, Baldo MP, Peotta VA (2019) Gut microbiota, diet, and chronic diseases: the role played by oxidative stress. Oxid Med Cell Longev 2019:7092032
Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ (2011) Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J 5:220–230
Wang AR, Ran C, Ringø E, Zhou ZG (2018) Progress in fish gastrointestinal microbiota research. Reviews in Aquaculture 10:626–640
Wang Z, Du J, Lam SH, Mathavan S, Matsudaira P, Gong Z (2010) Morphological and molecular evidence for functional organization along the rostrocaudal axis of the adult zebrafish intestine. BMC Genomics 11:392
Wei J, Guo X, Liu H, Chen Y, Wang W (2018) The variation profile of intestinal microbiota in blunt snout bream (Megalobrama amblycephala) during feeding habit transition. BMC Microbiol 18:99
Wong, S., ; Rawls, J. F. , 2012. Intestinal microbiota composition in fishes is influenced by host ecology and environment. Mol Ecol 21, 3100-3102.
Wu S, Wang G, Angert ER, Wang W, Li W, Zou H (2012) Composition, diversity, and origin of the bacterial community in grass carp intestine. Plos One 7:e30440
Yang K, Zhong Q, Qin H, Long Y, Ou H, Ye J, Qu Y (2020) Molecular response mechanism in Escherichia coli under hexabromocyclododecane stress. Sci Total Environ 708:135199
Yi Y, Yang Z, Zhang S (2010) Ecological influence of dam construction and river-lake connectivity on migration fish habitat in the Yangtze River basin, China. Procedia Environmental Sciences 2:1942–1954
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glockner FO (2014) The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42:D643–D648
Yin S, Zhang J, Guo F, Poma G, Covaci A, Liu W (2020) Transplacental transfer mechanism of organochlorine pesticides: an in vitro transcellular transport study. Environ Int 135:105402
Yu Y, Raka F, Adeli K (2019) The Role of the Gut Microbiota in Lipid and Lipoprotein Metabolism. J Clin Med 8. https://doi.org/10.3390/jcm8122227
Yuan XY, Liu WB, Wang CC, Huang YY, Dai YJ, Cheng HH, Jiang GZ (2020) Evaluation of antioxidant capacity and immunomodulatory effects of cottonseed meal protein hydrolysate and its derivative peptides for hepatocytes of blunt snout bream (Megalobrama amblycephala). Fish Shellfish Immunol 98:10–18
Yukgehnaish, K., Kumar, P., Sivachandran, P., Marimuthu, K., Arshad, A., Paray, B.A., Arockiaraj, J., 2020. Gut microbiota metagenomics in aquaculture: factors influencing gut microbiome and its physiological role in fish. Reviews in Aquaculture.
Zeng A, Tan K, Gong P Lei P, Guo ZH, Wang SP, Gao SF, Zhou YH, Shu Y, Zhou, XL, Miao D, Zeng FJ, Liu HZ (2020) Correlation of microbiota in the gut of fish species and water. 3 Biotech 10. https://doi.org/10.1007/s13205-020-02461-5
Acknowledgements
We would like to thank the members of Shanghai Ocean University and Taizhou Institute of Agricultural Sciences for their help. We also thank Novogene Co., Ltd. (Beijing, China) for providing sequencing services. We are grateful to Editage (www.editage.cn) for English language editing.
Funding
Jiangsu Agricultural Science and Technology Independent Innovation Fund “Study on the key technology of artificial larger-scale seedling breeding of Parabramis pekinensis” [No. CX(19)3011].
Author information
Authors and Affiliations
Contributions
HLG and YMF conceived and designed the study. HLG, YZ, DHY, and ZJY collected the fish and prepared the samples. HLG drafted the manuscript. YMF performed the manuscript revision. WQT critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed were in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Experimental Animal Ethics Committee of Shanghai Ocean University (Shanghai, China). Consent to participate is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Rarefaction curve: Estimating the richness (at a 97%similarity level) of intestinal microbiota.
Additional file 2.
Shannon curves: Determining whether the sequencing depth can reflect the majority of microbial species information in samples.
Additional file 3.
Species accumulation curve: Determining whether the number of sequenced samples is sufficient.
Additional file 4
Phylum-level gut microbiota composition of Parabramis pekinensis shows the abundance of bacteria in samples from different habitats.
Additional file 5
Variation in the α-diversity of the gut microbiota between different habitats of Parabramis pekinensis. TheChao1 richness estimator is presented. Statistically significant differences are indicated with an asterisk, P < 0.05 (*), P < 0.01 (**).
Additional file 6
Variation in the α-diversity of the gut microbiota between different habitats of Parabramis pekinensis. The Shannon diversity index is presented. Statistically significant differences are indicated with an asterisk, P < 0.05 (*), P < 0.01 (**).
Additional file 7.
Difference in the intestinal microbiota between the different habitats. Significantly discriminative taxa with absolute LDA score ≥ 2.0.
Additional file 8
Phylum-level gut microbiota composition of Parabramis pekinensis shows the abundance of bacteria in different sections of the intestine.
Additional file 9
Variation in α-diversity of the gut microbiota in different sections of the intestine of Parabramis pekinensis. TheChao1 richness estimator is presented. Statistically significant differences are indicated with an asterisk, P < 0.05 (*), P < 0.01 (**).
Additional file 10
Variation in the α-diversity of the gut microbiota in different sections of the intestine of Parabramis pekinensis. The Shannon diversity index is presented. Statistically significant differences are indicated with an asterisk, P < 0.05 (*), P < 0.01 (**).
Additional file 11.
Difference in intestinal microbiota between the different sections of the intestine. Significantly discriminative taxa with absolute LDA score ≥ 2.0.
Additional file 12.
Functional predictions of intestinal microbiota using PICRUSt, showing the 50 dominant pathways(level1 and level3 KEGG orthology groups).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gu, H., Feng, Y., Zhang, Y. et al. Differential study of the Parabramis pekinensis intestinal microbiota according to different habitats and different parts of the intestine. Ann Microbiol 71, 5 (2021). https://doi.org/10.1186/s13213-020-01614-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01614-4