
Abstract
We envisage the development of new Brain Health Services to achieve primary and secondary dementia prevention. These services will complement existing memory clinics by targeting cognitively unimpaired individuals, where the focus is on risk profiling and personalized risk reduction interventions rather than diagnosing and treating late-stage disease. In this article, we review key potentially modifiable risk factors and genetic risk factors and discuss assessment of risk factors as well as additional fluid and imaging biomarkers that may enhance risk profiling. We then outline multidomain measures and risk profiling and provide practical guidelines for Brain Health Services, with consideration of outstanding uncertainties and challenges. Users of Brain Health Services should undergo risk profiling tailored to their age, level of risk, and availability of local resources. Initial risk assessment should incorporate a multidomain risk profiling measure. For users aged 39–64, we recommend the Cardiovascular Risk Factors, Aging, and Incidence of Dementia (CAIDE) Dementia Risk Score, whereas for users aged 65 and older, we recommend the Brief Dementia Screening Indicator (BDSI) and the Australian National University Alzheimer’s Disease Risk Index (ANU-ADRI). The initial assessment should also include potentially modifiable risk factors including sociodemographic, lifestyle, and health factors. If resources allow, apolipoprotein E ɛ4 status testing and structural magnetic resonance imaging should be conducted. If this initial assessment indicates a low dementia risk, then low intensity interventions can be implemented. If the user has a high dementia risk, additional investigations should be considered if local resources allow. Common variant polygenic risk of late-onset AD can be tested in middle-aged or older adults. Rare variants should only be investigated in users with a family history of early-onset dementia in a first degree relative. Advanced imaging with 18-fluorodeoxyglucose positron emission tomography (FDG-PET) or amyloid PET may be informative in high risk users to clarify the nature and burden of their underlying pathologies. Cerebrospinal fluid biomarkers are not recommended for this setting, and blood-based biomarkers need further validation before clinical use. As new technologies become available, advances in artificial intelligence are likely to improve our ability to combine diverse data to further enhance risk profiling. Ultimately, Brain Health Services have the potential to reduce the future burden of dementia through risk profiling, risk communication, personalized risk reduction, and cognitive enhancement interventions.
Similar content being viewed by others

Background
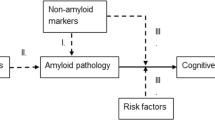
Frisoni and colleagues [1] recently highlighted the rationale for primary and secondary dementia prevention and the need for new services aimed at cognitively unimpaired individuals. Primary dementia prevention strategies for individuals with unknown disease markers include modifiable risk factors, lifestyle, and multiple domain interventions to reduce disease incidence. Secondary prevention targets high risk cognitively unimpaired individuals with biomarker evidence of disease pathology, to prevent or delay symptom onset.
Current memory clinics are ill-equipped to deal with the number of cognitively unimpaired individuals seeking help in memory clinics who believe they may be at increased risk of dementia ([2], this issue). For this reason, we envision the development of new Brain Health Services (BHSs) with specific missions including dementia risk profiling, risk communication ([3], this issue), risk reduction ([4], this issue), and cognitive enhancement ([5], this issue). Admittedly, BHSs pose specific societal challenges ([6], this issue).
This review focuses on the first principle of risk profiling and is the second part of a Special Issue series of six articles, published in Alzheimer’s Research & Therapy, which together provide a user manual for BHSs. We provide an overview of modifiable and genetic risk factors, before discussing best practices for the assessment of risk factors in a BHS setting. We also consider the potential assessment of fluid and imaging biomarkers for risk profiling. We then outline multiple domain measures and risk profiling in the context of primary and secondary prevention services. Finally, we provide practical guidelines for BHSs, and consider possible uncertainties, inconsistencies, and challenges.
Risk factors
Overview of modifiable risk factors
The concept of prevention being better than cure underpins the growing interest in the role of modifiable risk factors for cognitive impairment and dementia [7]. The 2020 Report of the Lancet Commission identified 12 modifiable risk factors, which, with appropriate interventions, could prevent up to 40% of dementia cases worldwide [8]. This may particularly benefit low- and middle-income countries where the prevalence of dementia is thought to be rising faster than in higher income countries [8].
Education is an early life potentially modifiable risk factor linked to late-life dementia risk [8], either by exerting a direct effect on brain structure by, for example, improving vascularization contributing to cognitive reserve, or by shaping healthier behaviors that reduce cardiovascular and cerebrovascular damage [9]. If causality is assumed and low levels of education were eliminated, then it has been estimated this would lead to a 7% reduction in dementia prevalence [8].
Hearing loss, traumatic brain injury (TBI), hypertension, alcohol consumption (> 21 units per week), and obesity have been identified as key potentially modifiable midlife dementia risk factors [8]. Poor encoding of sound may affect brain structure and function, impose higher cognitive load, and reduce social interaction [10]. Oxidative stress, inflammatory effects, and reduced cerebral flow contribute to brain pathology associated with factors such as TBI and hypertension [11]. The percentage reduction in dementia prevalence if these risk factors were eliminated ranges from 8% for hearing loss to 1% for alcohol and obesity [8].
Diabetes, smoking, air pollution, depression, social isolation, and physical inactivity have been identified as potentially modifiable late life dementia risk factors [8]. Diabetes [12] and physical inactivity [13] are associated with an adverse vascular profile, which itself is associated with an increased dementia risk [13]. Diabetes increases inflammation and oxidative stress on the brain [14]. Smoking and air pollution enhance reactive oxygen and inflammatory responses [11, 15]. Depression and social isolation are associated with accelerated brain and cardiovascular aging [16] and poor health behaviors [17]. The reduction in dementia prevalence associated with elimination of these risk factors ranges from 5% for smoking to 1% for diabetes [8].
As our understanding of modifiable risk factors improves, this list of “key” risk factors will need to be updated. Guidelines for risk reduction of cognitive decline and dementia published by the World Health Organization (WHO) had a greater focus on interventions and present building evidence for additional modifiable risk factors such as diet [18]. For example, emerging evidence suggests that diet, cognitive stimulation, vitamin D, and pesticide exposure may also be important [19, 20]. Based upon current evidence, the 12 potentially modifiable dementia risk factors identified in the 2020 Report of the Lancet Commission should be incorporated into BHS assessments and prioritized in personalized interventions.
Genetic risk factors and interactions with modifiable risk factors
AD is the most common type of dementia and has a strong genetic component, involving both common and rare genetic variants [21] as illustrated in the discovery timeline summarized in Fig. 1. To date, 34 genetic variants have been associated with AD [22]. Specifically, PSEN1, PSEN2, and APP mutations cause AD dementia in virtually 100% of carriers (autosomal dominant AD [23]), with a mean age at dementia onset of 35–65 years [24] and symptom duration of about 10 years [25]. Nevertheless, the major driver of AD in the general population is the common apolipoprotein E ɛ4 variant (APOE ɛ4), which is associated with an elevated risk of developing dementia, i.e., 51–95% in APOE ɛ4/ɛ4 and 22–90% in APOE ɛ4/- [26, 27] and a mean age at dementia onset of 73–74 years in APOE ɛ4/ɛ4 and 75–82 in APOE ɛ4/- [28, 29]. Although common variants often have very small effects on a person’s AD risk, jointly they may modify the risk and age at onset of Alzheimer's disease and dementia significantly [22].

Timeline of the discovery of frequent and rare variants in Alzheimer’s disease. Frequent/common variants discovered in GWAS are shown above the horizontal date axis, and rare variants discovered using sequencing and arrays are shown below the date axis
Our knowledge of the genes implicated in non-AD dementias is less comprehensive. Mutations in microtubule associated protein tau gene and the gene encoding progranulin are specific for frontotemporal dementia. Other variants including the intronic expansion of a hexanucleotide repeat in C9orf72 and SERPINA1 are also implicated, though few common variants have been identified (see Fig. 1). For dementia with Lewy bodies, only APOE, Glucocerebrosidase, and Synuclein Alpha have been replicated. Lastly, for vascular dementia there are no consistent findings for common variants.
It is unknown whether genetic risk modifies the influence of life-style on dementia. Four large multidomain trials of dementia prevention have been conducted. However, only the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability found a significant difference in the primary outcome following a lifestyle, metabolic, and vascular intervention [30]. The beneficial effect was only observed in APOE ɛ4 carriers [31]. Null group level findings in other trials may therefore mask effects in genetic subgroups. Comparable findings from observational studies are mixed. There was no interaction between lifestyle factors and polygenic risk score in relation to all-cause dementia risk in the UK Biobank [32]. However, there was a significant interaction between a composite of lifestyle and health factors and APOE/polygenic risk in relation to all-cause dementia in the Rotterdam Study [33]. Taken together, these findings provide suggestive rather than conclusive evidence that, contrary to expectation, those with a high genetic risk may be more likely to benefit from targeted dementia prevention interventions.
Assessments in the clinic
Assessing risk factors in the clinic
When assessing cognitively unimpaired users in BHSs, consideration of their modifiable lifestyle and clinical risk factors and their genetic profile can inform personalized and targeted dementia prevention interventions.
Assessing potentially modifiable risk factors
The potentially modifiable risk factors to be assessed in BHSs are shown in Table 1, along with examples of methods to assess these factors in a clinical setting.
Assessing genetic risk factors
Combining effects of APOE*4 and common variants allows a precise prediction of the risk and age of onset of AD [22, 45, 46] and pathology in the brain [47]. Age specific risk curves may have clinical utility in BHS, allowing determination of future risk of AD at different stages of the life course [22]. These estimates can be provided using polygenic risk scores based on replicated variants [22] assessed by dedicated AD chips or putative genome wide variants that are marginally associated to the disease that can be assessed by general genetics arrays [45,46,47]. Although in many countries testing for rare variants conferring a high risk of AD (see Fig. 1) is the domain of clinical genetics, routine testing for such variants in a BHS may be useful for users with a family history of AD. Within a BHS, testing for known major genes that harbor rare variants may be done in collaboration with clinical geneticists. As is the case with many complex disorders, rare variants in genes will be encountered for which the functional effects and the risk of AD is yet unknown in archives such as Omim and ClinVar [48]. However, collaboration between the genomics and clinical community could facilitate genetic counseling in the setting of a BHS [48].
Assessing additional biomarkers
Fluid and neuroimaging biomarkers can be used to differentiate between asymptomatic individuals with and without underlying pathology. This can be used to target people who are particularly likely to benefit from interventions. Assessment of pathology also provides a baseline for disease burden that can be subsequently used to assess rates of progression. Nevertheless, the use of biomarkers in BHSs depends on local facilities and resources and on the context of BHS implementation ([2], this issue).
Assessing fluid (CSF and plasma) biomarkers
Many studies have consistently shown that core AD cerebrospinal fluid (CSF) biomarkers amyloid β (Aβ42 and Aβ42/Aβ40 ratio), total-tau (T-tau), and phosphorylated tau (P-tau) reflect key elements of AD pathophysiology and have high diagnostic value and high concordance with amyloid positron emission tomography (PET) [49]. However, there are currently no disease specific fluid biomarkers for non-AD dementia. Furthermore, a lumbar puncture is often regarded as complicated and invasive and subjects may have side-effects in the form of transient headache. Thus, for a BHS clinical setting, blood biomarkers are likely to be more practical and acceptable to users than CSF biomarkers.
Technical developments have allowed for quantification of brain-specific proteins in blood samples. For amyloidosis, the plasma Aβ42/Aβ40 ratio shows high concordance with amyloid PET [50], and can be measured on fully automatized instruments [51]. Blood biomarkers for tau pathology include P-tau181, which shows a marked increase in AD and high concordance with tau PET [52, 53], while levels are normal in other tauopathies, such as frontotemporal dementia. Importantly, plasma P-tau181 is increased in unimpaired elderly having brain amyloidosis, but still a negative tau PET scan [54]. This suggests it may be sensitive to pathological change at an earlier stage. Studies of other tau variants, specifically P-tau217, show encouraging results [55]. Neurofilament light (NFL) is a well-validated neurodegeneration biomarker showing increases in several neurodegenerative disorders, including AD [56], and predicts future rate of cognitive decline [57]. Importantly, plasma NFL increases early in the preclinical phase of AD [58, 59]. While blood biomarkers are very promising, they need further real-world validation before they can be recommended for use in BHSs [60, 61].
Assessing imaging biomarkers
Magnetic resonance imaging (MRI) promises to be a sensitive early biomarker of neurodegeneration given that genetic cohorts of dementia demonstrate structural MRI changes many years before symptom onset [62,63,64]. The finding of selective early hippocampal atrophy is well established in AD [64, 65] and is an accepted biomarker for clinical trials [66, 67], yet translating this into detecting early AD for clinical use requires further work [68]. However, there are a number of potential methodological developments in artificial intelligence, PET, and MRI technology that may lead to more specific and biologically relevant neuroimaging biomarkers [69].
Cerebrovascular risk is a particular focus for neuroimaging studies and impacts on cognition in healthy aging [70]. While silent territorial infarcts are relatively rare, cerebral small vessel disease is extremely common, encompassing white matter hyperintensities, lacunes, widened Virchow-Robin spaces, and cerebral microbleeds [71]. White matter hyperintensities are a frequent finding associated with cardiovascular risk factors such as hypertension and smoking. They significantly increase the risk of stroke, dementia, and overall mortality [72], especially when lesions become confluent [73]. Stroke itself is strongly associated with incident all-cause dementia [74]. Lacunes are found more frequently in individuals with atrial fibrillation and present an independent risk factor for cognitive decline. Cerebral microbleeds can be due to cardiovascular risk factors deep in the basal nuclei, while lobar cerebral microbleeds are reflective of amyloid-angiopathy; they only carry a weak risk for dementia on a population level [75].
Current consensus practice for assessing MRI scans is to use visual rating scales, such as the medial temporal lobe atrophy scale [76], the parietal atrophy scale [77], the global cortical atrophy scale [78], the age-related white matter changes [79], and the Fazekas scale for white matter lesions [80, 81]. Measurement of regional cortical thickness can also identify presymptomatic amyloid positive individuals [82]. However, with the advent of artificial intelligence, new neuroimaging tools for diagnosis and prognosis are emerging [83,84,85] that may provide more sensitive assessments in the near future.
PET has provided a suite of tools for assessing people with cognitive impairment using specific ligands that bind to physiological targets. The most well established in clinical practice is 18-Fluorodeoxyglucose (FDG) PET which has proved useful for predicting cognitive impairment in Parkinson’s disease [86]. Ligands for beta-amyloid have found the presence of beta-amyloid increases with age, reaching 65% in health over 80s [87]. However, a positive beta-amyloid PET did not correlate to cognition, so the implications of this finding remains uncertain for predicting risk. It has been shown in genetic forms of AD that amyloid accumulates 15–20 years prior to symptom onset and it is thought to be an early critical factor in disease, although changes in amyloid load do not reliably correlate with cognitive change [62]. Other ligands for tau [88] inflammation [89] or synaptic integrity [90] exist, but remain in the research domain. The cost and availability of PET imaging may limit its applicability to BHSs but could have a role in selected high risk individuals.
Risk profiling
Multidomain measures and risk profiling
A number of dementia risk prediction models have been developed to determine dementia risk in middle-aged or older adults [91, 92]. The validity of most risk models is unknown, as is the degree to which they can be appropriately used in different populations. Prediction models which have been validated in multiple samples include the Cardiovascular Risk Factors, Aging and Dementia (CAIDE) score [93], the Australian National University Alzheimer’s Disease Risk Index (ANU-ADRI) [94, 95], and the Brief Dementia Screening Indicator (BDSI) [96]. Basic characteristics of these models are shown in Table 2. The CAIDE score assesses long-term risk of dementia in middle-aged adults, whereas the ANU-ADRI and the BDSI predict medium-term AD and dementia risk respectively in older adults. The overall accuracy of these risk prediction models is moderate (range 0.64–0.78), indicating that, although they can be improved upon, they can also generate useful predictions. It is notable that 10 of the 12 modifiable risk factors for dementia included in the 2020 Report of the Lancet Commission are included in these models [8]. The only modifiable risk factors identified in that report which are not currently included are hearing loss and air pollution.
The CAIDE score has a moderate level of discriminative accuracy over 20 years follow-up (area under the curve (AUC) = 0.77, 95% CI = 0.71–0.83). The addition of APOE e4 did not substantially increase accuracy (AUC = 0.78, 95% CI = 0.72–0.84). When this model was externally validated, it performed similarly in a midlife population (AUC = 0.75) [97] but poorly in late-life populations with shorter follow-up times (AUC range 0.49–0.57) [95]. When tested in three cohorts, the ANU-ADRI was found to have moderate levels of discriminative accuracy for Alzheimer’s disease: Rush Memory and Aging Project study AUC = 0.64 (95% CI = 0.60–0.68), Kungsholmen Project study AUC = 0.74 (95% CI 0.71–0.77), and Cardiovascular Health Cognition study AUC = 0.73 (95% CI = 0.69–0.78). The BDSI was tested in four cohorts including the Cardiovascular Health Study (CHS), The Framingham Heart Study (FHS), the Health and Retirement Study (HRS), and the Sacramento Area Latino Study on Aging (SALSA). The discrimination accuracy of the final model was moderate across cohorts: CHS AUC = 0.68 (95% CI = 0.65–0.72), FHS AUC = 0.77 (95% CI = 0.73–0.82), HRS AUC = 0.76 (95% CI = 0.74–0.77), and SALSA AUC = 0.78 (95% CI = 0.72–0.83).
There have also been attempts to develop new models in at-risk subpopulations. For example, the Diabetes-Specific Dementia Risk Score (DSDRS) is a model for type 2 diabetics. The DSDRS was found to have reasonable accuracy in the development (AUC = 0.74) and validation (AUC = 0.75) cohorts [97]. Disease-specific predictive models may be important as generic dementia risk prediction models may not work well in specific subpopulations [98]. Further, not all prediction models for dementia developed in high-income countries are necessarily applicable to low- and middle-income countries [99].
Discussion
Summary
We now have a reasonable idea what the “key” potentially modifiable dementia risk factors are in early-, mid-, and late- life. It is also likely that further risk factors will be added to this list in the future. Of course, optimal risk profiling for identification of patients at high risk of dementia may not necessarily be synonymous with optimal modifiable risk factor profiling for targeted interventions. For risk profiling, rare genetic variants for early-onset dementia have been identified, and common variants for late-onset dementia, particularly AD, are now known. Further research is needed to investigate possible gene-environment interactions. CSF biomarkers are not very practical in the context of BHSs; however, blood-based biomarkers may be useful subject to further real-world validation. Structural MRI is becoming established as a clinically useful imaging biomarker of dementia pathologies, and advanced imaging may be a useful supplement to this if available. Existing dementia risk prediction models offer a practical way of risk profiling individual users, though there is room for improvement and they have not yet been optimized for use in BHSs.
Practical guidelines
The assessment of risk factors and risk profiling in BHSs will require a multidisciplinary team, and a balance between precision and practicality. Individual assessment of modifiable risk factors is likely to involve multiple measures and may prove to be time consuming. Some assessments may be completed in advance of the appointment and a specialist nurse appointment may also enrich the information available. The individual will undergo an assessment tailored to their age, level of risk following an initial assessment, and local resources available (see Fig. 2). Comprehensive recommendations for BHSs, including risk profiling outlined here, will be presented in a separate article. A follow-up communication of the user’s results will be required ([3], this issue), followed by the proposal of an individualized plan for risk reduction ([4], this issue) and/or cognitive enhancement interventions ([5], this issue) and/or clinical trial opportunities.

Proposed workflow for dementia risk profiling in BHSs
The initial assessment should include the exclusion of pre-existing dementia. Risk profiling should incorporate a multidomain risk profiling measure validated for use with the relevant age group, assessment of additional risk factors, APOE ɛ4 status if possible, and structural MRI. To assess long-term dementia risk in middle-aged individuals aged 39–64, we recommend that BHSs use the CAIDE score. The CAIDE should not be used for anyone younger than 39 whose dementia risk will be negligible over 20 years or in those aged 65 years or older as accuracy is poor in older adults and better alternatives are available. APOE ɛ4 genotyping should be undertaken if possible to permit use of the full CAIDE model and as multimodal interventions may be more effective in e4 carriers [31]. This will allow for targeted allocation of limited resources when attempting dementia prevention. To assess medium-term dementia risk in individuals aged 65 and older, we recommend that BHSs use the BDSI and the ANU-ADRI which produces a comparable risk prediction for AD specifically. The ANU-ADRI also has the practical advantage of incorporating a larger number of modifiable risk factors such as smoking and physical activity which can inform targeted interventions. Additional risk factor assessment should focus on those factors with the strongest evidence base and greatest opportunity to intervene, particularly those outlined in the 2020 Report of the Lancet Commission [8]. These can be divided into early life (education), midlife (hearing loss, TBI, hypertension, alcohol consumption, and obesity), and late-life (diabetes, smoking, air pollution, depression, social isolation and physical inactivity) and should be assessed routinely as appropriate for the user’s age. Some, but not all, of these risk factors are incorporated into existing dementia risk scores (see Table 2). Structural MRI should be used in BHSs to enable the assessment of non-degenerative pathologies (e.g., inflammation, tumors), cerebrovascular burden (particularly cerebral small vessel disease including white matter hyperintensities and lacunes), and neurodegenerative processes (generalized, medial temporal lobe and hippocampal atrophy). Determination of whether a user has high dementia risk may be made on the basis of clinical judgment of the initial assessment results or by formally combining the information using a computerized decision support system, if available.
If initial BHS dementia risk profiling indicates that the user has a low risk of dementia, then low intensity interventions can be implemented, for example signposting to relevant risk reduction public health information and resources or low-cost non-specialist guidance. If however the initial risk profiling indicates that the user has a high risk of dementia then additional optional investigations should be considered if local facilities and resources allow. Common variant polygenic risk of late-onset AD can be tested in middle-aged or older adults. However, rare variants should only be investigated in users with a family history of early-onset dementia (< 65 years) in a first-degree relative in collaboration with clinical geneticists. Advanced imaging with FDG-PET or amyloid PET may also prove to be informative in high risk patients to clarify their burden of underlying pathology. Plasma biomarkers may prove to be a useful additional optional investigation in high risk users in the near future, though they are not currently recommended for use in BHSs before further real-world validation. Future risk profiling tools may therefore benefit from incorporating richer genetic information using polygenic risk scores and more advanced biomarker and imaging findings.
Conclusions
Risk profiling in BHS involves a core assessment comprised of multidomain risk prediction models in combination with additional risk factors, APOE ɛ4 status if possible, and structural MRI. If resources allow, then additional investigations including more comprehensive genetic testing and advanced neuroimaging can be undertaken in high risk users. Further research is necessary to refine risk profiling and risk reduction approaches in low- and middle-income countries. Results can then be communicated to users, a personalized risk reduction and cognitive enhancement plan formulated, and clinical trial opportunities identified.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Change history
23 October 2021
The paper was amended to add a DOI in references part of the Brain Health Services series.
Abbreviations
- AD:
-
Alzheimer’s disease
- ANU-ADRI:
-
Australian National University Alzheimer’s Disease Risk Index
- APOE :
-
Apolipoprotein E
- AUC:
-
Area under the curve
- BDSI:
-
Brief Dementia Screening Indicator
- BHS:
-
Brain health services
- CAIDE:
-
Cardiovascular Risk Factors, Aging and Dementia
- CHS:
-
Cardiovascular Health Study
- CSF:
-
Cerebrospinal fluid
- DSDRS:
-
Diabetes-Specific Dementia Risk Score
- FDG:
-
18-Fluorodeoxyglucose
- FHS:
-
Framingham Heart Study
- HRS:
-
Health and Retirement Study
- MRI:
-
Magnetic resonance imaging
- Nfl:
-
Neurofilament light
- PET:
-
Positron emission tomography
- P-tau:
-
Phosphorylated tau
- SALSA:
-
Sacramento Area Latino Study on Aging
- TBI:
-
Traumatic brain injury
References
Frisoni GB, Molinuevo JL, Altomare D, Carrera E, Barkhof F, Berkhof J, et al. Precision prevention of Alzheimer’s and other dementias: Anticipating future needs in the control of risk factors and implementation of disease-modifying therapies. Alzheimers Dement. 2020;16(10):1457–68. https://doi.org/10.1002/alz.12132.
Altomare D, Molinuevo JL, Ritchie C, Ribaldi F, Carrera E, Dubois B, Jessen F, McWhirter L, Scheltens P, van der Flier WM, Vellas B, Démonet JF, Frisoni GB. Brain Health Services: Organization, structure and challenges for implementation. A user manual for Brain Health Services – Part 1 of 6. Alzheimers Res Ther. 2021. https://doi.org/10.1186/s13195-021-00827-2.
Visser LNC, Minguillon C, Sánchez-Benavides G, Abramowicz M, Altomare D, Fauria K, Frisoni GB, Georges J, Ribaldi F, Scheltens P, van der Schaar J, Zwan M, van der Flier WM, Molinuevo JL. Dementia risk communication. A user manual for Brain Health Services – Part 3 of 6. Alzheimers Res Ther. 2021. https://doi.org/10.1186/s13195-021-00840-5.
Solomon A, Stephen R, Altomare D, Carrera E, Frisoni GB, Kulmala J, Molinuevo JL, Nilsson P, Ngandu T, Ribaldi F, Vellas B, Scheltens P, Kivipelto M. Multidomain interventions: state-of-the-art and future directions for protocols to implement precision dementia risk reduction. A user manual for Brain Health Services – Part 4 of 6. Alzheimers Res Ther. 2021. https://doi.org/10.1186/s13195-021-00875-8.
Brioschi Guevara A, Bieler M, Altomare D, Berthier M, Csajka C, Dautricourt S, Démonet JF, Dodich A, Frisoni GB, Miniussi C, Molinuevo JL, Ribaldi F, Scheltens P, Chételat G. Protocols for cognitive enhancement. A user manual for Brain Health Services – Part 5 of 6. Alzheimers Res Ther. 2021. https://doi.org/10.1186/s13195-021-00844-1.
Milne R, Altomare D, Ribaldi F, Molinuevo JL, Frisoni GB, Brayne C. Societal and equity challenges for Brain Health Services. A user manual for Brain Health Services – Part 6 of 6. Alzheimers Res Ther. 2021. https://doi.org/10.1186/s13195-021-00885-6.
Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673–734. https://doi.org/10.1016/S0140-6736(17)31363-6.
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413–46. https://doi.org/10.1016/S0140-6736(20)30367-6.
Arenaza-Urquijo EM, Wirth M, Chételat G. Cognitive reserve and lifestyle: moving towards preclinical Alzheimer’s disease. Front Aging Neurosci. 2015;7:134. https://doi.org/10.3389/fnagi.2015.00134.
Peelle JE, Troiani V, Grossman M, Wingfield A. Hearing loss in older adults affects neural systems supporting speech comprehension. J Neurosci. 2011;31(35):12638–43. https://doi.org/10.1523/JNEUROSCI.2559-11.2011.
Hughes TF, Ganguli M. Modifiable midlife risk factors for late-life cognitive impairment and dementia. Curr Psychiatry Rev. 2009;5(2):73–92. https://doi.org/10.2174/157340009788167347.
Leon BM, Maddox TM. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J Diabetes. 2015;6:1246–58.
Kivimäki M, Singh-Manoux A, Pentti J, Sabia S, Nyberg ST, Alfredsson L, et al. Physical inactivity, cardiometabolic disease, and risk of dementia: an individual-participant meta-analysis. BMJ. 2019;365:l1495. https://doi.org/10.1136/bmj.l1495.
Shalev D, Arbuckle MR. Metabolism and memory: obesity, diabetes, and dementia. Biol Psychiatry. 2017;82(11):e81–3. https://doi.org/10.1016/j.biopsych.2017.09.025.
Peters R, Ee N, Peters J, Booth A, Mudway I, Anstey KJ. Air pollution and dementia: a systematic review. J Alzheimers Dis. 2019;70(s1):S145–s163. https://doi.org/10.3233/JAD-180631.
Cacioppo JT, Hawkley LC, Norman GJ, Berntson GG. Social isolation. Ann N Y Acad Sci. 2011;1231(1):17–22. https://doi.org/10.1111/j.1749-6632.2011.06028.x.
Kobayashi LC, Steptoe A. Social isolation, loneliness, and health behaviors at older ages: longitudinal cohort study. Ann Behav Med. 2018;52(7):582–93. https://doi.org/10.1093/abm/kax033.
World Health Organization. Risk reduction of cognitive decline and dementia. Geneva: WHO guidelines; 2019.
Deckers K, van Boxtel MP, Schiepers OJ, et al. Target risk factors for dementia prevention: a systematic review and Delphi consensus study on the evidence from observational studies. Int J Geriatr Psychiatry. 2015;30(3):234–46. https://doi.org/10.1002/gps.4245.
Killin LOJ, Starr JM, Shiue IJ, Russ TC. Environmental risk factors for dementia: a systematic review. BMC Geriatr. 2016;16(1):175. https://doi.org/10.1186/s12877-016-0342-y.
Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
van der Lee SJ, Wolters FJ, Ikram MK, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 2018;17:434–44.
Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011;3(1):1–1. https://doi.org/10.1186/alzrt59.
Rossor MN, Fox NC, Mummery CJ, Schott JM, Warren JD. The diagnosis of young-onset dementia. Lancet Neurol. 2010;9(8):793–806. https://doi.org/10.1016/S1474-4422(10)70159-9.
Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253–60. https://doi.org/10.1212/WNL.0000000000000596.
Reiman EM, Arboleda-Velasquez JF, Quiroz YT, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667. https://doi.org/10.1038/s41467-019-14279-8.
Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–7. https://doi.org/10.1038/mp.2011.52.
Myers RH, Schaefer EJ, Wilson PW, et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology. 1996;46(3):673–7. https://doi.org/10.1212/WNL.46.3.673.
Slooter AJ, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol. 1998;55(7):964–8. https://doi.org/10.1001/archneur.55.7.964.
Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255–63. https://doi.org/10.1016/S0140-6736(15)60461-5.
Solomon A, Turunen H, Ngandu T, Peltonen M, Levälahti E, Helisalmi S, et al. Effect of the Apolipoprotein E Genotype on cognitive change during a multidomain lifestyle intervention: a subgroup analysis of a randomized clinical trial. JAMA Neurol. 2018;75(4):462–70. https://doi.org/10.1001/jamaneurol.2017.4365.
Lourida I, Hannon E, Littlejohns TJ, Langa KM, Hyppönen E, Kuzma E, et al. Association of lifestyle and genetic risk with incidence of dementia. JAMA. 2019;322(5):430–7. https://doi.org/10.1001/jama.2019.9879.
Licher S, Ahmad S, Karamujić-Čomić H, Voortman T, Leening MJG, Ikram MA, et al. Genetic predisposition, modifiable-risk-factor profile and long-term dementia risk in the general population. Nat Med. 2019;25(9):1364–9. https://doi.org/10.1038/s41591-019-0547-7.
Sharp ES, Gatz M. Relationship between education and dementia: an updated systematic review. Alzheimer Dis Assoc Disord. 2011;25(4):289–304. https://doi.org/10.1097/WAD.0b013e318211c83c.
Corrigan JD, Bogner J. Initial reliability and validity of the Ohio State University TBI Identification Method. J Head Trauma Rehabil. 2007;22(6):318–29. https://doi.org/10.1097/01.HTR.0000300227.67748.77.
Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018;39(33):3021–104. https://doi.org/10.1093/eurheartj/ehy339.
McKenna H, Treanor C, O’Reilly D, Donnelly M. Evaluation of the psychometric properties of self-reported measures of alcohol consumption: a COSMIN systematic review. Subst Abuse Treat Prev Policy. 2018;13(1):6. https://doi.org/10.1186/s13011-018-0143-8.
Razi S, Manish G, Keshav G, Sukriti K, Gupta A. Site or size of waist circumference, which one is more important in metabolic syndrome? Int J Med Public Health. 2016;6:69–72.
Lycke M, Lefebvre T, Cool L, van Eygen K, Boterberg T, Schofield P, et al. Screening methods for age-related hearing loss in older patients with cancer: a review of the literature. Geriatrics (Basel). 2018;3(3):48. https://doi.org/10.3390/geriatrics3030048.
Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41(2):255–323. https://doi.org/10.1093/eurheartj/ehz486.
Levis B, Sun Y, He C, et al. Accuracy of the PHQ-2 alone and in combination with the PHQ-9 for screening to detect major depression: systematic review and meta-analysis. JAMA. 2020;323:2290–300.
Lubben JE. Assessing social networks among elderly populations. Fam Community Health. 1988;11(3):42–52. https://doi.org/10.1097/00003727-198811000-00008.
Koenig HG, Westlund RE, George LK, Hughes DC, Blazer DG, Hybels C. Abbreviating the Duke Social Support Index for use in chronically ill elderly individuals. Psychosomatics. 1993;34(1):61–9. https://doi.org/10.1016/S0033-3182(93)71928-3.
Sylvia LG, Bernstein EE, Hubbard JL, Keating L, Anderson EJ. Practical guide to measuring physical activity. J Acad Nutr Diet. 2014;114(2):199–208. https://doi.org/10.1016/j.jand.2013.09.018.
Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14(3):e1002258. https://doi.org/10.1371/journal.pmed.1002258.
Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain. 2015;138(12):3673–84. https://doi.org/10.1093/brain/awv268.
Tan CH, Bonham LW, Fan CC, Mormino EC, Sugrue LP, Broce IJ, et al. Polygenic hazard score, amyloid deposition and Alzheimer’s neurodegeneration. Brain. 2019;142(2):460–70. https://doi.org/10.1093/brain/awy327.
Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JGJ, Weiss M, et al. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer’s disease: a clinical interpretation strategy. Eur J Hum Genet. 2017;25(8):973–81. https://doi.org/10.1038/ejhg.2017.87.
Shaw LM, Arias J, Blennow K, Galasko D, Molinuevo JL, Salloway S, et al. Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer’s disease. Alzheimers Dement. 2018;14(11):1505–21. https://doi.org/10.1016/j.jalz.2018.07.220.
Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6(1):26801. https://doi.org/10.1038/srep26801.
Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related beta-amyloid status. JAMA Neurol. 2019;76(9):1060–9. https://doi.org/10.1001/jamaneurol.2019.1632.
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26(3):379–86. https://doi.org/10.1038/s41591-020-0755-1.
Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387–97.
Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19(5):422–33. https://doi.org/10.1016/S1474-4422(20)30071-5.
Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772–81. https://doi.org/10.1001/jama.2020.12134.
Lewczuk P, Riederer P, O’Bryant SE, Verbeek MM, Dubois B, Visser PJ, et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J Biol Psychiatry. 2018;19(4):244–328. https://doi.org/10.1080/15622975.2017.1375556.
Mattsson N, Andreasson U, Zetterberg H, Blennow K, Alzheimer’s Disease Neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557–66.
Weston PSJ, Poole T, Ryan NS, Nair A, Liang Y, Macpherson K, et al. Serum neurofilament light in familial Alzheimer disease: a marker of early neurodegeneration. Neurology. 2017;89(21):2167–75. https://doi.org/10.1212/WNL.0000000000004667.
Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med. 2019;25(2):277–83. https://doi.org/10.1038/s41591-018-0304-3.
Chhatwal JP, Schultz AP, Dang Y, Ostaszewski B, Liu L, Yang HS, et al. Plasma N-terminal tau fragment levels predict future cognitive decline and neurodegeneration in healthy elderly individuals. Nat Commun. 2020;11(1):6024. https://doi.org/10.1038/s41467-020-19543-w.
Cullen NC, Leuzy A, Palmqvist S, et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nature Aging. 2020;1:114–23. https://doi.org/10.1038/s43587-020-00003-5.
Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804.
Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative ( GENFI ) study: a cross-sectional analysis. Lancet Neurol. 2015;14:253–62.
Schott JM, Fox NC, Frost C, Scahill RI, Janssen JC, Chan D, et al. Assessing the onset of structural change in familial Alzheimer’s disease. Ann Neurol. 2003;53(2):181–8. https://doi.org/10.1002/ana.10424.
Fox NC, Freeborough PA, Rossor MN. Visualisation and quantification of rates of atrophy in Alzheimer’s disease. Lancet. 1996;348(9020):94–7. https://doi.org/10.1016/S0140-6736(96)05228-2.
Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6(2):67–77. https://doi.org/10.1038/nrneurol.2009.215.
Hill DLG, Schwarz AJ, Isaac M, et al. Coalition Against Major Diseases/European Medicines Agency biomarker qualification of hippocampal volume for enrichment of clinical trials in predementia stages of Alzheimer’s disease. Alzheimers Dement. 2014;10:421–429.e423.
Lombardi G, Crescioli G, Cavedo E, et al. Structural magnetic resonance imaging for the early diagnosis of dementia due to Alzheimer’s disease in people with mild cognitive impairment. Cochrane Database Syst Rev. 2020;3(3):CD009628. https://doi.org/10.1002/14651858.CD009628.pub2.
Rittman T. Neurological update: neuroimaging in dementia. J Neurol. 2020;267(11):3429–35. https://doi.org/10.1007/s00415-020-10040-0.
Veldsman M, Tai X-Y, Nichols T, Smith S, Peixoto J, Manohar S, et al. Cerebrovascular risk factors impact frontoparietal network integrity and executive function in healthy ageing. Nat Commun. 2020;11(1):4340. https://doi.org/10.1038/s41467-020-18201-5.
Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12(8):822–38. https://doi.org/10.1016/S1474-4422(13)70124-8.
Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341(jul26 1):c3666. https://doi.org/10.1136/bmj.c3666.
Inzitari D, Pracucci G, Poggesi A, Carlucci G, Barkhof F, Chabriat H, et al. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of LADIS (leukoaraiosis and disability) study cohort. Bmj. 2009;339(jul06 1):b2477. https://doi.org/10.1136/bmj.b2477.
Kuźma E, Lourida I, Moore SF, Levine DA, Ukoumunne OC, Llewellyn DJ. Stroke and dementia risk: a systematic review and meta-analysis. Alzheimers Dement. 2018;14(11):1416–26. https://doi.org/10.1016/j.jalz.2018.06.3061.
Bos D, Wolters FJ, Darweesh SKL, Vernooij MW, de Wolf F, Ikram MA, et al. Cerebral small vessel disease and the risk of dementia: a systematic review and meta-analysis of population-based evidence. Alzheimers Dement. 2018;14(11):1482–92. https://doi.org/10.1016/j.jalz.2018.04.007.
Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992;55(10):967–72. https://doi.org/10.1136/jnnp.55.10.967.
Koedam EL, Lehmann M, van der Flier WM, et al. Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol. 2011;21(12):2618–25. https://doi.org/10.1007/s00330-011-2205-4.
Pasquier F, Leys D, Weerts JG, Mounier-Vehier F, Barkhof F, Scheltens P. Inter- and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol. 1996;36(5):268–72. https://doi.org/10.1159/000117270.
Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, Sjögren M, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. 2001;32(6):1318–22. https://doi.org/10.1161/01.STR.32.6.1318.
Vernooij MW, Pizzini FB, Schmidt R, Smits M, Yousry TA, Bargallo N, et al. Dementia imaging in clinical practice: a European-wide survey of 193 centres and conclusions by the ESNR working group. Neuroradiology. 2019;61(6):633–42. https://doi.org/10.1007/s00234-019-02188-y.
Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149(2):351–6. https://doi.org/10.2214/ajr.149.2.351.
Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19(3):497–510. https://doi.org/10.1093/cercor/bhn113.
Giorgio J, Landau SM, Jagust WJ, Tino P, Kourtzi Z. Alzheimer’s Disease Neuroimaging I. Modelling prognostic trajectories of cognitive decline due to Alzheimer’s disease. Neuroimage Clin. 2020;26:102199. https://doi.org/10.1016/j.nicl.2020.102199.
Archetti D, Ingala S, Venkatraghavan V, Wottschel V, Young AL, Bellio M, et al. Multi-study validation of data-driven disease progression models to characterize evolution of biomarkers in Alzheimer’s disease. NeuroImage Clin. 2019;24:101954. https://doi.org/10.1016/j.nicl.2019.101954.
Fisher CK, Smith AM, Walsh JR. Machine learning for comprehensive forecasting of Alzheimer’s disease progression. Sci Rep. 2019;9(1):1–14. https://doi.org/10.1038/s41598-019-49656-2.
Pilotto A, Premi E, Paola Caminiti S, Presotto L, Turrone R, Alberici A, et al. Single-subject SPM FDG-PET patterns predict risk of dementia progression in Parkinson disease. Neurology. 2018;90(12):e1029–37. https://doi.org/10.1212/WNL.0000000000005161.
Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83.
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4(1):58. https://doi.org/10.1186/s40478-016-0315-6.
Endres CJ, Pomper MG, James M, Uzuner O, Hammoud DA, Watkins CC, et al. Initial evaluation of 11C-DPA-713, a novel TSPO PET ligand, in humans. J Nuclear Med. 2009;50(8):1276–82. https://doi.org/10.2967/jnumed.109.062265.
Nabulsi NB, Mercier J, Holden D, Carré S, Najafzadeh S, Vandergeten MC, et al. Synthesis and preclinical evaluation of 11C-UCB-J as a PET Tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nuclear Med. 2016;57(5):777–84. https://doi.org/10.2967/jnumed.115.168179.
Hou XH, Feng L, Zhang C, Cao XP, Tan L, Yu JT. Models for predicting risk of dementia: a systematic review. J Neurol Neurosurg Psychiatry. 2019;90(4):373–9. https://doi.org/10.1136/jnnp-2018-318212.
Tang EY, Harrison SL, Errington L, et al. Current developments in dementia risk prediction modelling: an updated systematic review. PLoS One. 2015;10:e0136181.
Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol. 2006;5(9):735–41. https://doi.org/10.1016/S1474-4422(06)70537-3.
Anstey KJ, Cherbuin N, Herath PM. Development of a new method for assessing global risk of Alzheimer’s disease for use in population health approaches to prevention. Prev Sci. 2013;14(4):411–21. https://doi.org/10.1007/s11121-012-0313-2.
Anstey KJ, Cherbuin N, Herath PM, Qiu C, Kuller LH, Lopez OL, et al. A self-report risk index to predict occurrence of dementia in three independent cohorts of older adults: the ANU-ADRI. PLoS One. 2014;9(1):e86141. https://doi.org/10.1371/journal.pone.0086141.
Barnes DE, Beiser AS, Lee A, et al. Development and validation of a brief dementia screening indicator for primary care. Alzheimers Dement. 2014;10:656–665.e651.
Exalto LG, Biessels GJ, Karter AJ, et al. Risk score for prediction of 10 year dementia risk in individuals with type 2 diabetes: a cohort study. Lancet Diabetes Endocrinol. 2013;1(3):183–90. https://doi.org/10.1016/S2213-8587(13)70048-2.
Tang EYH, Price CI, Robinson L, Exley C, Desmond DW, Köhler S, et al. Assessing the predictive validity of simple dementia risk models in harmonised stroke cohorts. Stroke. 2020;51(7):2095–102. https://doi.org/10.1161/STROKEAHA.120.027473.
Stephan BCM, Pakpahan E, Siervo M, Licher S, Muniz-Terrera G, Mohan D, et al. Prediction of dementia risk in low-income and middle-income countries (the 10/66 Study): an independent external validation of existing models. Lancet Glob Health. 2020;8(4):e524–35. https://doi.org/10.1016/S2214-109X(20)30062-0.
Acknowledgements
European Task Force for Brain Health Services (in alphabetical order): Marc ABRAMOWICZ, Daniele ALTOMARE, Frederik BARKHOF, Marcelo BERTHIER, Melanie BIELER, Kaj BLENNOW, Carol BRAYNE, Andrea BRIOSCHI, Emmanuel CARRERA, Gael CHÉTELAT, Chantal CSAJKA, Jean-François DEMONET, Alessandra DODICH, Bruno DUBOIS, Giovanni B. FRISONI, Valentina GARIBOTTO, Jean GEORGES, Samia HURST, Frank JESSEN, Miia KIVIPELTO, David LLEWELLYN, Laura McWHIRTER, Richard MILNE, Carolina MINGUILLÓN, Carlo MINIUSSI, José Luis MOLINUEVO, Peter M NILSSON, Janice RANSON, Federica RIBALDI, Craig RITCHIE, Philip SCHELTENS, Alina SOLOMON, Wiesje VAN DER FLIER, Cornelia VAN DUIJN, Bruno VELLAS, Leonie VISSER.
Funding
This paper was the product of a workshop funded by the Swiss National Science Foundation entitled “Dementia Prevention Services” (grant number: IZSEZ0_193593).
JMR is supported by Alzheimer’s Research UK.
DJL is supported by the National Institute for Health Research (NIHR) Applied Research Collaboration (ARC) South West Peninsula, Alzheimer’s Research UK, National Health and Medical Research Council (NHMRC), JP Moulton Foundation, National Institute on Aging/National Institutes of Health (RF1AG055654), and Alan Turing Institute/Engineering and Physical Sciences Research Council (EP/N510129/1).
CJM is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
GBF received funding from the EU-EFPIA Innovative Medicines Initiatives 2 Joint Undertaking (IMI 2 JU) “European Prevention of Alzheimer’s Dementia consortium” (EPAD, grant agreement number: 115736) and “Amyloid Imaging to Prevent Alzheimer’s Disease” (AMYPAD, grant agreement number: 115952); the Swiss National Science Foundation: “Brain connectivity and metacognition in persons with subjective cognitive decline (COSCODE): correlation with clinical features and in vivo neuropathology” (grant number: 320030_182772).
KB is supported by the Swedish Research Council (#2017-00915), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), and the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986).
CD is supported by the CoSTREAM project (European Union‘s Horizon 2020 research and innovation program under grant agreement number 667375).
ET is funded by the National Institute for Health Research (NIHR) School for Primary Care Research. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Consortia
Contributions
Janice M. Ranson and David J Llewellyn contributed to the drafting and revision of the manuscript for intellectual content. Timothy Rittman, Shabina Hayat, Carol Brayne, Frank Jessen, Kaj Blennow, Cornelia van Duijn, Frederik Barkhof, Eugene Tang, Blossom Stephan, and Catherine Mummery contributed to the drafting and revision of the manuscript for intellectual content. José Luis Molinuevo and Philip Scheltens contributed to the conception of the work. Daniele Altomare, Giovanni B. Frisoni, and Federica Ribaldi conceived and organized the workshop whence the Papers of the BHS series in this issue of Alzheimer’s Research & Therapy originated, conceived the related editorial initiative, revised this manuscript for intellectual content, harmonized the manuscript with the other Papers of the BHS series, and approved the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
GBF reports grants from Alzheimer Forum Suisse, Académie Suisse des Sciences Médicales, Avid Radiopharmaceuticals, Biogen, GE International, Guerbert, Association Suisse pour la Recherche sur l’Alzheimer, IXICO, Merz Pharma, Nestlé, Novartis, Piramal, Roche, Siemens, Teva Pharmaceutical Industries, Vifor Pharma, and Alzheimer’s Association; he has received personal fees from AstraZeneca, Avid Radiopharmaceuticals, Elan Pharmaceuticals, GE International, Lundbeck, Pfizer, and TauRx Therapeutics.
KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program.
JLM is currently a full time employee of Lundbeck and has previously served as a consultant or at advisory boards for the following for-profit companies, or has given lectures in symposia sponsored by the following for-profit companies: Roche Diagnostics, Genentech, Novartis, Lundbeck, Oryzon, Biogen, Lilly, Janssen, Green Valley, MSD, Eisai, Alector, BioCross, GE Healthcare, ProMIS Neurosciences.
All other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ranson, J.M., Rittman, T., Hayat, S. et al. Modifiable risk factors for dementia and dementia risk profiling. A user manual for Brain Health Services—part 2 of 6. Alz Res Therapy 13, 169 (2021). https://doi.org/10.1186/s13195-021-00895-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-021-00895-4