
Abstract
Background
According to new diagnostic guidelines for Alzheimer’s disease (AD), biomarkers enable estimation of the individual likelihood of underlying AD pathophysiology and the associated risk of progression to AD dementia for patients with mild cognitive impairment (MCI). Nonetheless, how conflicting biomarker constellations affect the progression risk is still elusive. The present study explored the impact of different cerebrospinal fluid (CSF) biomarker constellations on the progression risk of MCI patients.
Methods
A multicentre cohort of 469 patients with MCI and available CSF biomarker results and clinical follow-up data was considered. Biomarker values were categorized as positive for AD, negative or borderline. Progression risk differences between patients with different constellations of total Tau (t-Tau), phosphorylated Tau at threonine 181 (p-Tau) and amyloid-beta 1–42 (Aβ42) were studied. Group comparison analyses and Cox regression models were employed.
Results
Patients with all biomarkers positive for AD (N = 145) had the highest hazard for progression to dementia due to AD, whilst patients with no positive biomarkers (N = 111) had the lowest. The risk of patients with only abnormal p-Tau and/or t-Tau (N = 49) or with positive Aβ42 in combination with positive t-Tau or p-Tau (N = 119) is significantly lower than that of patients with all biomarkers positive.
Conclusions
The risk of progression to dementia due to AD differs between patients with different CSF biomarker constellations.
Similar content being viewed by others


Background
An increasing body of evidence suggests that Alzheimer’s disease (AD) pathophysiology can be identified using biomarkers [1, 2]. AD is characterized by abnormal patterns in structural and functional imaging as well as by a pathological cerebrospinal fluid (CSF) signature [3]. The pathological CSF signature is defined by decreased CSF concentrations of the peptide amyloid-beta 1–42 (Aβ42) and increased levels of the proteins total Tau (t-Tau) and Tau phosphorylated at threonine 181 (p-Tau). It is of note that biomarkers reflect AD neuropathological changes with relatively high accuracy [4, 5]. In clinical practice, CSF biomarkers aid clinicians with decision-making, embody a key tool in the differential diagnosis especially of atypical dementia syndromes and increase diagnostic confidence [4, 6–9].
Biomarkers enable the identification of AD pathophysiology in pre-dementia stages of the disease, such as the stage of mild cognitive impairment (MCI) [10]. MCI is a clinical entity characterized by cognitive deficits which are so mild that activities of daily living remain largely unaffected [10]. It is a heterogeneous clinical syndrome with regard to aetiology, clinical appearance and prognosis. MCI can be caused by different diseases (e.g. AD, cerebrovascular disease, depression, frontotemporal lobar degenerations, etc.). As a consequence, only some patients with MCI progress to dementia due to AD or to other dementias within limited time periods, while cognitive functioning remains stable or even reverses to normal in others [11–15]. Biomarkers embody a valuable instrument in estimating the likelihood that MCI is engendered by AD. Interestingly, biomarkers are an integral part of the recently proposed National Institute on Aging–Alzheimer’s Association (NIA-AA) guidelines for the diagnosis of MCI in clinical research settings [16]. These guidelines suggest categorizing MCI according to the individual likelihood of underlying AD pathophysiology and the associated risk of developing AD dementia in the future; the highest likelihood category is characterized by biomarker findings pointing to the presence of AD pathophysiology, whereas the lowest likelihood category is characterized by findings not typical for AD. However, the criteria do not consider conflicting biomarker constellations, although they are very common in MCI [17–19].
A large number of studies have investigated the prognostic utility of established AD biomarkers (for instance [10, 20–28]), but none of them has taken into account the NIA-AA guidelines in combination with exclusive consideration of all three established CSF biomarkers. To shed light on this grey area, we conducted a retrospective study focused on differences in progression to dementia due to AD of patients with MCI and different CSF biomarker constellations.
Methods
Participants
The study sample encompassed data from all phases of the AD Neuroimaging Initiative (ADNI) (ADNI 1, Go and 2), a collaborative project of academic institutions and private corporations across the USA and Canada which began in October 2004. The study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. The ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California. Data used in this study were obtained from the ADNI database (www.adni-info.org) on 27 August 2014. Patients with MCI, fulfilling international diagnostic criteria [16], and with available CSF Aβ42, t-Tau and p-Tau values at baseline and clinical follow-up data were included. In the ADNI, patients with MCI had Mini-Mental-State Examination (MMSE) scores between 24 and 30, a Clinical Dementia Rating (CDR) score of 0.5, memory complaints and objective memory deficits on the Wechsler Memory-Scale-Logical Memory II test. They were not significantly impaired in their activities of daily living. Patients with diagnoses other than MCI at baseline, controls and patients with MCI but not all CSF biomarker findings available at baseline were excluded from the study. Patients diagnosed with AD dementia at follow-up met the NIA-AA diagnostic guidelines for dementia due to probable AD [1]. Regarding MCI patients who had not progressed to dementia but discontinued participation in follow-up visits or died, the data of their last follow-up visit were considered in the analysis.
CSF collection and analysis
CSF collection, shipping, aliquoting, storage and analysis took place according to ADNI standard operating procedures (SOPs) [29]. It is noteworthy that some early CSF samples were mistakenly collected into inappropriate CSF collection tubes at the ADNI sites. However, this was corrected rapidly and the exposure time to any inappropriate CSF collection tube was of limited significance due to the short time the CSF was in contact with the transfer tubes (approximately 25.7 min) [29]. ADNI baseline CSF samples were analysed at the ADNI biomarker core laboratory at University of Pennsylvania according to published methods [5, 30]. CSF samples were put into the freezer at –80 °C. The CSF concentrations of Aβ42, t-Tau and p-Tau were measured using the multiplex xMAP Luminex platform with Innogenetics immunoassay kit-based reagents (INNO-BIA AlzBio 3; Ghent, Belgium) [30].
APOE genotyping
APOE genotypes were determined for all ADNI participants through analysis of blood samples using standard polymerase chain reaction methods [31].
Classification of patients with MCI
In line with the NIA-AA algorithm, each patient’s biomarker values were categorized as either positive for AD, negative for AD or borderline. The definition of the range of borderline values was based on biomarker cut-off values and standard deviations (SDs) selected from previous reports on ADNI MCI patients [5]. The range of borderline values was specified with the aim to reach a reasonable compromise between minimizing the chance of an artificial categorization as positive and at the same time classifying less than 20% of the measured values of each biomarker as borderline. Values within 20% of the SD from the respective cut-off point were classified as borderline [32]. Aβ42 concentrations lower than the defined range of Aβ42 borderline values and t-Tau and p-Tau levels higher than the respective borderline ranges were assumed to be AD positive. All other biomarker values were considered negative. Aβ42, t-Tau and p-Tau concentrations <181 pg/ml, >105.2 pg/ml, and >26.6 pg/ml, respectively, were thus regarded as positive for AD. CSF levels of Aβ42 > 203 pg/ml and t-Tau and p-Tau concentrations <80.8 pg/ml and <19.4 pg/ml, respectively, were considered negative for AD.
Patients were classified according to their fluid biomarker profile into the following subgroups:
-
MCI with no positive biomarkers (MCINon+).
-
MCI with all biomarkers positive (MCIAll+).
-
MCI with positive Aβ42 but negative or borderline p-Tau and t-Tau (MCIAβ+).
-
MCI with positive Aβ42 and positive p-Tau or t-Tau (MCIAβ+T+).
-
MCI with negative or borderline Aβ42 but positive p-Tau and/or t-Tau (MCIT+).
The biomarker constellations of the three latter subgroups are so far not being considered in the NIA-AA diagnostic guidelines, because for MCI patients with such biomarker constellations no likelihood grade for the presence of AD pathology is assigned by the NIA-AA criteria [16].
Statistical analysis
The statistical analyses were performed in SPSS v19.0 for Windows (IBM Corp., Somers, NY, USA). Normal distribution of data was checked using the Kolmogorov–Smirnov test. The raw biomarker data of study participants were graphically presented by means of non-negative matrix factorization (NNMF) [32, 33], a data-learning technique particularly suited for analysing positive valued data so that the available information is condensed in a low-dimensional (2D) space. The overall set of measurements:
X i = {Aβ42, t-Tau, p-Tau}unlikely i , i = 1, 2, …, N , where N is the total number of participants, was approximated as:
X [N×3] ≈ W [N×2] B [2×3]
in order to minimize the reconstruction error induced by the Frobenius norm: ||X-WB||2. In this way, the vector of measurements X i associated with the ith participant took the form of:
X i = w i1 B 1 + w i2 B 2,
where B 1 and B 2 were the unit length vectors for a parsimonious 2D representation and w i1 and w i2 were the corresponding components. Differences between the MCI subgroups regarding demographic and CSF data, MMSE scores, follow-up duration, presence of the APOE ε4 allele and progression rates to dementia due to AD were tested by analysis of variance (ANOVA), Bonferroni post-hoc analysis, Kruskal–Wallis test, Mann–Whitney test and chi-square test as appropriate. Differences in the hazard of progression between the MCI subgroups were analysed using Cox regression models, adjusting for patient characteristics that significantly differed between the subgroups. Two-sided p < 0.05 was considered statistically significant.
Results
A total of 469 MCI patients out of 1729 ADNI participants with available baseline data fulfilled the inclusionary criteria of the study. APOE ε4 and sex distribution, as well as age and MMSE scores, significantly differed between the subgroups (Table 1). In particular, the MMSE scores of the MCINon+ subgroup were significantly higher compared with the scores of the MCIAll+ (p < 0.001) and MCIAβ+T+ (p < 0.01) subgroups. MMSE scores in the MCIAll+ subgroup were significantly lower in comparison with those of the MCIAβ+ (p = 0.02), MCIAβ+T+ (p = 0.03) and MCIT+ (p < 0.01) subgroups. Across the five studied MCI subgroups, approximately 45% of patients had conflicting CSF biomarker constellations. Figure 1, a graphical presentation of participants’ Aβ42, t-Tau and p-Tau CSF levels using NNMF, points to the high variability of the CSF biomarker findings in patients with MCI. Data were available from clinical follow-up visits conducted every 6 months up to 8 years after baseline. In total, 159 patients with MCI progressed to dementia due to AD. No patient progressed to any other form of dementia. The difference between the MCI subgroups in the proportions of patients who developed dementia due to AD within the follow-up period attained statistical significance (p < 0.001), whilst the duration of the follow-up period did not differ.

Condensed representation, as a 2D scatter plot, of the CSF biomarker values of the study participants. The ensemble of trivariate measurements of CSF Aβ42, p-Tau and t-Tau for all participants was analysed via NNMF and approximated by means of a bivariate data swarm that conveniently represents the total variation in the original data. Labels indicate the different groups and lend semantics to the plot. MCI Non+ MCI without positive CSF biomarkers, MCI Aβ+ MCI with positive Aβ42 and negative or borderline p-Tau and t-Tau, MCI Aβ+T+ MCI with positive Aβ42 and positive t-Tau or p-Tau, MCI All+ MCI with Aβ42 and both t-Tau and p-Tau positive, MCI T+ MCI with negative or borderline Aβ42 and at least p-Tau or t-Tau positive
Cox regression analyses unveiled a significant association between group membership and risk of progression to AD dementia (P < 0.001), whilst sex, age and the presence of the APOE ε4 allele did not exert such an influence. As expected, lower MMSE scores were related to a higher hazard of developing dementia. The differences in progression risk between patients in the MCINon+ subgroup and those with positive Aβ42 values (MCIAβ+, MCIAβ+T+, MCIAll+ subgroups) reached statistical significance. In addition, the MCIAll+ subgroup was at significantly higher risk for progression compared with both the MCIAβ+T+ and MCIT+ subgroups. No further significant differences with regard to progression risk were detected between any other of the compared MCI subgroups (Table 2, Fig. 2). The risk pattern in relation to different biomarker constellations in MCI is presented in Fig. 3.

Cox regression survival curves for patients with MCI and different constellations of CSF Aβ42 and neuronal injury markers (p-Tau and t-Tau). MCI Non+ MCI without positive CSF biomarkers, MCI Aβ+ MCI with positive Aβ42 and negative or borderline p-Tau and t-Tau, MCI Aβ+T+ MCI with positive Aβ42 and positive t-Tau or p-Tau, MCI All+ MCI with Aβ42 and both t-Tau and p-Tau positive, MCI T+ MCI with negative or borderline Aβ42 and at least p-Tau or t-Tau positive

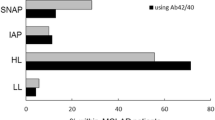
Risk for progression to dementia due to AD of patients with MCI and different constellations of CSF Aβ42 and neuronal injury markers (p-Tau and t-Tau). AD Alzheimer’s disease, MCI Non+ MCI without positive CSF biomarkers, MCI Aβ+ MCI with positive Aβ42 and negative or borderline p-Tau and t-Tau, MCI Aβ+T+ MCI with positive Aβ42 and positive t-Tau or p-Tau, MCI All+ MCI with Aβ42 and both t-Tau and p-Tau positive, MCI T+ MCI with negative or borderline Aβ42 and at least p-Tau or t-Tau positive
Discussion
In line with a number of previous reports [17–19], but in contrast to others [8, 9, 34, 35], approximately half of the MCI cases in our study had conflicting CSF biomarker constellations. This discrepancy in the frequency of patients with conflicting CSF biomarker results could be possibly attributed to differences in study design. For instance, not all studies considered all three CSF AD biomarkers. In addition, past studies implemented a dichotomization strategy in interpreting biomarker findings, whilst in the present study biomarker values were categorized as positive, negative or borderline in line with the NIA-AA guidelines. Moreover, it is possible that academic, research centres recruit more patients with complex constellations of biomarker findings, whilst more patients with AD-typical CSF profiles and consequently more advanced neuropathology are recruited in non-academic, clinical settings. Interestingly, it has been shown that patients of a non-academic memory clinic suffered from more severe clinical symptoms in comparison with the patients of an academic memory clinic [36].
Constellations with conflicting CSF biomarker findings are not currently being considered by the NIA-AA criteria for MCI [16], and our study provides initial evidence on the role of conflicting CSF biomarker constellations for dementia risk estimation. Our findings indicate that compared with the constellation without positive biomarkers, the presence of Aβ42 positivity confers a higher risk for future AD dementia irrespective of t-Tau and p-Tau levels. Thus, it seems that Aβ42 is not only the first marker to become positive in the course of AD [37], but also the decisive marker to determine dementia risk. Despite the absence of general consensus, because a number of past reports point to a higher or almost similar prognostic utility of tau peptides compared with Aβ42 [20, 35, 38–42], our observations are in line with several previous studies which showed that Aβ42 has a higher prognostic utility in comparison with tau or failed to find an association between tau and cognitive deterioration [5, 22, 43, 44]. It is important to mention, however, that our results cannot necessarily be generalized to all patients with MCI, because the ADNI MCI cohort is deliberately limited to those with prominent memory deficits in order to enrich the sample with pre-dementia AD cases. MCI is heterogeneous by definition, and the studied biomarkers may react differently in early non-AD cases. Hence, the confounding effects of other brain pathologies associated with increased p-Tau and/or t-Tau levels (such as cerebrovascular changes, Lewy bodies, etc.) are minimized in ADNI, so that the role of Aβ42 as an indicator of AD pathophysiology may be exaggerated. Furthermore, it should be underscored that our findings could have been biased by the artificial definition of the range of borderline biomarker values as well as by the fact that, due to sample size reasons, borderline values were not considered separately from negative values in our analyses. Interestingly, an alternative analysis considering borderline and positive values together resulted in an amelioration of the significance of the role of Aβ42 (the results of the alternative analysis are presented in Additional file 1). Hence, further studies with larger samples enabling the separate consideration of borderline values are warranted before definite conclusions can be drawn.
The higher dementia risk in the MCIAll+ subgroup compared with the other subgroups is in line with the proposed model of a temporal evolution of AD biomarkers as well as with a recently published, probabilistic, data-driven model of biomarker changes in sporadic AD, independent of a-priori patient staging and biomarker cut-off points [37, 45]. Our observation supports the assumption that as clinical symptoms advance and the threshold to dementia is reached, the abnormality of biomarkers becomes evident [46, 47]. The lack of significant difference in terms of dementia risk between the MCIAll+ and MCIAβ+ subgroups is probably a spurious finding, related to the relatively small size of the MCIAβ+ subgroup and/or the definition of the cut-off values. This assumption is supported by the highly significant difference between the MCIAll+ and MCIAβ+T+ subgroups, although the latter subgroup has a higher dementia risk compared with the MCIAβ+ subgroup as illustrated in Fig. 2. Moreover, the alternative analysis, in which borderline and positive biomarker values were treated as one group, unveiled a significant difference in progression risk between the MCI subgroup with all markers non-negative (positive or borderline) and the subgroup with only Aβ42 non-negative (Additional file 1). Thus, the observed lack of significant difference in dementia risk between the MCIAll+ and MCIAβ+ subgroups should be treated with caution. In addition, it is noteworthy that our results confirm the approach of the NIA-AA algorithm to assign the highest likelihood of AD to MCI patients with all biomarkers positive.
Our findings suggest that MCI patients with positive Aβ42 values are at the same risk for AD dementia whether or not they have one positive Tau marker (either t-Tau or p-Tau). Hence, patients with positive Aβ42 and non-positive or conflicting p-Tau/t-Tau levels, who cannot be categorized according to the current NIA-AA algorithm, seem to have the same dementia risk, which lies between that of the lowest and highest risk groups (MCINon+ and MCIAll+ respectively). However, this observation is in contrast with the findings of a large number of previous reports which have shown that MCI patients with two positive CSF markers have a higher risk to progress to dementia compared with MCI patients with only one positive biomarker [20, 22, 25, 28, 40, 44, 48–51]. As a result, this finding should be treated with caution. It cannot be precluded that the observation of the present study has been biased by the definition of the cut-off points and/or by the relatively small size of the MCIAβ+ subgroup. Nonetheless, the alternative analysis in which borderline and positive biomarker values were treated as one group (non-negative values) did not reveal significant differences in the progression risk between the MCI subgroup with only Aβ42 non-negative and that with both Aβ42 and p-Tau or t-Tau non-negative (Additional file 1). Further studies are thus required in order to shed more light on the progression risk of the MCIAβ+ and MCIAβ+T+ subgroups.
In terms of dementia risk, MCI patients with one or two positive Tau markers but negative or borderline Aβ42 values (MCIT+ subgroup) may be placed between patients without positive biomarkers (MCINon+ subgroup) and those with positive Aβ42 and non-positive or conflicting p-Tau/t-Tau levels (MCIAβ+ and MCIAβ+T+ subgroup, respectively). The term “suspected non-AD pathophysiology (SNAP)” has recently been proposed to designate individuals with abnormal markers of neuronal injury without evidence of amyloid accumulation [10, 18, 52]. According to our findings, the progression risk of the MCIT+ subgroup does not significantly differ from that of the MCINon+ subgroup. Simultaneously, it is no different from that of the MCIAβ+ and MCIAβ+T+ subgroups. However, the dementia risk of the two latter subgroups does in fact differ from the MCINon+ subgroup. As a consequence, the MCIT+ risk can be placed between that of MCINon+ and MCIAβ+, MCIAβ+T+ subgroups (Fig. 3). Nonetheless, this finding should also be treated with caution due to the limited size of the ADNI MCIT+ subgroup and because a prior study showed that the highest proportion of subjects who progressed to dementia was observed not only in the MCI subgroup with both amyloid and neuronal injury markers positive for AD but also in the MCI subgroup with only neuronal injury biomarkers positive [10]. This discrepancy could be explained by the different markers of neuronal injury considered in the two studies (neurochemical vs imaging), especially in light of the pathophysiological character of the former and the downstream topographical character of the latter [53].
The present study should be viewed in the light of some limitations. Owing to the lack of histopathological verification, the main outcome measure was based purely on clinical diagnoses, which are not always confirmed at autopsy [54]. Moreover, the ADNI encompasses individuals recruited at specialized research centres and does not mirror constellations in the community. This is clearly illustrated by the fact that within the follow-up period only conversion to dementia due to AD and not to other forms of dementia was observed. Furthermore, in building the MCI subgroups with distinct biomarker constellations we did not consider borderline values separately from negative values, because such an approach would have expanded the number of MCI subgroups and reduce their size. This limitation could explain the relatively high proportion—in comparison with previous reports—of patients in the MCINon+ subgroup as well as in the MCIT+ subgroup who progressed to dementia due to AD [55], because it is possible that borderline biomarker values became positive shortly after baseline. In addition, it can be reckoned that our observations are biased by the artificial definition of the range of borderline biomarker values. In light of the lack of empirical data with regard to definitions of the range of borderline values, our findings should be treated with caution. Nonetheless, the NIA-AA guidelines clearly specify the presence of borderline biomarker values. As a consequence, further studies considering borderline biomarker values are warranted. A further shortcoming of the study is the collection of some early CSF samples into inappropriate tubes at the ADNI sites. Although the error was corrected rapidly and despite the relatively limited exposure time to any inappropriate CSF collection tube, this error could embody a source of bias because the use of different collection tubes increases intra-laboratory variability [34, 56–58]. Moreover, we did not take into account imaging biomarker data. However, it should be underscored that while combining imaging with neurochemical biomarker data may be relevant for research settings, it is rarely applicable to clinical settings because of limitations related to scanner equipment and sophisticated image analyses expertise.
Conclusions
The present study provides a further piece of evidence for the prognostic differences between MCI subgroups with distinct neurochemical biomarker constellations. The study reveals significant differences between subgroups with conflicting biomarkers, on the one hand, and patients with all neurochemical biomarkers positive or non-positive (borderline or negative) for AD on the other. Even though our observations exclusively refer to neurochemical biomarkers and do not consider imaging markers, they point to the necessity of modifying/refining the NIA-AA algorithms for categorizing MCI.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- ANOVA:
-
Analysis of variance
- APOE:
-
Apolipoprotein E
- Aβ42 :
-
Amyloid-beta 1–42
- CDR:
-
Clinical Dementia Rating
- CSF:
-
Cerebrospinal fluid
- MCI:
-
Mild cognitive impairment
- MCIAll+ :
-
Mild cognitive impairment with all biomarkers positive
- MCIAβ+ :
-
Mild cognitive impairment with positive Aβ42 but negative or borderline p-Tau and t-Tau
- MCIAβ+T+ :
-
Mild cognitive impairment with positive Aβ42 and positive p-Tau or t-Tau
- MCINon+ :
-
Mild cognitive impairment with no positive biomarkers
- MCIT+ :
-
Mild cognitive impairment with negative or borderline Aβ42 but p-Tau and/or t-Tau positive
- MMSE:
-
Mini-Mental State Examination
- NIA-AA:
-
National Institute on Aging–Alzheimer Association
- p-Tau:
-
Phosphorylated tau at threonine 181
- SD:
-
Standard deviation
- SNAP:
-
Suspected non-AD pathophysiology
- t-Tau:
-
Total tau
References
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi:10.1016/j.jalz.2011.03.005.
McKhann GM. Changing concepts of Alzheimer disease. JAMA. 2011;305:2458–9. doi:10.1001/jama.2011.810.
Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, Van der Flier, Wiesje M. Alzheimer’s disease. Lancet. 2016. doi:10.1016/S0140-6736(15)01124-1.
Schoonenboom NSM, Reesink FE, Verwey NA, Kester MI, Teunissen CE, van de Ven PM, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology. 2012;78:47–54. doi:10.1212/WNL.0b013e31823ed0f0.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi:10.1002/ana.21610.
Troussiere AC, Wallon D, Mouton-Liger F, Yatimi R, Robert P, Hugon J, et al. Who needs cerebrospinal biomarkers? A national survey in clinical practice. J Alzheimers Dis. 2014;40:857–61. doi:10.3233/JAD-132672.
Kester MI, Boelaarts L, Bouwman FH, Vogels RL, Groot ER, van Elk EJ, et al. Diagnostic impact of CSF biomarkers in a local hospital memory clinic. Dement Geriatr Cogn Disord. 2010;29:491–7. doi:10.1159/000313534 .
Mouton-Liger F, Wallon D, Troussiere A-C, Yatimi R, Dumurgier J, Magnin E, et al. Impact of cerebro-spinal fluid biomarkers of Alzheimer’s disease in clinical practice: a multicentric study. J Neurol. 2014;261:144–51. doi:10.1007/s00415-013-7160-3.
Duits FH, Prins ND, Lemstra AW, Pijnenburg YAL, Bouwman FH, Teunissen CE, et al. Diagnostic impact of CSF biomarkers for Alzheimer’s disease in a tertiary memory clinic. Alzheimers Dement. 2015;11:523–32. doi:10.1016/j.jalz.2014.05.1753.
Petersen RC, Aisen P, Boeve BF, Geda YE, Ivnik RJ, Knopman DS, et al. Mild cognitive impairment due to Alzheimer disease in the community. Ann Neurol. 2013;74:199–208. doi:10.1002/ana.23931.
Alexopoulos P, Grimmer T, Perneczky R, Domes G, Kurz A. Do all patients with mild cognitive impairment progress to dementia? J Am Geriatr Soc. 2006;54:1008–10. doi:10.1111/j.1532-5415.2006.00752.x.
Alexopoulos P, Grimmer T, Perneczky R, Domes G, Kurz A. Progression to dementia in clinical subtypes of mild cognitive impairment. Dement Geriatr Cogn Disord. 2006;22:27–34. doi:10.1159/000093101.
Maioli F, Coveri M, Pagni P, Chiandetti C, Marchetti C, Ciarrocchi R, et al. Conversion of mild cognitive impairment to dementia in elderly subjects: a preliminary study in a memory and cognitive disorder unit. Arch Gerontol Geriatr. 2007;44 Suppl 1:233–41. doi:10.1016/j.archger.2007.01.032.
Ravaglia G, Forti P, Montesi F, Lucicesare A, Pisacane N, Rietti E, et al. Mild cognitive impairment: epidemiology and dementia risk in an elderly Italian population. J Am Geriatr Soc. 2008;56:51–8. doi:10.1111/j.1532-5415.2007.01503.x.
Rozzini L, Chilovi BV, Conti M, Bertoletti E, Delrio I, Trabucchi M, Padovani A. Conversion of amnestic Mild Cognitive Impairment to dementia of Alzheimer type is independent to memory deterioration. Int J Geriatr Psychiatry. 2007;22:1217–22. doi:10.1002/gps.1816.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9. doi:10.1016/j.jalz.2011.03.008.
Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–9. doi:10.1212/WNL.0b013e3181bc010c.
Alexopoulos P, Kriett L, Haller B, Klupp E, Gray K, Grimmer T, et al. Limited agreement between biomarkers of neuronal injury at different stages of Alzheimer’s disease. Alzheimers Dement. 2014;10:684–9. doi:10.1016/j.jalz.2014.03.006.
Yakushev I, Muller MJ, Buchholz H-G, Lang U, Rossmann H, Hampel H, et al. Stage-dependent agreement between cerebrospinal fluid proteins and FDG-PET findings in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:241–7.
Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–8. doi:10.1212/WNL.0b013e3181e8e8b8.
Vos SJB, van Rossum IA, Verhey F, Knol DL, Soininen H, Wahlund L-O, et al. Prediction of Alzheimer disease in subjects with amnestic and nonamnestic MCI. Neurology. 2013;80:1124–32. doi:10.1212/WNL.0b013e318288690c .
Heister D, Brewer JB, Magda S, Blennow K, McEvoy LK. Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology. 2011;77:1619–28. doi:10.1212/WNL.0b013e3182343314.
van Rossum IA, Vos SJB, Burns L, Knol DL, Scheltens P, Soininen H, et al. Injury markers predict time to dementia in subjects with MCI and amyloid pathology. Neurology. 2012;79:1809–16. doi:10.1212/WNL.0b013e3182704056.
Prestia A, Caroli A, van der Flier WM, Ossenkoppele R, van Berckel B, Barkhof F, et al. Prediction of dementia in MCI patients based on core diagnostic markers for Alzheimer disease. Neurology. 2013;80:1048–56. doi:10.1212/WNL.0b013e3182872830.
Prestia A, Caroli A, Wade SK, van der Flier WM, Ossenkoppele R, van Berckel B, et al. Prediction of AD dementia by biomarkers following the NIA-AA and IWG diagnostic criteria in MCI patients from three European memory clinics. Alzheimers Dement. 2015;11:1191–201. doi:10.1016/j.jalz.2014.12.001.
Jack CR, Therneau TM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, et al. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol. 2015. doi:10.1016/S1474-4422(15)00323-3.
Callahan BL, Ramirez J, Berezuk C, Duchesne S, Black SE. Predicting Alzheimer’s disease development: a comparison of cognitive criteria and associated neuroimaging biomarkers. Alzheimers Res Ther. 2015;7:68. doi:10.1186/s13195-015-0152-z.
Vos SJ, Verhey F, Frolich L, Kornhuber J, Wiltfang J, Maier W, et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain. 2015;138:1327–38. doi:10.1093/brain/awv029.
Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6:230–8. doi:10.1016/j.jalz.2010.03.008.
Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, Foroud T, et al. Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology. 2011;76:69–79. doi:10.1212/WNL.0b013e318204a397.
Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement. 2010;6:265–73. doi:10.1016/j.jalz.2010.03.013.
Alexopoulos P, Roesler J, Thierjung N, Werle L, Buck D, Yakushev I, et al. Mapping CSF biomarker profiles onto NIA-AA guidelines for Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2015. doi:10.1007/s00406-015-0628-7.
Lee DD, Seung HS. Learning the parts of objects by non-negative matrix factorization. Nature. 1999;401:788–91. doi:10.1038/44565 .
Dumurgier J, Schraen S, Gabelle A, Vercruysse O, Bombois S, Laplanche J-L, et al. Cerebrospinal fluid amyloid-beta 42/40 ratio in clinical setting of memory centers: a multicentric study. Alzheimers Res Ther. 2015;7:30. doi:10.1186/s13195-015-0114-5.
Kester MI, van der Vlies AE, Blankenstein MA, Pijnenburg YAL, van Elk EJ, Scheltens P, van der Flier WM. CSF biomarkers predict rate of cognitive decline in Alzheimer disease. Neurology. 2009;73:1353–8. doi:10.1212/WNL.0b013e3181bd8271.
Weih M, Sulimma A-K, Lehfeld H, Niklewski G, Sonnenberg M, Richter-Schmidinger T, et al. Comparison of patient therapy adherence of two structural different memory clinics. Fortschr Neurol Psychiatr. 2009;77:212–7. doi:10.1055/s-0028-1109069.
Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi:10.1016/S1474-4422(12)70291-0.
Degerman Gunnarsson M, Lannfelt L, Ingelsson M, Basun H, Kilander L. High tau levels in cerebrospinal fluid predict rapid decline and increased dementia mortality in alzheimer’s disease. Dement Geriatr Cogn Disord. 2014;37:196–206. doi:10.1159/000355556.
Prestia A, Caroli A, Herholz K, Reiman E, Chen K, Jagust WJ, Frisoni GB. Diagnostic accuracy of markers for prodromal Alzheimer’s disease in independent clinical series. Alzheimers Dement. 2013;9:677–86. doi:10.1016/j.jalz.2012.09.016.
Mattsson N. CSF Biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385. doi:10.1001/jama.2009.1064 .
Ravaglia S, Bini P, Sinforiani E, Franciotta D, Zardini E, Tosca P, et al. Cerebrospinal fluid levels of tau phosphorylated at threonine 181 in patients with Alzheimer’s disease and vascular dementia. Neurol Sci. 2008;29:417–23. doi:10.1007/s10072-008-1023-1.
Sämgård K, Zetterberg H, Blennow K, Hansson O, Minthon L, Londos E. Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int J Geriatr Psychiatry. 2010;25:403–10. doi:10.1002/gps.2353.
Sunderland T, Wolozin B, Galasko D, Levy J, Dukoff R, Bahro M, et al. Longitudinal stability of CSF tau levels in Alzheimer patients. Biol Psychiatry. 1999;46:750–5.
Duits FH, Teunissen CE, Bouwman FH, Visser P-J, Mattsson N, Zetterberg H, et al. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimers Dement. 2014;10:713–23.e2. doi:10.1016/j.jalz.2013.12.023.
Young AL, Oxtoby NP, Daga P, Cash DM, Fox NC, Ourselin S, et al. A data-driven model of biomarker changes in sporadic Alzheimer’s disease. Brain. 2014;137:2564–77. doi:10.1093/brain/awu176.
Jack CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58. doi:10.1016/j.neuron.2013.12.003.
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi:10.1016/S1474-4422(13)70044-9.
Leuzy A, Carter SF, Chiotis K, Almkvist O, Wall A, Nordberg A. Concordance and diagnostic accuracy of [11C]PIB PET and cerebrospinal fluid biomarkers in a sample of patients with mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2015;45:1077–88.
Perani D, Cerami C, Caminiti SP, Santangelo R, Coppi E, Ferrari L, et al. Cross-validation of biomarkers for the early differential diagnosis and prognosis of dementia in a clinical setting. Eur J Nucl Med Mol Imaging. 2016;43:499–508.
Gomar JJ. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer’s Disease Neuroimaging Initiative. Arch Gen Psychiatry. 2011;68:961. doi:10.1001/archgenpsychiatry.2011.96.
Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology. 2009;73:294–301. doi:10.1212/WNL.0b013e3181af79fb.
Jack Jr CR, Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, et al. An operational approach to National Institute on Aging–Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol. 2012;71:765–75. doi:10.1002/ana.22628.
Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29. doi:10.1016/S1474-4422(14)70090-0.
Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71:266–73. doi:10.1097/NEN.0b013e31824b211b.
Guo L-H, Alexopoulos P, Eisele T, Wagenpfeil S, Kurz A, Perneczky R. The National Institute on Aging–Alzheimer’s Association research criteria for mild cognitive impairment due to Alzheimer’s disease: predicting the outcome. Eur Arch Psychiatry Clin Neurosci. 2013;263:325–33. doi:10.1007/s00406-012-0349-0.
Vos SJB, Visser PJ, Verhey F, Aalten P, Knol D, Ramakers I, et al. Variability of CSF Alzheimer’s disease biomarkers: implications for clinical practice. PLoS ONE. 2014;9, e100784. doi:10.1371/journal.pone.0100784.
del Campo M, Mollenhauer B, Bertolotto A, Engelborghs S, Hampel H, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: an update. Biomark Med. 2012;6:419–30.
Lehmann S, Schraen S, Quadrio I, Paquet C, Bombois S, Delaby C, et al. Impact of harmonization of collection tubes on Alzheimer’s disease diagnosis. Alzheimers Dement. 2014;10:S390–4.e2. doi:10.1016/j.jalz.2013.06.008.
Acknowledgements
None.
Funding
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI, adni.loni.usc.edu) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). The ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private-sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education. Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). Ethics approval was obtained from the institutional review boards of each institution involved: Oregon Health and Science University; University of Southern California; University of California—San Diego; University of Michigan; Mayo Clinic, Rochester; Baylor College of Medicine; Columbia University Medical Center; Washington University, St. Louis; University of Alabama—Birmingham; Mount Sinai School of Medicine; Rush University Medical Center; Wien Center; Johns Hopkins University; New York University; Duke University Medical Center; University of Pennsylvania; University of Kentucky; University of Pittsburgh; University of Rochester Medical Center; University of California, Irvine; University of Texas Southwestern Medical School; Emory University; University of Kansas, Medical Center; University of California, Los Angeles; Mayo Clinic, Jacksonville; Indiana University; Yale University School of Medicine; McGill University, Montreal-Jewish General Hospital; Sunnybrook Health Sciences, Ontario; U.B.C. Clinic for AD & Related Disorders; Cognitive Neurology—St. Joseph’s, Ontario; Cleveland Clinic Lou Ruvo Center for Brain Health; Northwestern University; Premiere Research Inst (Palm Beach Neurology); Georgetown University Medical Center; Brigham and Women’s Hospital; Stanford University; Banner Sun Health Research Institute; Boston University; Howard University; Case Western Reserve University; University of California, Davis—Sacramento; Neurological Care of CNY; Parkwood Hospital; University of Wisconsin; University of California, Irvine—BIC; Banner Alzheimer’s Institute; Dent Neurologic Institute; Ohio State University; Albany Medical College; Hartford Hospital, Olin Neuropsychiatry Research Center; Dartmouth-Hitchcock Medical Center; Wake Forest University Health Sciences; Rhode Island Hospital; Butler Hospital; UC San Francisco; Medical University South Carolina; St. Joseph’s Health Care Nathan Kline Institute; University of Iowa College of Medicine; Cornell University and University of South Florida: USF Health Byrd Alzheimer’s Institute. The investigators within the ADNI contributed to the design and implementation of the ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found online (http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf).
Availability of supporting data
Not applicable.
Authors’ contributions
PA conceived of the study, participated in its design and coordination and in data analysis and interpretation, and helped to draft the manuscript. LW participated in study design, in data analysis and interpretation, and in drafting the manuscript. JR participated in data interpretation and critically revised the manuscript for important intellectual content. NT participated in data analysis and interpretation, and critically revised the manuscript for important intellectual content. LSG participated in data interpretation and critically revised the manuscript for important intellectual content. IY participated in data interpretation and critically revised the manuscript for important intellectual content. NL participated in data analysis and interpretation, and critically revised the manuscript for important intellectual content. SW participated in data analysis and interpretation, and critically revised the manuscript for important intellectual content. PG participated in data interpretation and critically revised the manuscript for important intellectual content. AK participated in data interpretation and helped to draft the manuscript. RP participated in study design and data interpretation, and helped to draft the manuscript. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethical approval and consent to participate, consent for publication
The study procedures were approved by the institutional review boards of all participating centres (see Additional file 2) and written informed consent was obtained from all participants or authorized representatives.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional files
Additional file 1:
Presents the results of the alternative analysis based on consideration of borderline and positive biomarker values together. (DOCX 27 kb)
Additional file 2:
Is an acknowledgements list for ADNI publications: ADNI infrastructure and site investigators. (PDF 661 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Alexopoulos, P., Werle, L., Roesler, J. et al. Conflicting cerebrospinal fluid biomarkers and progression to dementia due to Alzheimer’s disease. Alz Res Therapy 8, 51 (2016). https://doi.org/10.1186/s13195-016-0220-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-016-0220-z