
Abstract
Background
Genome-wide association studies have identified more than 30 Alzheimer’s disease (AD) risk genes, although the detailed mechanism through which all these genes are associated with AD pathogenesis remains unknown. We comprehensively evaluate the roles of the variants in top 30 non-APOE AD risk genes, based on whether these variants were associated with altered mRNA transcript levels, as well as brain amyloidosis, tauopathy, and neurodegeneration.
Methods
Human brain gene expression data were obtained from the UK Brain Expression Consortium (UKBEC), while other data used in our study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. We examined the association of AD risk allele carrier status with the levels of gene expression in blood and brain regions and tested the association with brain amyloidosis, tauopathy, and neurodegeneration at baseline, using a multivariable linear regression model. Next, we analyzed the longitudinal effects of these variants on the change rates of pathology using a mixed effect model.
Results
Altogether, 27 variants were detected to be associated with the altered expression of 21 nearby genes in blood and brain regions. Eleven variants (especially novel variants in ADAM10, IGHV1-68, and SLC24A4/RIN3) were associated with brain amyloidosis, 7 variants (especially in INPP5D, PTK2B) with brain tauopathy, and 8 variants (especially in ECHDC3, HS3ST1) with brain neurodegeneration. Variants in ADAMTS1, BZRAP1-AS1, CELF1, CD2AP, and SLC24A4/RIN3 participated in more than one cerebral pathological process.
Conclusions
Genetic variants might play functional roles and suggest potential mechanisms in AD pathogenesis, which opens doors to uncover novel targets for AD treatment.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is highly heritable, with late-onset AD (LOAD) showing heritability of 58–79% [1]. Previous large-scale genome-wide association studies (GWASs) have discovered more than 20 AD gene variants that confer risk for LOAD [2,3,4,5,6,7]. APOE is still the strongest genetic risk factor for LOAD, responsible for a 3- to 15-fold increase in risk [8]. Yet these confirmed variants only account for a small portion of disease heritability. In the search for additional LOAD risk variants, recent GWAS meta-analyses [9,10,11] identified totally more than 30 AD risk genes. The identification of these novel gene variants might provide valuable insights into the molecular mechanisms with important roles in AD pathogenesis.
Although most of the non-APOE AD-associated genetic variants described to date are located in intronic or noncoding regions, these variants still could affect the nearby gene expression and exert protective or disease-inducing effects [12, 13]. In addition, AD develops following a long pre-clinical phase with abnormal neuropathological biomarkers [14]. Recently, the National Institute on Aging and Alzheimer’s Association (NIA-AA) published an updated research framework [15] that provides a flexible platform to generate or test hypotheses concerning different pathologic processes of AD, which defines AD by neuropathological biomarkers in three categories: amyloidosis [A] biomarkers, particularly cortical amyloid-PET ligand binding or low CSF Aβ42; tauopathy [T] biomarkers, particularly elevated CSF phosphorylated tau (pTau) or cortical tau-PET ligand binding; and neurodegeneration [N] biomarkers, particularly elevated CSF total tau (tTau), diminished 18F-fluorodeoxyglucose (FDG)-PET, or brain structural atrophy on MRI.
Many of the early identified AD variants have been associated with expression levels of their nearby genes and implicated in brain amyloidosis and neurodegeneration [12, 16, 17]. Relatively, fewer studies have reported associations of these top AD risk gene variants with brain tauopathy [18,19,20], and most of these studies focused on a single variant, or a few variants. More importantly, the precise disease-associated mechanisms of the novel genetic variants identified in the recent GWAS meta-analysis remain unknown. Here, we report a comprehensive analysis of the associations of the variants in the top 30 non-APOE AD risk genes from current large-scale GWAS studies of the transcript expression levels, and the pathological processes of brain amyloidosis, tauopathy, and neurodegeneration, using the baseline and follow-up data from AD-related CSF, PET, and MRI measures. Understanding the mechanisms by which these variants contribute to AD risk will lead to a better understanding of the disease-associated mechanisms and help uncover novel therapeutic avenues.
Methods
Study design and participants
Human brain gene expression data were obtained from Braineac dataset, UK Brain Expression Consortium (UKBEC), and included 10 brain regions from 134 neuropathologically normal individuals of European descent. The 10 brain regions were the cerebellar cortex (CRBL), frontal cortex (FCTX), hippocampus (HIPP), medulla (MEDU), occipital cortex (OCTX), putamen (PUTM), substantia nigra (SNIG), temporal cortex (TCTX), thalamus (THAL), and intralobular white matter (WHMT). In the Braineac dataset, Affymetrix GeneChip Human exon 1.0ST arrays were used to measure the gene expression in transcript levels. Detailed information on these methods is described in the Braineac database [21]. Genomic DNA for individuals from UKBEC was extracted from sub-dissected samples (100–200 mg) of human post-mortem brain tissue using Gentra Puregene Kit (Qiagen, UK). Samples from every individual were run on the genotype chip (the Illumina Infinium Omni1-Quad BeadChip). The BeadChips were scanned using an iScan (Illumina, USA) with an AutoLoader (Illumina, USA). GenomeStudio v.1.8.X (Illumina, USA) was used for analyzing the data and generating SNP calls. All other data used in this study were obtained from Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD, VA Medical Center and University of California-San Francisco. Our analyses included all individuals with diagnosed AD, mild cognitive impairment (MCI), and normal cognition (NC), with clinical information, GWAS data, and gene expression data from peripheral blood (Affymetrix Human Genome U219 Array platform), or longitudinal AD-related CSF, PET, and MRI data. Furthermore, we selected only non-Hispanic white individuals to avoid population stratification effects which can lead to spurious findings. In total, 1183 individuals at baseline were included in our study (Table 1). Among them, 739 individuals had gene expression data (Additional file 1: Figure S1).
Gene variant selection and imputation
The ADNI-1, ADNI-2, and ADNI–Grand Opportunity (GO) participants were genotyped according to the manufacturer’s protocol. We focused on well-established AD risk genes identified in AD GWASs available to date (Additional file 1: Table S1), which yielded a total of 68 variants. Missing genotypes were imputed using the Beagle software with the HapMap GRCh37 as a reference. Among them, 19 of our genes were represented by more than one variant. We performed linkage disequilibrium (LD) analyses followed by Cohen kappa (κ) statistics. When choosing between 2 variants with significant overlap (high r2 and high κ), we retained the variant with the smallest amount of missing data. Our final number of non-APOE AD variants was thus reduced to 42 (Additional file 1: Table S2). More detailed information is described in Additional file 1 .
CSF measurements
CSF Aβ42 and pTau were measured at the ADNI Biomarker Core Laboratory (University of Pennsylvania) using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium; for research use only reagents) immunoassay kit-based reagents. All CSF biomarker assays were performed in duplicate and averaged.
AV45-PET/AV1415-PET data acquisition and analyses
A detailed description of PET image acquisition and processing can be found at http://adni.loni.usc.edu/datasamples/pet/. The AV45-PET (amyloid-PET) and AV1415-PET (tau-PET) standardized uptake value ratios (SUVRs) were formed by normalizing composite multi-region target regions of interest (ROIs) to the cerebellar crus gray matter. The amyloid-PET target meta-ROI included the frontal, anterior cingulate, precuneus, and parietal cortex [22]. The tau-PET target meta-ROI used in the primary analysis included the amygdala, entorhinal cortex, fusiform, parahippocampal, and inferior temporal and middle temporal gyri [23].
FDG-PET data acquisition and analyses
The cerebral metabolic rate for glucose (CMRgl) data on FDG-PET was downloaded from the ADNI dataset. Mean FDG uptake was averaged from 5 meta-ROIs including the right and left angular gyri, right and left inferior temporal regions, and bilateral posterior cingulate. PET images were spatially normalized in Statistical Parametric Mapping (SPM) to the MNI PET template. We intensity-normalized each meta-ROI mean by dividing it by the pons/vermis reference region mean.
Structural MRI data
Hippocampal volume (HV) and estimated intracranial volume (eICV) were performed from T1-weighted MRI acquired with a Siemens Trio 3.0 T or 1.5 T scanner. Regional volume estimates were processed using the Freesurfer software (https://surfer.nmr.mgh.harvard.edu). HV was adjusted for eICV using the following equation: Adjusted HV (HVa) = Raw HV – b (eICV – Mean eICV), where b is the regression coefficient when HV is regressed against eICV.
Statistical analyses
Clinical and demographic characteristics for each variant were compared using t tests or χ2 tests with 2-sided P values, as appropriate. First, a multivariable linear regression model was used to analyze the association of AD risk allele carrier status with the gene expression levels, using the genes that have been annotated to those variants by GWAS. Age and gender as covariates were included in the model and tested for statistical significance. A multivariable linear regression model was also used to analyze the association of AD risk allele carrier status with brain amyloidosis, tauopathy, and neurodegeneration, based on AD-related CSF, PET, and MRI data at baseline. Age, gender, educational level, APOEε4 genotype, and diagnosis were included as covariates. Then, we analyzed the longitudinal effects of these variants on the change rates of pathologically characteristic data above, using a multivariable linear mixed effect model with fixed effects of time (year) from baseline, AD risk allele carrier status, and interaction between time from baseline and AD risk allele carrier status. The model included random slope and intercept terms for each participant with age, gender, educational level, APOEε4 genotype, and diagnosis as covariates. All statistical analyses were conducted using R statistical software. We defined associations with false discovery rate (FDR)-adjusted P values < 0.05 as statistically significant using the Benjamini-Hochberg procedure.
Results
Association of variants with gene expression in the blood
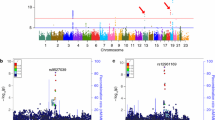
The associations between AD risk allele carrier status and gene expression in the blood are shown in Additional file 1: Table S3. Figure 1 shows those associated variants that reached FDR-adjusted significance level. In total, 15 variants were detected to be associated with the altered expression in blood of 10 nearby genes. The minor alleles of 5 variants in CR1, ECHDC3, MS4A6A, and NME8 were associated with decreased expressions, while the other 10 variants in BIN1, EPHA1, HLA-DRB1, PTK2B, SLC24A4/RIN3, and ZCWPW1 associated with increased expressions.

Association of the variants with gene expression in peripheral blood and brain regions. The significant associations between the variants and the levels of gene expression in blood were identified from the ADNI database, and the significant associations in specific brain regions were obtained from the Braineac dataset. FDR-adjusted P values with statistical significance are shown. Abbreviations: CRBL, cerebellar cortex; FCTX, frontal cortex; HIPP, hippocampus; MEDU, medulla; OCTX, occipital cortex; PUTM, putamen; SNIG, substantia nigra; TCTX, temporal cortex; THAL, thalamus; WHMT, intralobular white matter
Association of variants with gene expression in the brain
The associations between AD risk allele carrier status and brain gene expression are shown in Additional file 1: Table S4. Figure 1 shows the significantly associated variants in 10 different brain regions. In total, 18 variants were detected to be associated with the altered expression of 15 nearby genes in specific brain regions. Especially in AD-related TCTX, HIPP, and FCTX regions, the minor alleles of rs11136000 in CLU and rs6701713 in CR1 were associated with increased expressions of CLU and CR1. The minor alleles of rs2526378 in BZRAP1-AS1 and rs12590654 in SLC24A4/RIN3 were also associated with increased expression especially in TCTX/FCTX and FCTX, respectively, while the minor allele of rs3865444 in CD33 was associated with decreased expression in TCTX (Additional file 1: Figure S2).
Association of variants with brain amyloidosis
We tested for associations of AD risk allele carrier status with brain amyloidosis, based on CSF Aβ42 or amyloid-PET data at baseline and follow-up. Our results indicate that ABCA7 rs3752246 was significantly associated with CSF Aβ42 levels (FDR-adjusted P = 0.008) and amyloid-PET levels (FDR-adjusted P = 0.001) at baseline (Fig. 2a, b), suggesting the strongest association with brain amyloid pathology. In addition, FERMT2 rs17125944 was detected as associated with the altered CSF Aβ42 levels at baseline and follow-up. The risk C allele carrier of rs17125944 was associated with decreased CSF Aβ42 levels at baseline (FDR-adjusted P = 0.004), but these changes over time were not clear from the longitudinal analysis (Fig. 3a).

Association of the variants with brain amyloidosis, tauopathy, and neurodegeneration (FDG-PET levels or MRI hippocampal volumes) at baseline. We tested for significant associations of AD risk allele carrier status with brain amyloidosis, based on CSF Aβ42 or amyloid-PET data; the associations with brain neurodegeneration, based on FDG-PET or MRI HVa data; and the associations with brain tauopathy, based on CSF pTau or tau-PET data. a The minor allele carriers of ABCA7 rs3752246, BZRAP1-AS1 rs2526378, FERMT2 rs17125944, SLC24A4/RIN3 rs10498633, and SLC24A4/RIN3 rs12590654 were significantly associated with decreased CSF Aβ42 levels, and ADAM10 rs593742, IGHV1-68 rs79452530, and CELF1 rs3740688 associated with increased CSF Aβ42 levels. b The minor allele carriers of ABCA7 rs3752246 were associated with increased amyloid-PET levels, and ADAMTS1 rs2830500, CLU rs11136000, and EPHA1 rs11771145 associated with decreased levels of amyloid-PET. c The minor allele carriers of BZRAP1-AS1 rs2526378 and HS3ST1 rs6448807 were associated with decreased FDG-PET levels. d The minor allele carriers of ECHDC3 rs11257242 were associated with increased MRI HVa levels. e The minor allele carriers of BIN1 rs744373 and BIN1 rs6733839 had lower CSF pTau levels, and CD2AP rs9381563 and INPP5D rs10933431 had higher CSF pTau levels. f The minor allele carriers of BIN1 rs744373 and BIN1 rs7561528 had higher tau-PET levels, and SLC24A4/RIN3 rs10498633 had lower tau-PET levels

Longitudinal effects of the variants on the change rates of brain amyloidosis, tauopathy, and neurodegeneration. a The risk C allele carrier of FERMT2 rs17125944 was associated with decreased CSF Aβ42 levels at baseline, although these changes over time were not obvious from the longitudinal analysis. b The longitudinal effect of PTK2B rs28834970 on the change rate of CSF pTau levels was significant. The minor allele carriers of PTK2B rs28834970 exhibited faster rise in CSF pTau levels. c Longitudinal effects of CELF1 rs3740688 and PICALM rs3851179 on the change rate of MRI HVa levels were significant. The minor allele carriers of CELF1 rs3740688 and PICALM rs3851179 exhibited slower decline in MRI HVa levels. d Longitudinal effects of ADAMTS1 rs2830500, CD2AP rs9381563, and CD33 rs3865444 on the change rate of FDG-PET levels were significant. The minor allele carriers of CD2AP rs9381563 exhibited faster decline in FDG-PET levels, and the minor allele carriers of ADAMTS1 rs2830500 and CD33 rs3865444 exhibited slower decline in FDG-PET levels. We analyzed the longitudinal effects of these variants on the change rates of pathology using a linear mixed effect model. The shaded regions refer to 95% confidence intervals
There were 9 other variants associated with brain amyloidosis. FDR-adjusted P values are shown in Additional file 1: Table S5. Among them, ADAM10 rs593742, BZRAP1-AS1 rs2526378, CELF1 rs3740688, IGHV1-68 rs79452530, SLC24A4/RIN3 rs10498633, and SLC24A4/RIN3 rs12590654 were associated with altered CSF Aβ42 levels at baseline (Fig. 2a), while ADAMTS1 rs2830500, CLU rs11136000, and EPHA1 rs11771145 were associated with altered amyloid-PET levels at baseline (Fig. 2b). Remarkably, there was no evidence for an effect of these variants on CSF Aβ42 levels or amyloid-PET levels from the longitudinal analysis.
Association of variants with brain tauopathy
We examined the associations of AD risk allele carrier status with brain tauopathy, based on CSF pTau or tau-PET data at baseline and follow-up. Our results indicate that BIN1 rs744373 was significantly associated with CSF pTau levels (FDR-adjusted P = 0.004) and tau-PET levels (FDR-adjusted P = 0.045) at baseline (Fig. 2e, f), and thus, there is a strong association with brain tau pathology. In addition, another two variants rs6733839 and rs7561528 in BIN1 were detected to be associated with tau pathology. The minor allele carriers of rs744373 and rs6733839 in BIN1 had lower CSF pTau levels at baseline (Fig. 2e), and rs744373 and rs7561528 in BIN1 had higher tau-PET levels (Fig. 2f).
Furthermore, CD2AP rs9381563 and INPP5D rs10933431 were discovered to be associated with altered CSF pTau levels, and SLC24A4/RIN3 rs10498633 was associated with altered tau-PET levels at baseline (Fig. 2e, f). Additional file 1: Table S5 lists the FDR-corrected P results for these associations. Based on the longitudinal follow-up data, although there was no association of PTK2B rs28834970 with brain tauopathy at baseline, the longitudinal effect of this variant on the change rate of CSF pTau levels was significant (FDR-adjusted P = 0.018). The minor allele carrier of PTK2B rs28834970 was associated with the rapid growth rate of CSF pTau levels (Fig. 3b; Additional file 1: Figure S3a).
Association of variants with brain neurodegeneration
We analyzed the associations of AD risk allele carrier status with brain neurodegeneration, based on FDG-PET or MRI HVa data at baseline and follow-up. The newly discovered variants BZRAP1-AS1 rs2526378 and HS3ST1 rs6448807 were associated with altered FDG-PET levels (FDR-adjusted P = 0.046 and P = 0.007, respectively; Fig. 2c), and ECHDC3 rs11257242 associated with altered MRI HVa levels (FDR-adjusted P = 0.045, Fig. 2d) at baseline. Based on the longitudinal follow-up data, the longitudinal effects of CELF1 rs3740688 and PICALM rs3851179 on the change rate of MRI HVa levels were significant, and the effects of ADAMTS1 rs2830500, CD2AP rs9381563, and CD33 rs3865444 on the change rate of FDG-PET levels were also significant, although there was no association of these variants with brain neurodegeneration at baseline. Figure 3c, d shows these longitudinal associations (also in Additional file 1: Figure S3b,c), and Additional file 1: Table S5 lists the FDR-corrected P results.
It is worth noting that, according to our current set of analyses, AD-associated genetic variants ADAMTS1 rs2830500, BZRAP1-AS1 rs2526378, and CELF1 rs3740688 might affect the mechanisms involved both in brain amyloidosis and neurodegeneration, implying the commonalities or convergence in function. AD-associated genetic variants CD2AP rs9381563 might affect the mechanisms involved both in brain tauopathy and neurodegeneration, and SLC24A4/RIN3 rs10498633 in brain amyloidosis and tauopathy (Fig. 4).

Role of variants in top 30 non-APOE AD risk genes in AD pathogenesis. Although most AD-associated genetic variants described to date are located in intronic or noncoding regions, these variants still could affect the nearby gene expression and exert protective or disease-inducing effects in AD-related amyloidosis, tauopathy, or neurodegeneration. Based on our analysis of all currently available data in ADNI, 11 variants were associated with brain amyloidosis, 7 variants associated with brain tauopathy, and 8 variants associated with brain neurodegeneration. Among them, variants in ADAMTS1, BZRAP1-AS1, and CELF1 affect the mechanisms involved both in brain amyloidosis and neurodegeneration, CD2AP in brain tauopathy and neurodegeneration, and SLC24A4/RIN3 in brain amyloidosis and tauopathy, implying these genes might contribute to AD risk via either common or distinct mechanisms
Discussion
Although large GWAS has recently identified novel variants that are associated with altered AD risk, we have a relatively poor understanding of their functional impact. In this study, we comprehensively analyze possible functional effects of the variants in top 30 non-APOE AD risk genes, based on whether (1) these variants are associated with altered expression levels and (2) the variants are associated with brain amyloidosis, tauopathy, and neurodegeneration.
Firstly, 27 variants were detected as associated with the altered expression of 21 nearby genes in blood and brain regions. Among them, variants in ABCA7, ADAM10, ADAMTS1, BZRAP1-AS1, CASS4, CD2AP, CD33, CELF1, CLU, HS3ST1, and PICALM were associated with the altered gene expressions in different brain regions alone, and variants in BIN1, ECHDC3, EPHA1, HLA-DRB1, NME8, and PTK2B were associated with the altered gene expressions only in the blood, while only 6 variants in CR1, MS4A6A, SLC24A4/RIN3, and ZCWPW1 affected these 4 nearby gene expressions both in the blood and in the brain, which perhaps become promising key biomarkers for AD diagnosis. Previous studies have shown that the overlap of genomic variants influencing transcript expression levels in both the human brain and blood is relatively low [24]. Of course, the smaller sample sizes from human brain tissues might be one possible reason for an underestimation of the true level. Notably, although 6 variants are associated with both brain and blood expression, the association was not always in the same direction. For example, CR1 rs6701713 was associated with decreased expression in blood but increased expression in specific brain regions. The direction of association of the same variant in different brain regions was also different (Additional file 1: Figure S2). Therefore, genetic variants may require tissue-, cell-, region-, and disease-specific factors to exert their influences on gene expression. In any case, the variants associated with AD susceptibility are more likely to affect the expression levels in a tissue-specific and region-specific manner, and provide important regulating mechanisms of genetic variants in AD risk.
AD research has mainly focused in brain TCTX, HIPP, and FCTX regions. Consistent with previous studies [12, 25,26,27], our study showed that AD risk variants in CR1, CD33, CLU, and SLC24A4/RIN3 affect their gene expression levels in the above key regions for brain regulatory effects. Remarkably, the novel variant rs2526378 in BZRAP1-AS1 was found to influence its expression levels in TCTX/FCTX. Besides the changes in cortical regions, some genetic variants were associated with the altered expression in subcortical brain structures (THAL, PUTM) or WHMT (Fig. 1). Because the degenerative processes in these regions might contribute to cognitive decline and are mechanistically important in AD [28,29,30], more attention should be paid to these associations in future research, which might become novel potential treatment targets. In general, evaluating the potential associations using large-scale expression GWAS datasets for multiple brain regions and peripheral blood in the same individuals would provide more valuable information.
Based on the baseline and longitudinal follow-up data, we were able to confirm the previously reported associations of genetic variants in ABCA7, CELF1, CLU, EPHA1, and FREMT2 with brain amyloidosis as previously described [16, 20, 31]. The novel genome-wide variants in ADAM10 (most important α-secretase in the process of amyloid-β protein precursor (APP) cleavage) [32, 33] and ADAMTS1 (within 665 kb of APP on chromosome 21) [34] were also found to affect brain amyloidosis. Additionally and to the best of our knowledge, we are the first to report the associations for novel variants in IGHV1-68, BZRAP1-AS1, and SLC24A4/RIN3 with brain amyloidosis.
Previous studies have shown that BIN1 rs744373 was associated with altered tau-PET and CSF pTau levels [18, 35]. Our study supports the suggestion that other variants in BIN1 are also significantly associated with tau biomarkers. Similarly, we confirm the previously reported association of INPP5D, CD2AP, and PTK2B with brain tauopathy [19, 20, 36, 37]. Conversely, the relationship between the variant in SLC24A4/RIN3 and tau pathology is a new discovery. SLC24A4 CpG methylation sites were found associated with Aβ burden and tau pathology previously [38] and SLC24A4 also appeared to take part in lipid metabolism [39] and brain glucose metabolism [17].
As most of the earlier studies were cross-sectional analyses, a stage-specific association might occur for genetic variants that influence the course of neurodegeneration [17]. Using longitudinal follow-up data in our analysis, it is possible to identify the effects of variants in CD2AP, CD33, CELF1, and PICALM on the changes in brain metabolism or atrophy over time, supporting the reported associations of these genes with brain neurodegeneration [17, 40,41,42]. However, the associations of variants in ADAMTS1, ECHDC3, BZRAP1-AS1, and HS3ST1 with neurodegeneration biomarkers at baseline are novel findings of this research. Aligned with this, HS3ST1 was reported as significantly associated with working memory in probable-MCI patients [43].
According to our results, variants in ADAMTS1, BZRAP1-AS1, and CELF1 affect the mechanisms involved both in brain amyloidosis and neurodegeneration, CD2AP in brain tauopathy and neurodegeneration, and SLC24A4/RIN3 in brain amyloidosis and tauopathy (Fig. 4), implying these genes might contribute to AD risk via either common or distinct mechanisms. Previous studies have shown that ADAMTS1, within 665 kb of APP on chromosome 21, has elevated expression in Down’s syndrome and LOAD brain [34, 44] and is a potential neuroprotective gene or neuroinflammatory gene important to microglial response [45]. BZRAP1 (also known as TSPOAP1) is a subunit of the benzodiazepine receptor complex in mitochondria and a marker of neuroinflammation. Previous studies have demonstrated that the TSPO ligand can reverse Aβ accumulation and behavioral impairment in transgenic mice [46]. Moreover, the CELF1 variant has been shown to affect cognition and CSF Aβ42 levels by modifying expression [31, 47]. And the fly homolog of CELF1, aret, also shows mediation of tau toxicity [48]. Much more research on the roles of these genes and how they relate to each other is very important.
It is worth noting that, in our study, we place CSF and PET imaging biomarkers all into analysis; that is because the fundamental difference between the two should be recognized. CSF biomarkers are measures of the concentrations of proteins in CSF from the lumbar sac that reflect the rates of both production (protein expression or release/secretion from neurons or other brain cells) and clearance (degradation or removal) at a given point in time. While imaging measures, on the other hand, represent the magnitude of the neuropathologic load or damage accumulated over time. Low CSF Aβ42 is therefore best considered a biomarker of a pathologic state that is associated with amyloid plaque formation and not a measure of amyloid plaque load as amyloid-PET is. Similarly, CSF pTau is best considered a biomarker of a pathologic state that is associated with PHF tau formation and not a measure of pathologic tau deposits as tau-PET is [15]. In addition, FDG-PET was labeled as neurodegeneration biomarkers in our current study, because our selection and classification of biomarkers is based on the 2018 NIA-AA research framework. However, a growing number of studies, including the recent article published by our team [49], suggest FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis, because FDG hypometabolism is a summation of multiple biological processes, not just neuronal hypometabolism and neurodegeneration. We sincerely hope more practitioners and academia to deep study in this field to further improve the understanding of AD biomarkers.
Generally, the ultimate goal of understanding the genetic architecture of AD is to enhance the understanding of disease mechanisms. Based on the above A/T/N classification system, our identification of many risk genes suggests their shared function in brain amyloidosis, tauopathy, or neurodegeneration, which might provide interesting targets for future functional follow-up and biological interpretation.
Limitation
Our current study has limitations. Firstly, the subjects with tau-PET data, especially the longitudinal tau-PET data, are limited. Secondly, the follow-up CSF data are limited because of the invasiveness of the procedure to acquire samples. With the rapid development and wide application of PET technology, future studies will validate our findings in a large, independent, longitudinal cohort with a greater number of individuals and time points and with a longer follow-up time to provide more statistically powerful results.
Conclusions
In summary, our current study provides new insights into the variants in the top 30 non-APOE AD risk genes associated with transcript expression levels and involved in the pathological processes of brain amyloidosis, tauopathy, and neurodegeneration. This evidence increases the possibility that genetic variants might play functional roles and suggest potential mechanisms in AD pathogenesis. Further studies are needed to fully understand their roles in AD process, and our research opens doors to the investigation of novel targets for AD treatment.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- CSF:
-
Cerebrospinal fluid
- FDG:
-
Fluorodeoxyglucose
- GWASs:
-
Genome-wide association studies
- HV:
-
Hippocampal volume
- LOAD:
-
Late-onset Alzheimer’s disease
- MCI:
-
Mild cognitive impairment
- NC:
-
Normal cognition
- NIA-AA:
-
National Institute on Aging and Alzheimer’s Association
- PET:
-
Positron-emission tomography
- pTau:
-
Phosphorylated tau
- tTau:
-
Total tau
- UKBEC:
-
UK Brain Expression Consortium
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41.
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–40.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Witoelar A, Rongve A, Almdahl IS, Ulstein ID, Engvig A, White LR, et al. Meta-analysis of Alzheimer’s disease on 9,751 samples from Norway and IGAP study identifies four risk loci. Sci Rep. 2018;8:18088.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13.
Katsumata Y, Nelson PT, Estus S, Fardo DW. Translating Alzheimer’s disease-associated polymorphisms into functional candidates: a survey of IGAP genes and SNPs. Neurobiol Aging. 2019;74:135–46.
Jiang Q, Hu Y, Liu G. Association of Alzheimer disease susceptibility variants and gene expression in the human brain. JAMA Neurol. 2016;73:1255.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Apostolova LG, Risacher SL, Duran T, Stage EC, Goukasian N, West JD, et al. Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 2018;75:328–41.
Stage E, Duran T, Risacher SL, Goukasian N, Do TM WJD, et al. The effect of the top 20 Alzheimer disease risk genes on gray-matter density and FDG PET brain metabolism. Alzheimers Dement (Amst). 2016;5:53–66.
Franzmeier N, Rubinski A, Neitzel J, Ewers M. The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat Commun. 2019;10:1766.
Ramos de Matos M, Ferreira C, Herukka SK, Soininen H, Janeiro A, Santana I, et al. Quantitative genetics validates previous genetic variants and identifies novel genetic players influencing Alzheimer’s disease cerebrospinal fluid biomarkers. J Alzheimers Dis. 2018;66:639–52.
Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–56.
Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–28.
Yu JT, Li JQ, Suckling J, Feng L, Pan A, Wang YJ, et al. Frequency and longitudinal clinical outcomes of Alzheimer’s AT(N) biomarker profiles: a longitudinal study. Alzheimers Dement. 2019;15:1208–17.
Jack CR Jr, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13:205–16.
McKenzie M, Henders AK, Caracella A, Wray NR, Powell JE. Overlap of expression quantitative trait loci (eQTL) in human brain and blood. BMC Med Genet. 2014;7:31.
Roshchupkin GV, Adams HH, van der Lee SJ, Vernooij MW, van Duijn CM, Uitterlinden AG, et al. Fine-mapping the effects of Alzheimer’s disease risk loci on brain morphology. Neurobiol Aging. 2016;48:204–11.
Allen M, Kachadoorian M, Carrasquillo MM, Karhade A, Manly L, Burgess JD, et al. Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol Genet. 2015;1:e15.
Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79:221–8.
Cho H, Kim JH, Kim C, Ye BS, Kim HJ, Yoon CW, et al. Shape changes of the basal ganglia and thalamus in Alzheimer’s disease: a three-year longitudinal study. J Alzheimers Dis. 2014;40:285–95.
de Jong LW, van der Hiele K, Veer IM, Houwing JJ, Westendorp RG, Bollen EL, et al. Strongly reduced volumes of putamen and thalamus in Alzheimer’s disease: an MRI study. Brain. 2008;131:3277–85.
Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22.
Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–61.
Yuan XZ, Sun S, Tan CC, Yu JT, Tan L. The role of ADAM10 in Alzheimer’s disease. J Alzheimers Dis. 2017;58:303–22.
Vassar R. ADAM10 prodomain mutations cause late-onset Alzheimer’s disease: not just the latest FAD. Neuron. 2013;80:250–3.
Miguel RF, Pollak A, Lubec G. Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold overexpressed in neurodegenerative disorders as Down syndrome, Alzheimer’s and Pick’s disease. Brain Res Mol Brain Res. 2005;133:1–5.
Wang HF, Wan Y, Hao XK, Cao L, Zhu XC, Jiang T, et al. Bridging integrator 1 (BIN1) genotypes mediate Alzheimer’s disease risk by altering neuronal degeneration. J Alzheimers Dis. 2016;52:179–90.
McMillan CT, Lee EB, Jefferson-George K, Naj A, Van Deerlin VM, Trojanowski JQ, et al. Alzheimer’s genetic risk is reduced in primary age-related tauopathy: a potential model of resistance? Ann Clin Transl Neurol. 2018;5:927–34.
Dourlen P, Fernandez-Gomez FJ, Dupont C, Grenier-Boley B, Bellenguez C, Obriot H, et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of tau pathology. Mol Psychiatry. 2017;22:874–83.
Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J, et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015;72:15–24.
Kraja AT, Borecki IB, Tsai MY, Ordovas JM, Hopkins PN, Lai CQ, et al. Genetic analysis of 16 NMR-lipoprotein fractions in humans, the GOLDN study. Lipids. 2013;48:155–65.
Furney SJ, Simmons A, Breen G, Pedroso I, Lunnon K, Proitsi P, et al. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer’s disease. Mol Psychiatry. 2011;16:1130–8.
Chauhan G, Adams HHH, Bis JC, Weinstein G, Yu L, Toglhofer AM, et al. Association of Alzheimer’s disease GWAS loci with MRI markers of brain aging. Neurobiol Aging. 2015;36:1765 e7–e16.
Li JQ, Wang HF, Zhu XC, Sun FR, Tan MS, Tan CC, et al. GWAS-linked loci and neuroimaging measures in Alzheimer’s disease. Mol Neurobiol. 2017;54:146–53.
Espinosa A, Hernandez-Olasagarre B, Moreno-Grau S, Kleineidam L, Heilmann-Heimbach S, Hernandez I, et al. Exploring genetic associations of Alzheimer’s disease loci with mild cognitive impairment neurocognitive endophenotypes. Front Aging Neurosci. 2018;10:340.
Medoro A, Bartollino S, Mignogna D, Marziliano N, Porcile C, Nizzari M, et al. Proteases upregulation in sporadic Alzheimer’s disease brain. J Alzheimers Dis. 2019;68:931–8.
Wilcock DM. Neuroinflammation in the aging Down syndrome brain; lessons from Alzheimer’s disease. Curr Gerontol Geriatr Res. 2012;2012:170276.
Barron AM, Garcia-Segura LM, Caruso D, Jayaraman A, Lee JW, Melcangi RC, et al. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2013;33:8891–7.
Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age (Dordr). 2016;38:41.
Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, Devenport D, et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum Mol Genet. 2014;23:870–7.
Ou YN, Xu W, Li JQ, Guo Y, Cui M, Chen KL, et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: a longitudinal study. Alzheimers Res Ther. 2019;11:57.
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
This study was supported by grants from the National Natural Science Foundation of China (91849126), the National Key R&D Program of China (2018YFC1314700), Taishan Scholars Program of Shandong Province (tsqn20161078), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University.
Author information
Authors and Affiliations
Consortia
Contributions
MST and JTY conceptualized the study, analyzed and interpreted the data, and drafted and revised the manuscript. YXY, WX, HFW, and LT analyzed and interpreted the data. CTZ, QD, LT, and JS interpreted the data and revised the manuscript. JS and JTY had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to the writing and revisions of the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by institutional review boards of all participating institutions, and written informed consent was obtained from all participants or their guardians according to the Declaration of Helsinki (consent for research).
Consent for publication
Not applicable.
Competing interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tan, MS., Yang, YX., Xu, W. et al. Associations of Alzheimer’s disease risk variants with gene expression, amyloidosis, tauopathy, and neurodegeneration. Alz Res Therapy 13, 15 (2021). https://doi.org/10.1186/s13195-020-00755-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-020-00755-7