
Abstract
Background
Approximately a third of frontotemporal dementia (FTD) is genetic with mutations in three genes accounting for most of the inheritance: C9orf72, GRN, and MAPT. Impaired synaptic health is a common mechanism in all three genetic variants, so developing fluid biomarkers of this process could be useful as a readout of cellular dysfunction within therapeutic trials.
Methods
A total of 193 cerebrospinal fluid (CSF) samples from the GENetic FTD Initiative including 77 presymptomatic (31 C9orf72, 23 GRN, 23 MAPT) and 55 symptomatic (26 C9orf72, 17 GRN, 12 MAPT) mutation carriers as well as 61 mutation-negative controls were measured using a microflow LC PRM-MS set-up targeting 15 synaptic proteins: AP-2 complex subunit beta, complexin-2, beta-synuclein, gamma-synuclein, 14–3-3 proteins (eta, epsilon, zeta/delta), neurogranin, Rab GDP dissociation inhibitor alpha (Rab GDI alpha), syntaxin-1B, syntaxin-7, phosphatidylethanolamine-binding protein 1 (PEBP-1), neuronal pentraxin receptor (NPTXR), neuronal pentraxin 1 (NPTX1), and neuronal pentraxin 2 (NPTX2). Mutation carrier groups were compared to each other and to controls using a bootstrapped linear regression model, adjusting for age and sex.
Results
CSF levels of eight proteins were increased only in symptomatic MAPT mutation carriers (compared with controls) and not in symptomatic C9orf72 or GRN mutation carriers: beta-synuclein, gamma-synuclein, 14–3-3-eta, neurogranin, Rab GDI alpha, syntaxin-1B, syntaxin-7, and PEBP-1, with three other proteins increased in MAPT mutation carriers compared with the other genetic groups (AP-2 complex subunit beta, complexin-2, and 14–3-3 zeta/delta). In contrast, CSF NPTX1 and NPTX2 levels were affected in all three genetic groups (decreased compared with controls), with NPTXR concentrations being affected in C9orf72 and GRN mutation carriers only (decreased compared with controls). No changes were seen in the CSF levels of these proteins in presymptomatic mutation carriers. Concentrations of the neuronal pentraxins were correlated with brain volumes in the presymptomatic period for the C9orf72 and GRN groups, suggesting that they become abnormal in proximity to symptom onset.
Conclusions
Differential synaptic impairment is seen in the genetic forms of FTD, with abnormalities in multiple measures in those with MAPT mutations, but only changes in neuronal pentraxins within the GRN and C9orf72 mutation groups. Such markers may be useful in future trials as measures of synaptic dysfunction, but further work is needed to understand how these markers change throughout the course of the disease.
Similar content being viewed by others

Background
Frontotemporal dementia (FTD) is the most common cause of dementia affecting people under the age of 60. Clinically, it presents heterogeneously, manifesting as a behavioural variant (bvFTD), language impairment (primary progressive aphasia, PPA), or with a motor presentation (either amyotrophic lateral sclerosis, FTD-ALS, or an atypical parkinsonian disorder). The FTD spectrum is characteristically associated with neuronal dysfunction and loss in the frontal and temporal lobes, but more widespread cortical, subcortical, cerebellar, and brainstem involvement is now also recognized [1]. Around a third of people with FTD have a genetic cause, with the most common mutations occurring in three genes: GRN (progranulin), C9orf72 (chromosome 9 open reading frame 72), and MAPT (microtubule-associated protein tau) [2, 3]. Lastly, the underlying pathology of FTD can be one of three forms: cellular inclusions containing abnormal forms of tau, TAR DNA-binding protein 43 (TDP-43), or FET proteins (fused in sarcoma (FUS), Ewing sarcoma (EWS), and TATA-binding associated factor 15 (TAF15)) [4].
The interaction between clinical phenotype, neuroanatomical features, genotype, and pathology is complex and means that FTD can be hard to diagnose (particularly its specific pathological form during life) and difficult to track over time. To further examine some of these outstanding issues in the FTD field, researchers have aimed to develop fluid biomarkers, measured typically in the cerebrospinal fluid (CSF), serum, or plasma using a variety of different techniques. Biomarkers can provide an insight into the underlying pathophysiology of FTD and in the context of clinical trials could offer a direct experimental medicine approach to understanding the molecular mechanisms through measurement of biofluids pre- and post-intervention [5].
Whilst some pathways are specific to certain pathogenetic forms of FTD, studies in recent years have particularly highlighted the importance of synaptic health [6,7,8,9,10] as one of the major pathophysiological mechanisms across the FTD spectrum. Progressive synaptic dysfunction and loss have been shown to occur in FTD, raising the hypothesis that any changes in synaptic proteins in brain tissue may also be reflected in their concentrations within the CSF (and potentially the blood) of people with FTD. In this study, we investigated a panel of CSF synaptic markers in presymptomatic and symptomatic people with genetic FTD from the GENetic Frontotemporal dementia Initiative (GENFI), hypothesizing that we would find differential abnormalities across MAPT, GRN, and C9orf72 mutation carriers.
Methods
Participants and sample collection
Participants were recruited from the GENFI study, which follows patients with FTD due to a pathogenic mutation in MAPT, GRN, or C9orf72 (symptomatic mutation carriers) and healthy at-risk first-degree relatives (either presymptomatic mutation carriers or non-carriers) [11]. We included 77 presymptomatic mutation carriers (31 C9orf72, 23 GRN, 23 MAPT), 55 symptomatic mutation carriers (26 C9orf72, 17 GRN, 12 MAPT), and 61 non-carriers. Age at the time of CSF sample collection was not statistically different within each group, and a similar percentage of males and females was included (Table 1). Participants were assessed using a standardized history and examination and classified as symptomatic if they met consensus diagnostic criteria [12, 13]. The CDR Dementia Staging Instrument with National Alzheimer Coordinating Centre Frontotemporal Lobar Degeneration component (CDR® plus NACC FTLD) was used to assess disease severity. Local ethics committees at each site approved the study, and all participants provided written informed consent.
CSF collection and LC–MS/MS analysis
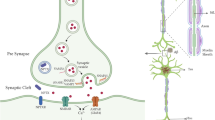
CSF was collected in polypropylene tubes through a lumbar puncture and centrifuged to remove insoluble material and cells. Supernatants were aliquoted and stored at − 80 °C within 2 h after withdrawal. For the mass spectrometry analysis, sample preparation was performed as described previously [14]. Briefly, to 100 µL of CSF, a mixture of stable isotope-labeled peptides (internal standard) was added (25 µL, 0.032 pmol/µL, JPT Peptide Technologies, Berlin, Germany; SpikeTides L). This was then followed by a stepwise protocol of reduction, alkylation, and tryptic digestion and lastly solid-phase extraction for purification purposes (for detailed sample preparation, refer to Additional file 3: Appendix 2). LC–MS/MS analysis was performed using a microflow HPLC, equipped with a Hypersil Gold reversed-phase column (100 × 2.1 mm, particle size 1.9 µm, Thermo Fisher Scientific), and a Triple Quadrupole Mass Spectrometer (6495 Triple Quadrupole LC/MS system, Agilent Technologies). LC–MS settings are shown in Additional file 3: Appendix 2. To monitor the performance of the assay over time, quality control (QC) sample replicates were injected at regular intervals during runs. The panel of synaptic markers included (Fig. 1) AP-2 complex subunit beta, complexin-2, beta-synuclein, gamma-synuclein, 14–3-3 proteins (eta, epsilon, zeta/delta), neurogranin, Rab GDP dissociation inhibitor alpha (Rab GDI alpha), syntaxin-1B, syntaxin-7, phosphatidylethanolamine-binding protein 1 (PEBP-1), neuronal pentraxin receptor (NPTXR), neuronal pentraxin 1 (NPTX1), and neuronal pentraxin 2 (NPTX2). Table 2 shows the proteins and their respective proteotypic peptides targeted in the multiple reaction monitoring mass spectrometry analysis [14] as well as their analytical performance. For the proteins for which more than one peptide was quantified, the peptide with the best analytical performance (lowest coefficient of variation) is discussed in the main manuscript and shown in Fig. 2.

Diagrammatic representation of the synapse and the role of the different synaptic proteins included within the mass spectrometry panel. Adapted from Nilsson et al. [14]

Cerebrospinal fluid (CSF) concentrations of the synaptic panel proteins in the GENFI cohort including 23 presymptomatic MAPT (PS MAPT), 31 C9orf72 (PS C9), and 23 GRN (PS GRN) mutation carriers and 12 symptomatic MAPT (S MAPT), 26 C9orf72 (S C9), and 17 GRN (S GRN) mutation carriers and 61 non-carriers. The results are shown in fmol/μL. p-values: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. The bars indicate the median and the IQR. Only one peptide per protein is shown as discussed in the “Methods” section. Specific means, IC, and p-values are shown in Additional file 1: Table S2
Other biomarkers
Participants underwent volumetric T1-weighted magnetic resonance imaging according to the harmonized GENFI protocol on a 3T scanner. All images underwent a quality control check, and scans with movement or artefacts were removed from the analysis. Only scans from mutation carriers were included in the correlative analysis: of the 132 participants, 111 scans were available for the analysis: 49 C9orf72, 34 GRN, and 28 MAPT mutation carriers. Neuroanatomical regions of interest were generated as previously described using an automated atlas segmentation propagation and label fusion strategy called geodesic information flow [11]. Specifically, total brain volume and volumes of the frontal, temporal, and parietal cortices were calculated and expressed as a percentage of total intracranial volume (TIV), computed with SPM12 (Statistical Parametric Mapping, Welcome Trust Centre for Neuroimaging, London, UK) running under Matlab R2014b.
Participants also had plasma samples collected as part of the GENFI protocol. Plasma was collected, processed, and stored in aliquots at − 80 °C according to standardized procedures. Plasma neurofilament light chain (NfL) levels were correlated with synaptic proteins with only measures from mutation carriers included: of 132 participants, 108 plasma NfL values were available for the analysis: 47 C9orf72, 34 GRN, and 27 MAPT mutation carriers. Plasma NfL concentration was measured with single molecule array (Simoa) technology using the Neurology 4-Plex A kit (Quanterix, Billerica, USA) on an HD-X Analyzer following the manufacturer’s instructions (Quanterix, Billerica, USA). Measurements were completed in duplicate (all CVs below 15%) over a total of 3 batches, each with an 8-point calibration curve tested in triplicate and 2 controls tested in duplicate, as reported before [15].
Data processing and statistical analysis
Mass spectrometer data processing was performed in Skyline 20.1 (MacCoss Lab Software). All peaks were visually inspected and adjusted if required for optimal peak area calculation. The relative peptide concentration (fmol/µL) was obtained by the ratio of the total area for each peptide against the total area of the corresponding internal standard (IS) multiplied by the amount of IS added per volume of CSF.
All statistical analyses were performed in STATA (v.16) and RStudio (R version 4.0.2). The Shapiro–Wilk test was performed to determine the normality of distribution of each synaptic marker in each group. The levels of each synaptic protein were compared between the groups using a linear regression model adjusting for age at CSF sample collection and sex; bootstrapping with 2000 repetitions was used if the synaptic measures were not normally distributed.
Spearman correlation coefficients were assessed for the synaptic markers between their values and other biomarker data including normalized volumes of total brain, frontal cortex, temporal cortex, and parietal cortex; plasma NfL; and the CDR plus NACC FTLD sum of boxes score.
Results
Concentrations of synaptic markers by genotype
Significant increases in CSF levels of several synaptic proteins were seen in symptomatic MAPT mutation carriers compared with controls (Fig. 2, Additional file 1: Table S1): beta-synuclein, gamma-synuclein, 14–3-3 eta, neurogranin, Rab GDI alpha, syntaxin 1B, syntaxin-7, and PEBP-1. CSF levels of all of these proteins except 14–3-3 eta were increased in the symptomatic MAPT group compared with the symptomatic C9orf72 group (as was AP-2 complex subunit beta). Similarly, levels of all of these proteins except 14–3-3 eta and both beta and gamma-synuclein were increased in the symptomatic MAPT group compared with the symptomatic GRN group (as was complexin-2). Furthermore, 14–3-3 zeta/delta was additionally increased in the symptomatic MAPT group compared with both C9orf72 and GRN symptomatic mutation carriers. CSF levels of all of these proteins except syntaxin-7 were increased in the symptomatic MAPT mutation carriers compared with the presymptomatic MAPT mutation carriers (Fig. 2).
In contrast, CSF concentrations of the neuronal pentraxins were found to be decreased in most of the mutation carrier groups compared with controls. At least one of the peptides measured for NPTXR, NPTX1, and NPTX2 were decreased in symptomatic C9orf72 and GRN mutation carriers compared to controls and to their respective presymptomatic group. One NPTX1 and one NPTX2 peptide were also decreased in the symptomatic MAPT group compared to controls and the presymptomatic MAPT mutation carriers (Fig. 2).
Correlations of synaptic markers with other biomarkers
For the synaptic markers that had increased CSF concentrations in MAPT mutation carriers, no significant correlations were seen with brain volumes, NfL, or CDR plus NACC FTLD.
However, a number of significant correlations were seen with the CSF levels of neuronal pentraxins across the genetic groups (Table 3). In presymptomatic C9orf72 mutation carriers, there were significant positive correlations of total brain volume with NPTXR and NPTX2 (r = 0.42 and 0.38, respectively). Additionally, there were significant positive correlations in this group with temporal cortex volume for NPTXR (r = 0.50, p = 0.006) and NPTX2 (0.49, 0.007) and with parietal cortex volume for NPTX1 (0.41, 0.029). Two of the neuronal pentraxins (NPTXR and NPTX2) were significantly negatively correlated with CDR plus NACC FTLD. In the presymptomatic GRN group, there were significant positive correlations with the frontal lobe (r = 0.52 to 0.53) and parietal lobe (r = 0.45 to 0.59) for almost all of the measures. There were no correlations with any of the imaging measures in the presymptomatic MAPT group, but there was a significant negative correlation with NfL for NPTX1 (r = − 0.46, p = 0.040).
In the symptomatic C9orf72 group, NPTXR was significantly negatively correlated with NfL concentration (r = − 0.68), whilst in the symptomatic GRN group, NPTXR and NPTX2 positively correlated with both frontal (r = 0.73 and 0.80, respectively) and temporal (r = 0.77 and 0.65, respectively) lobe volumes. In the symptomatic MAPT mutation carriers, there was a significant positive correlation of total brain volume with NPTX2 (r = 0.95, p = 0.004).
Discussion
In this study, we showed an increased CSF concentration of multiple synaptic markers in symptomatic MAPT mutation carriers. In contrast, concentrations of the neuronal pentraxins were decreased in all three symptomatic genetic groups. Although no group-wise differences in CSF levels were seen presymptomatically, correlations with brain volumes in the C9orf72 and GRN groups suggest that the neuronal pentraxins change in the lead up to symptom onset as the brain volume starts to decrease.
For the proteins found to have abnormal CSF levels in MAPT mutations, little is known previously about their involvement in the pathophysiology of FTD. Beta- and gamma-synucleins are present in the proteinaceous aggregates characteristic of the alpha-synucleinopathies [16] although their normal function is still unclear. Previous studies have shown an increase in these markers in the CSF of people with Alzheimer’s disease (AD) [14, 17, 18], but in one prior study of beta-synuclein in the CSF of people with undifferentiated FTD, the levels were normal [17]. The results in this study therefore represent a novel association with MAPT mutations.
14–3-3 proteins are highly expressed in the brain, particularly enriched in the presynaptic site and are implicated in synaptic plasticity by acting as modulators of neurotransmission [19]. Although they are established biomarkers for Creutzfeldt-Jakob disease, they have also been genetically linked to AD and found to colocalize with tau in the neurofibrillary tangles as well as in Lewy bodies in Parkinson’s disease [20]. Furthermore, increased levels of 14–3-3 protein have previously been reported in CSF from people with FTD (not differentiated into a specific form) in a single study, as well as in people with AD [14, 21]. Here, we show increased levels of 14–3-3 eta protein in the symptomatic MAPT mutation carriers when compared with the non-carrier group, and for 14–3-3 zeta/delta, when compared with the other symptomatic groups. These results could potentially indicate a specific relationship with tau pathology, related to the deposition in neurofibrillary tangles of 14–3-3 proteins. However, there are also trends to an increase in some of the 14–3-3 proteins in GRN mutations, so genotype differences may not be as clear here.
Neurogranin has been well-studied in the AD field as a fluid biomarker over recent years [22, 23]. It is a postsynaptic molecule involved in long-term potentiation and synaptic plasticity mediated by Ca2+ and calmodulin signalling pathways [24, 25]. In CSF, neurogranin shows an increase in people with AD compared with controls [14, 26,27,28]. Furthermore, increased concentrations of neurogranin in CSF predict cognitive decline from mild cognitive impairment (MCI) to AD [29, 30]. In a previous study, FTD levels of neurogranin were not significantly different to controls [31], although when stratified into those with tau and TDP-43 pathology, there was a trend for an increase in the tau group. A further study has also shown that neurogranin was significantly decreased in comparison with controls in plasma exosomes from people with FTD [32]. In this study, we show an increase in neurogranin levels in symptomatic MAPT mutation carriers, again suggesting a specific relationship with tau pathology.
AP-2 complex subunit beta, the syntaxins, Rab GDI alpha, and PEBP-1 are all implicated in the process of synapse vesicle exocytosis and neurotransmitter release at the synaptic cleft, and their CSF levels have previously been shown to be abnormal in AD [14, 33,34,35,36]. Syntaxins participate in the formation of the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) complex, where they participate in synapse vesicle exocytosis together with complexin-2, which modulates the function of the SNARE complex [33, 37]. The levels of syntaxin 1B have been shown to be increased at an early preclinical stage in the CSF of people likely to develop AD, even before core CSF biomarkers for neurodegeneration [34]. None of these proteins has been previously studied in FTD, but given their increase also in AD, it may be that these are all tau-specific markers of synaptic dysfunction, and further study in other primary tauopathies would be important.
Finally, we showed changes across all three genetic groups in the neuronal pentraxins. Pentraxins are multifunctional proteins divided into different groups according to their length. They are not exclusively localized in the central nervous system (CNS) and are involved in the inflammatory response as well as synaptic plasticity among other functions [35]. The sub-family of neuronal pentraxins includes the soluble neuronal pentraxins 1 (NPTX1) and 2 (NPTX2) and the transmembrane neuronal pentraxin receptor (NPTXR). NPTXs are implicated in synaptic plasticity, synapse formation, and remodelling [36]. Both the two secreted NPTX1 and NPTX2 and the transmembrane receptor NPTXR have been found in several studies to be decreased in CSF in AD compared with controls [14, 38,39,40,41] and appear to be markers of disease progression in AD [42,43,44]. Recent proteomic studies have shown decreased levels of NPTXR in symptomatic genetic FTD, in all three genetic groups [45], and in sporadic bvFTD and PPA [46]. Two further studies additionally showed that NPTX2 was decreased in symptomatic mutation carriers in all three groups compared with controls using antibody-based approaches [47, 48], and one of these studies showed that NPTX1 was decreased in C9orf72 and MAPT mutation carriers [48]. In our study, NPTX1 and NPTX2 were significantly decreased in all symptomatic groups, but NPTXR was only decreased in C9orf72 and GRN mutation carriers. In one prior study, levels of NPTX2 in CSF correlated with disease progression, with the suggestion also that NPTX2 levels change just prior to symptom onset [48, 49]. The correlations of the neuronal pentraxins with brain volumes in the presymptomatic C9orf72 and GRN mutation carriers suggest that for these two groups, NPTXR, NPTX1, and NPTX2 change in proximity to symptom onset as brain volumes start to decrease. In the symptomatic C9orf72 expansion carriers, the neuronal pentraxin levels correlated with NfL, which can be variable in concentration in this group [50]. However, it suggests that at least in C9orf72 expansion carriers, the neuronal pentraxins may be a measure of disease intensity and speed of progression. In contrast, in the GRN and MAPT mutation carriers, neuronal pentraxin concentrations correlated with brain volumes, suggesting that here they may be a measure of disease severity rather than intensity.
Limitations
Limitations of the study include the limited number of CSF samples in each group after stratification. However, this is the largest study so far of synaptic biomarkers in this uncommon disease and replicates prior work on neuronal pentraxins. Further work to replicate the findings in other MAPT mutation cohorts (and other primary tauopathies) as well as longitudinal analysis within the GENFI cohort will be important. Lastly, the specific synaptic markers panel used here was designed specifically to target AD pathology based on prior research and selected from a broad proteomic study in an AD cohort [51]. It may therefore be that this panel was more likely to find abnormalities in tauopathies and that other synaptic proteins not included in the panel might be better biomarkers for assessing synaptic dysfunction in FTD, particularly in those with TDP-43 pathology.
Conclusion
In this study, we show differential involvement of synaptic proteins in the three main genetic groups accounting for familial FTD. Our results suggest that different pathways may be related to synaptic health in relation to the underlying proteinopathy found in each mutation. Future studies will focus on replication of these findings, longitudinal analyses of these measures, and a broader proteomic study to better customise a synaptic biomarker panel targeted to different forms of FTD.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- FTD:
-
Frontotemporal dementia
- GENFI:
-
GENetic Frontrotemporal dementia Initiative
- bvFTD:
-
Behavioural variant FTD
- PPA:
-
Primary progressive aphasias
- FTD-ALS:
-
FTD with amyotrophic lateral sclerosis
- GRN :
-
Progranulin gene
- C9orf72 :
-
Chromosome 9 open reading frame 72 gene
- MAPT :
-
Microtubule-associated protein tau gene
- TDP-43:
-
TAR DNA-binding protein 43
- FET:
-
FUS, EWS, and TAF15 proteins
- FUS:
-
Fused in sarcoma
- EWS:
-
Ewing sarcoma
- TAF15:
-
TATA-binding associated factor 15
- CSF:
-
Cerebrospinal fluid
- CDR® plus NACC FTLD:
-
CDR Dementia Staging Instrument with National Alzheimer Coordinating Centre Frontotemporal Lobar Degeneration Component
- LC–MS/MS:
-
Liquid chromatography-mass spectrometry/mass spectrometry
- HPLC:
-
High-pressure liquid chromatography
- Rab GDI alpha:
-
Rab GDP dissociation inhibitor alpha
- PEBP-1:
-
Phosphatidylethanolamine-binding protein 1
- NPTXR:
-
Neuronal pentraxin receptor
- NPTX1:
-
Neuronal pentraxin 1
- NPTX2:
-
Neuronal pentraxin 2
- SPM12:
-
Statistical Parametric Mapping
- NfL:
-
Neurofilament light chain
- IS:
-
Internal standard
- AD:
-
Alzheimer’s disease
- MCI:
-
Mild cognitive impairment
- SNARE:
-
Soluble N-ethylmaleimide-sensitive factor attachment receptor
- CNS:
-
Central nervous system
References
Rohrer JD, Rosen HJ. Neuroimaging in frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):221–9.
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurol. 2009;73(18):1451–6.
Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075–86.
Mackenzie IRA, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochemistry. 2016;138:54–70.
Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harbor Perspect Med. 2012;2(9):a006221.
Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45(2):711–22.
Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, et al. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathologica Commun. 2014;1:34.
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–35.
Jiang S, Wen N, Li Z, Dube U, del Aguila J, Budde J, et al. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry. 2018;8(1):265.
Starr A, Sattler R. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018;1693:98–108.
Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14(3):253–62.
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–77.
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14.
Nilsson J, Gobom J, Sjödin S, Brinkmalm G, Ashton NJ, Svensson J, et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement (Amst). 2021;13(1):e12179.
Heller C, Foiani MS, Moore K, Convery R, Bocchetta M, Neason M, et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(3):263–70.
Galvin JE, Uryu K, Lee VMY, Trojanowski JQ. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc Natl Acad Sci U S A. 1999;96(23):13450–5.
Oeckl P, Metzger F, Nagl M, von Arnim CAF, Halbgebauer S, Steinacker P, et al. Alpha-, beta-, and gamma-synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in Alzheimer’s and Creutzfeldt-Jakob disease but no alteration in synucleinopathies. Mol Cell Proteomics. 2016;15(10):3126–38.
Oeckl P, Halbgebauer S, Anderl-Straub S, von Arnim CAF, Diehl-Schmid J, Froelich L, et al. Targeted mass spectrometry suggests beta-synuclein as synaptic blood marker in Alzheimer’s disease. J Proteome Res. 2020;19(3):1310–8.
Zhang J, Zhou Y. 14–3–3 proteins in glutamatergic synapses. Neural Plasticity. 2018;2018:8407609.
Foote M, Zhou Y. 14-3-3 proteins in neurological disorders. Int J Biochem Mol Biol. 2012;3(2):152.
Antonell A, Tort-Merino A, Ríos J, Balasa M, Borrego-Écija S, Auge JM, et al. Synaptic, axonal damage and inflammatory cerebrospinal fluid biomarkers in neurodegenerative dementias. Alzheimer’s & Dementia. 2020;16(2):262–72.
Xiang Y, Xin J, Le W, Yang Y. Neurogranin: a potential biomarker of neurological and mental diseases. Front Aging Neurosci. 2020;6:12.
Camporesi E, Nilsson J, Brinkmalm A, Becker B, Ashton NJ, Blennow K, et al. Fluid biomarkers for synaptic dysfunction and loss. Biomarker Insights. 2020;15:1177271920950319.
Gerendasy DD, Sutcliffe JG. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol. 1997;15(2):131–63.
Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28(19):3027.
de Vos A, Jacobs D, Struyfs H, Fransen E, Andersson K, Portelius E, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015;11(12):1461–9.
Tarawneh R, D’Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73(5):561.
Lista S, Toschi N, Baldacci F, Zetterberg H, Blennow K, Kilimann I, et al. Cerebrospinal fluid neurogranin as a biomarker of neurodegenerative diseases: a cross-sectional study. J Alzheimers Dis. 2017;59(4):1327–34.
Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Öhrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dementia. 2015;11(10):1180–90.
Sanfilippo C, Forlenza O, Zetterberg H, Blennow K. Increased neurogranin concentrations in cerebrospinal fluid of Alzheimer’s disease and in mild cognitive impairment due to AD. J Neural Transm (Vienna). 2016;123(12):1443–7.
Clarke MTM, Brinkmalm A, Foiani MS, Woollacott IOC, Heller C, Heslegrave A, et al. CSF synaptic protein concentrations are raised in those with atypical Alzheimer’s disease but not frontotemporal dementia. Alzheimer’s Res Ther. 2019;11(1):105.
Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach Iv, Goetzl L, Abner EL, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30(12):4141–8.
Trimbuch T, Rosenmund C. Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci. 2016;17(2):118–25.
Lleó A, Núñez-Llaves R, Alcolea D, Chiva C, Balateu-Paños D, Colom-Cadena M, et al. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid*. Mol Cell Proteomics. 2019;18(3):546–60.
Osera C, Pascale A, Amadio M, Venturini L, Govoni S, Ricevuti G. Pentraxins and Alzheimer’s disease: at the interface between biomarkers and pharmacological targets. Ageing Res Rev. 2012;11(2):189–98.
Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS. Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J Biol Chem. 2000;275(23):17786–92.
Mishima T, Fujiwara T, Sanada M, Kofuji T, Kanai-Azuma M, Akagawa K. Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PLoS ONE. 2014;9(2):e90004.
Galasko D, Xiao M, Xu D, Smirnov D, Salmon DP, Dewit N, et al. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimers Dement. 2019;5:871.
Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, et al. NPTX2 and cognitive dysfunction in Alzheimer’s disease. Elife. 2017;25(6):1–27.
Soldan A, Moghekar A, Walker KA, Pettigrew C, Hou X, Lu H, et al. Resting-state functional connectivity is associated with cerebrospinal fluid levels of the synaptic protein NPTX2 in non-demented older adults. Front Aging Neurosci. 2019;11:132.
Brinkmalm G, Sjödin S, Simonsen AH, Hasselbalch SG, Zetterberg H, Brinkmalm A, et al. A parallel reaction monitoring mass spectrometric method for analysis of potential CSF biomarkers for Alzheimer’s disease. Proteomics Clin Appl. 2018;12(1).
Swanson A, Willette AA. Neuronal Pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer’s disease spectrum. Brain Behav Immun. 2016;1(58):201–8.
Lim B, Sando SB, Grøntvedt GR, Bråthen G, Diamandis EP. Cerebrospinal fluid neuronal pentraxin receptor as a biomarker of long-term progression of Alzheimer’s disease: a 24-month follow-up study. Neurobiol Aging. 2020;1(93):97.e1-97.e7.
Begcevic I, Tsolaki M, Brinc D, Brown M, Martinez-Morillo E, Lazarou I, et al. Neuronal pentraxin receptor-1 is a new cerebrospinal fluid biomarker of Alzheimer’s disease progression. F1000Res. 2018;7:1012.
van der Ende EL, Meeter LH, Stingl C, van Rooij JGJ, Stoop MP, Nijholt DAT, et al. Novel CSF biomarkers in genetic frontotemporal dementia identified by proteomics. Ann Clin Transl Neurol. 2019;6(4):698–707.
Remnestål J, Öijerstedt L, Ullgren A, Olofsson J, Bergström S, Kultima K, et al. Altered levels of CSF proteins in patients with FTD, presymptomatic mutation carriers and non-carriers. Transl Neurodegener. 2020;9(1):27.
Bergström S, Öijerstedt L, Remnestål J, Olofsson J, Ullgren A, Seelaar H, et al. A panel of CSF proteins separates genetic frontotemporal dementia from presymptomatic mutation carriers: a GENFI study. Mol Neurodegener. 2021;16(1):79.
van der Ende EL, Xiao M, Xu D, Poos JM, Panman JL, Jiskoot LC, et al. Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(6):612-21.
van der Ende EL, Bron EE, Poos JM, Jiskoot LC, Panman JL, Papma JM, et al. A data-driven disease progression model of fluid biomarkers in genetic frontotemporal dementia. Brain. 2022;145(5):1805-17.
Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3(8):623–36.
Tijms B, Gobom J, Reus L, Jansen I, Hong S, Dobricic V, et al. Pathophysiological subtypes of Alzheimer’s disease based on cerebrospinal fluid proteomics. Brain. 2020;143(12):3776-92.
Acknowledgements
We thank the research participants for their contribution to the study.
Consortium members are shown in Additional file 2: Appendix 1.
Funding
The Dementia Research Centre is supported by Alzheimer’s Research UK, Alzheimer’s Society, Brain Research UK, and The Wolfson Foundation. This work was supported by the NIHR UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK. JDR is supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). This work was also supported by the MRC UK GENFI grant (MR/M023664/1), the Bluefield Project, and the JPND GENFI-PROX grant (2019–02248). Several authors of this publication are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510. ASE is supported by Race Against Dementia Fellowship by Alzheimer’s Research UK (ARUK-RADF2021A-003). ASE’s work is also supported by the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK. IS is supported by funding from the Alzheimer’s Society, UK. RC is supported by a Frontotemporal Dementia Research Studentships in Memory of David Blechner funded through The National Brain Appeal (RCN 290173). JCVS was supported by the Dioraphte Foundation grant 09–02-03–00, the Association for Frontotemporal Dementias Research Grant 2009, The Netherlands Organization for Scientific Research (NWO) grant HCMI 056–13-018, ZonMw Memorabel (Deltaplan Dementie, project number 733 051 042), Alzheimer Nederland, and the Bluefield project. FM received funding from the Tau Consortium and the Center for Networked Biomedical Research on Neurodegenerative Disease (CIBERNED). RS-V is supported by an Alzheimer’s Research UK Clinical Research Training Fellowship (ARUK-CRF2017B-2) and has received funding from Fundació Marató de TV3, Spain (grant no. 20143810). CG received funding from JPND-Prefrontals VR Dnr 529–2014-7504, VR 2015–02926, and 2018–02754; the Swedish FTD Inititative-Schörling Foundation; Alzheimer Foundation; Brain Foundation; and Stockholm County Council ALF. MM has received funding from a Canadian Institute of Health Research operating grant and the Weston Brain Institute and the Ontario Brain Institute. JBR has received funding from the Welcome Trust (220258), the Cambridge University Centre for Frontotemporal Dementia, the Medical Research Council (SUAG/051 G101400), and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (BRC-1215–20014). EF has received funding from a CIHR grant #327387. DG received support from the EU Joint Programme – Neurodegenerative Disease Research (JPND) and the Italian Ministry of Health (PreFrontALS) grant 733051042. RV has received funding from the Mady Browaeys Fund for Research into Frontotemporal Dementia. MO has received funding from BMBF (FTLDc). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018–02532); the European Research Council (#681712); Swedish State Support for Clinical Research (#ALFGBG-720931); the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809–2016862); the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21–831376-C, #ADSF-21–831381-C and #ADSF-21–831377-C); the Olav Thon Foundation; the Erling-Persson Family Foundation; Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228); the European Union’s Horizon 2020 research; and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), European Union Joint Program for Neurodegenerative Disorders (JPND2021-00694), and the UK Dementia Research Institute at UCL.
Author information
Authors and Affiliations
Consortia
Contributions
AS-E and JN have contributed equally to this paper. AS-E and JN have contributed to the experimental part of the work and the data generation. IS has contributed to the sample processing and database management. AS-E and JDR have contributed to the writing of the manuscript. JN, AB, HZ, and KB have contributed to the review of the manuscript. The rest of the authors who are part of the GENFI cohort have contributed samples for the study and have reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by all countries’ respective ethics committees, and all participants signed an informed consent to take part in the research. The participating centres in the GENFI study are University College London, Erasmus Medical Centre, University of Brescia, University of Milan, University of Barcelona, Université Laval, Donostia University Hospital, University of Tübingen, Karolinska Institutet, University of Toronto, University of Cambridge, University Hospitals Leuven, University of Western Ontario, Fondazione IRCCS Instituto Neurologico Carlo Besta, University of Coimbra, University of Oxford, McGill University, University of Manchester, LMU Klinikum, University of ULM IRCCSS Fondazione Don Carlo Gnocchi, Sorbonne Université, and University of Lille.
Consent for publication.
Not applicable.
Competing interests
KB has served as a consultant, at advisory boards or at data monitoring committees, for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineer, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper. HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. JDR has served as a consultant or on an advisory board for Alector, Prevail Therapeutics, Denali, Arkuda Therapeutics, Takeda, UCB, Wave Life Sciences, and Novartis. The other authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary tables.
Additional file 2: Appendix 1.
List of GENFI consortium authors.
Additional file 3: Appendix 2.
IS preparation and LC-MS settings for theanalysis of the synaptic protein panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sogorb-Esteve, A., Nilsson, J., Swift, I.J. et al. Differential impairment of cerebrospinal fluid synaptic biomarkers in the genetic forms of frontotemporal dementia. Alz Res Therapy 14, 118 (2022). https://doi.org/10.1186/s13195-022-01042-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-022-01042-3