
Abstract
Background
The potential for DNA methylation (DNAm) as an early marker for cardiovascular disease (CVD) and how such an association might differ by glycemic exposure has not been examined in type 1 diabetes, a population at increased CVD risk. We thus performed a prospective epigenome-wide association study of blood leukocyte DNAm (EPIC array) and time to CVD incidence over 28 years in a childhood-onset (< 17 years) type 1 diabetes cohort, the Pittsburgh Epidemiology of Diabetes Complications (EDC) study (n = 368 with DNA and no CVD at baseline), both overall and separately by glycemic exposure, as measured by HbA1c at baseline (split at the median: < 8.9% and ≥ 8.9%). We also assessed whether DNAm-CVD associations were independent of established cardiometabolic risk factors, including body mass index, estimated glucose disposal rate, cholesterol, triglycerides, blood pressure, pulse rate, albumin excretion rate, and estimated glomerular filtration rate.
Results
CVD (first instance of CVD death, myocardial infarction, coronary revascularization, ischemic ECG, angina, or stroke) developed in 172 participants (46.7%) over 28 years. Overall, in Cox regression models for time to CVD, none of the 683,597 CpGs examined reached significance at a false discovery rate (FDR) ≤ 0.05. In participants with HbA1c < 8.9% (n = 180), again none reached FDR ≤ 0.05, but three were associated at the a priori nominal significance level FDR ≤ 0.10: cg07147033 in MIB2, cg12324048 (intergenic, chromosome 3), and cg15883830 (intergenic, chromosome 1). In participants with HbA1c ≥ 8.9% (n = 188), two CpGs in loci involved in calcium channel activity were significantly associated with CVD (FDR ≤ 0.05): cg21823999 in GPM6A and cg23621817 in CHRNA9; four additional CpGs were nominally associated (FDR ≤ 0.10). In participants with HbA1c ≥ 8.9%, DNAm-CVD associations were only modestly attenuated after cardiometabolic risk factor adjustment, while attenuation was greater in those with HbA1c < 8.9%. No pathways were enriched in those with HbA1c < 8.9%, while pathways for calcium channel activity and integral component of synaptic membrane were significantly enriched in those with HbA1c ≥ 8.9%.
Conclusions
These results provide novel evidence that DNAm at loci involved in calcium channel activity and development may contribute to long-term CVD risk beyond known risk factors in type 1 diabetes, particularly in individuals with greater glycemic exposure, warranting further study.
Similar content being viewed by others


Background
Despite advances in treatment, people with type 1 diabetes continue to be at dramatically increased risk of cardiovascular disease (CVD) compared to people without diabetes [1]. Yet, no combination of genetic or clinical risk factors, including glycemic control and traditional cardiometabolic risk factors, fully explain the increased CVD risk associated with type 1 diabetes [2, 3]. Thus, it is likely that complex interactions between genetic factors and glycemic exposure, as well as other environmental and lifestyle exposures, influence earlier development of atherosclerosis in type 1 diabetes. DNA methylation (DNAm) provides a link between genetic susceptibility and risk factor exposure and its study has potential to uncover novel pathways to development of complex phenotypes, including CVD. Studies in the general population have shown differential patterns of methylation are associated with atherosclerosis, hypertension, lipids, and inflammation and such differential methylation may modify risk of clinical CVD events through those mechanisms [4]. Furthermore, a meta-analysis from the Cohorts for Heart and Aging Genetic Epidemiology (CHARGE) consortium identified 52 CpGs where DNAm was prospectively associated with incidence of coronary heart disease over a mean follow-up of 11 years [5]. However, to date there have been no published studies on methylation and CVD in type 1 diabetes despite the increased CVD burden carried by this population.
In type 1 diabetes, DNAm has been associated with glycemic exposure, as measured by HbA1c, in both the Diabetes Control and Complications Trial (DCCT)/ Epidemiology of Diabetes Interventions and Complications (EDIC) [6] and the Pittsburgh Epidemiology of Diabetes Complications (EDC) study [7]. In DCCT/EDIC, methylation at HbA1c-associated CpG sites explained much of the statistical association between HbA1c and risk of future microvascular disease, including proliferative retinopathy and diabetic kidney disease (DKD) [6]. Those results provide the strongest evidence thus far that past glycemic exposure is related to differential methylation, which may in turn influence future diabetes complication risk. A few other studies in type 1 diabetes have found associations between DNAm and proliferative retinopathy [8] and DKD [9,10,11,12] but those studies have been almost exclusively cross sectional and have not addressed differences in associations by glycemic control. Thus, given the limited prospective data on DNAm and diabetes complications in general and the lack of data on DNAm and CVD in type 1 diabetes specifically, our objective was to perform a prospective epigenome-wide association study (EWAS) of the association between whole blood-derived leukocyte DNAm and subsequent incidence of CVD over 28 years in type 1 diabetes. We hypothesized there are loci with DNAm associated with time-to-first CVD event in the Pittsburgh EDC study, a childhood-onset type 1 diabetes cohort. Additionally, as we have recently demonstrated risk factors for CVD differ by level of glycemic exposure [13], we further hypothesized that CVD-associated DNAm would differ by baseline HbA1c.
Results
Of the 368 individuals eligible for the primary analysis (Fig. 1), 172 (46.7%) developed CVD by 28 years. For the secondary outcomes, 28.0% (111 of 396 individuals) developed major adverse cardiovascular events (MACE) and 25.5% (102 of 400 individuals) major coronary artery disease (CAD). Kaplan Meier curves for time to total CVD, MACE, and major CAD over the 28-year follow-up are shown in Additional file 1: Fig. S2. Baseline characteristics overall and by total CVD incidence status are in Table 1.

Flowchart and timeline for the EDC DNA methylation sub-study. A Flowchart summarizing the determination of the analytic sample for DNA methylation and time to cardiovascular disease event in the EDC type 1 diabetes cohort. CVD = cardiovascular disease, MACE = major adverse cardiovascular event, CAD = coronary artery disease. B Study timeline. “Baseline” for the current analysis is the participant-specific time point at which the DNA specimen was collected
Total CVD, MACE, and major CAD EWAS: overall cohort
There were no significant associations (false discovery rate, FDR ≤ 0.05) in the overall cohort (Additional file 1: Fig. S3); three CpGs had FDR ≤ 0.20 and were included in post hoc functional analyses: cg02768721, PTPRN2 (p = 4.10 × 10–7), cg14524754, B3GNTL1 (p = 6.21 × 10–7), and cg06648759, intergenic CpG, Chr 13 (p = 8.27 × 10–7). The ten most significant CpGs for total CVD are shown in Table 2. A QQ plot is shown in Additional file 1: Fig. S4 (traditional genomic inflation λ = 0.91). For the secondary outcomes, MACE and major CAD, again, no CpGs were significantly associated with either outcome (Additional file 1: Table S1). Three CpGs with FDR ≤ 0.20 for MACE and 21 CpGs with FDR ≤ 0.20 for major CAD were included in post hoc analyses. Using the traditional genomic inflation factor, EWAS for MACE (λ = 1.44) and major CAD (λ = 1.51) both had evidence of significant inflation. After bacon correction, inflation was reduced for both outcomes (λ.bacon = 1.13 and 0.87, respectively).
HbA1c-stratified EWAS and cardiometabolic risk factor analysis
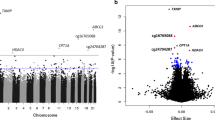
To assess our hypothesis that CVD-associated DNAm differs by HbA1c, we performed full EWAS stratified by median HbA1c at baseline (8.9%, 74 mmol/mol). Manhattan plots and gene track plots for CpGs with FDR ≤ 0.05 are in Fig. 2. In the n = 180 participants with HbA1c < 8.9%, 77 developed CVD (42.8%). No CpGs reached significance for CVD at FDR ≤ 0.05, but three were associated at the a priori nominal significance level (FDR ≤ 0.10) (Table 3). In the n = 188 participants with HbA1c ≥ 8.9%, 95 developed CVD (50.5%) and two CpGs were significantly associated with time to CVD (Table 3): cg21823999 in GPM6A (p = 7.03 × 10–8, FDR = 0.05) and cg23621817 in CHRNA9 (p = 1.40 × 10–7, FDR = 0.05) (Table 3). As shown in Fig. 2, panel C, cg21823999 is in the promotor/enhancer region GH04J176000 (chr4: 176000933–176002800) and interacts with WDR17. Four additional CpGs had nominally suggestive associations (FDR ≤ 0.10) in those with HbA1c ≥ 8.9% (Table 3).

Manhattan plots for the HbA1c-stratified epigenome-wide associations of DNA methylation with time-to-CVD event (A, B) and gene track plots for CpGs with FDR ≤ 0.05 in those with HbA1c ≥ 8.9% (cg21823999, C; cg23621817 D). On Manhattan plots, red line indicates the significance level FDR ≤ 0.05; blue line indicates nominal significance level FDR ≤ 0.10
We next examined cross sectional associations between CVD-associated CpGs and established cardiometabolic risk factors separately by HbA1c at study baseline, when all participants were free of CVD, to gain insight into pathophysiologic pathways by lower or higher glycemic exposure. Heat maps of cardiometabolic risk factor associations with suggestive CVD-associated CpGs (FDR ≤ 0.10) by HbA1c are in Fig. 3 (Panel A: HbA1c < 8.9%, Panel B: HbA1c ≥ 8.9%). Regression coefficients for the same associations are in Additional file 1: Table S3 (HbA1c < 8.9%) and Additional file 1: Table S4 (HbA1c ≥ 8.9%). In those with HbA1c < 8.9%, greater DNAm at cg07147033 in MIB2 was associated with a generally worse cardiometabolic risk factor pattern and there were significant (p < 0.0005) associations with BMI, eGDR, non-HDLc, triglycerides, AER, and eGFR. Also in those with HbA1c < 8.9%, greater DNAm at cg12323048 was significantly associated with higher non-HDLc and greater DNAm at cg15883830 was associated with higher non-HDLc and lower eGFR. In participants with HbA1c ≥ 8.9%, greater DNAm at cg01028223 (GET4) was significantly associated with lower non-HDLc only. After adjusting for cardiometabolic risk factors, associations between DNAm and time-to-CVD were attenuated to varying degrees (Fig. 4, Additional file 1: Table S5). Attenuation was greater in those with HbA1c < 8.9%, with 16–51% reduction in effect size after risk factor adjustment, compared to 7.5–26% reduction in those with HbA1c ≥ 8.9%.

Heat maps of cardiometabolic phenotype associations with CVD-associated CpGs (FDR ≤ 0.10) by median baseline HbA1c

Log hazard ratio (β) per 5% methylation at each CpG associated with total CVD, unadjusted (blue x) and adjusted (red circles) for traditional cardiometabolic risk factors (error bars are 95% confidence limits) by baseline HbA1c
Methylation quantitative trait loci (meQTLs)
To avoid overlooking potentially important functional relationships, we applied a more liberal significance cut-off of FDR ≤ 0.20 to select CpGs for inclusion in post hoc functional analyses. We performed GWAS using genotyping with imputation to identify variants associated with methylation of the CVD-associated CpGs with FDR ≤ 0.20. We identified 59 cis variants in the TBCD–B3GNTL1 region of chromosome 17 associated with cg14524754 methylation (Additional file 1: Table S6, Fig. S5) at genome-wide significance (p < 5 × 10–8); all 59 variants were confirmed to be significant meQTLs for cg14524754 in GoDMC (p < 5 × 10–8) and 29 were annotated as whole blood eQTLs (p < 0.0005) in GTEx. There were 31 cis variants in the FNDC10–MIB2–CDK11B–SLC35E2B region of chromosome 1 associated with cg07147033 methylation (Fig. 5); 20 were confirmed to be meQTLs for cg07147033 in GoDMC (p < 5 × 10–8) and 11 were whole blood eQTLs (p < 0.0005) in GTEx. Finally, there were 3 cis meQTLs for cg10296867 in GAS7 (Fig. 5); all 3 were meQTLs in GoDMC (p < 5 × 10–8), but none were eQTLs in GTEx. No significant meQTLs were detected for any of the other CpGs with FDR ≤ 0.20 for total CVD in EDC.

Locus Zoom plots for significant meQTLs for CpGs with DNAm associated with total CVD at FDR ≤ 0.20 in the EDC study. Each dot on the plot corresponds to a significant meQTL for DNAm at the corresponding CpG. The representative meQTLs are indicated on each plot
For each of the three CpGs with significant meQTLs, we selected the variant with the strongest statistical association as the representative meQTL to estimate its association with CVD risk. The representative variants were rs9909026 (B3GNTL1) for cg14524754, rs9442431 (MIB2) for cg07147033, and rs78106531 (GAS7) for cg10296867. None were associated with CVD risk overall or in those with HbA1c ≥ 8.9% at baseline; however, rs9442431 (T > C) in MIB2 was associated with CVD incidence in participants with HbA1c < 8.9% (HR = 2.20, 95% confidence interval: 1.44, 3.40, p = 0.00025).
Network and gene set enrichment analysis (GSEA)
To gain insight into potential pathways underlying DNAm-CVD associations, we performed GSEA of gene ontology (GO) terms [14] and KEGG pathways [15] and identified a Reactome Functional Interaction (FI) network [16]. In GSEA, in both the overall cohort and in the subset with HbA1c < 8.9%, there were no significantly enriched GO terms or KEGG pathways including ≥ 2 loci. For the subset with HbA1c ≥ 8.9%, GO terms calcium channel activity (GO: 0005262, p = 0.0015) and integral component of synaptic membrane (GO: 0099699, p = 0.0015), which both include GPM6A and CHRNA9, were enriched (Additional file 1: Table S7). There were no enriched KEGG pathways including ≥ 2 loci.
The candidate loci included in Reactome FI network analysis are listed in Additional file 1: Table S8. A FI network comprising seven modules was identified, three of which had significant pathway enrichment (Fig. 6). Significantly enriched top-level pathways (Additional file 1: Table S9) included DNA repair, DNA replication, Cell cycle, and Metabolism of proteins in Module 1 (FANCC, POLE, TBCD); Neuronal system and Protein localization in Module 2 (CHRNA9, GET4); and Signal transduction, Smooth muscle contraction, Developmental biology, and Vesicle-mediated transport in Module 5 (LINGO1, FCHO2, MYL9).

Reactome functional interaction network derived from the list of loci with DNA methylation associated with 28-year CVD incidence at FDR ≤ 0.20 in the EDC cohort. Candidate loci are indicated in black font (red font indicates linker genes used only to construct the network). Solid line = involved in same reaction as inputs or are components of a shared complex, \(\to\)=activator or catalyst, –|= inhibitor, dashed line = predicted interaction. Only modules with ≥ 2 candidate loci were analyzed for pathway enrichment
Discussion
In this EWAS examining DNAm and 28-year CVD risk in type 1 diabetes, we identified several CVD-associated CpGs in loci across the genome, including sites in GPM6A, CHRNA9, GAS7, GET4, MAD1L1, and MIB2. To our knowledge, this is the first report of a prospective EWAS of CVD in a type 1 diabetes cohort. Our results support our hypothesis that DNAm-CVD associations differ by glycemic exposure. Furthermore, adjustment for established cardiometabolic risk factors led to less attenuation of DNAm-CVD associations in participants with higher HbA1c compared to lower HbA1c, suggesting epigenetic regulation may make a greater independent contribution to CVD pathogenesis under conditions of higher glycemic exposure.
Hypermethylation of cg21823999 in GPM6A and hypomethylation of cg23621817 in CHRNA9, both loci involved in calcium channel activity, were associated with increased CVD risk only in participants with higher HbA1c at baseline. Calcium channel activity is responsible for vascular smooth muscle tone and reactivity via control of vasoconstriction and vasorelaxation [17]. While diabetes has been linked to aberrant calcium signaling [18], GPM6A and CHRNA9 have not previously been specifically implicated in contributing to CVD risk in diabetes. However, there is evidence GPM6A and CHRNA9 may be involved in myocardial development and repair [19, 20] and GPM6A is differentially expressed with respect to acute myocardial infarction [21]. Thus, it is plausible that glycemic exposure modifies epigenetic regulation of GPM6A and CHRNA9 expression to affect downstream CVD risk. Experimental studies on the impact of altered glucose state on GPM6A and CHRNA9 expression are needed to test that hypothesis.
We identified cis-meQTLs significantly associated with DNAm at three of the CVD-associated CpGs and validated those meQTLs using the GoDMC database. There were significant meQTLs for cg14524754 in B3GNTL1 which, while not well-characterized, has been linked to immunological changes related to lung and colorectal cancers [22], and in TBCD, which encodes tubulin-folding cofactor D, a chaperone protein necessary for β-tubulin folding [23]. In a heterodimer with α-tubulin, β-tubulin forms microtubules which not only play a key role in vascular remodeling under pathological conditions but also regulate inflammation [24]. Our findings support further investigation into the role of B3GNTL1 and TBCD in CVD pathogenesis in type 1 diabetes. We also identified significant meQTLs for cg07147033 in MIB2 and FNDC10. MIB2 is involved in Notch signaling, promoting cell proliferation and activation, protection against cytokine-induced cell death [25], and is in the lipid and atherosclerosis KEGG pathway [15]. In the current study, in participants with HbA1c < 8.9%, DNAm at cg07147033 in MIB2 was strongly associated with worse levels of several cardiometabolic risk factors, including BMI, eGDR, non-HDL cholesterol, triglycerides, AER, and eGFR. Additionally, much of the association between cg07147033 methylation and CVD risk was explained by adjustment for those risk factors, particularly non-HDL cholesterol.
In contrast, our results suggest DNAm may make a greater independent contribution to CVD risk under conditions of higher glycemic exposure. In those with HbA1c ≥ 8.9%, in addition to DNAm of GPM6A and CHRNA9 already described above, there were also associations between DNAm and CVD in loci involved in cellular response to stress (GAS7), protein localization (GET4), and cell cycle (MAD1L1) [16]. The direction of DNAm-CVD associations also differed by HbA1c. In those with lower HbA1c, for all top CpGs, hypermethylation was associated with increased CVD risk, while in those with higher HbA1c, we observed both hypo- and hypermethylation of CpGs associated with increased CVD risk. Prior studies have suggested that hypermethylation may be more characteristic of atherosclerosis, while hypomethylation may be more closely related to aging processes [26]. While more research is needed to support those hypotheses, our results could mean that epigenetically regulated accelerated aging pathways contribute to CVD risk under conditions of higher glycemic exposure. Indeed, accelerated aging related to hyperglycemia has previously been proposed as a potential explanation for the increased risk of vascular disease in type 1 diabetes [27].
We [7] and others [6] have previously shown DNAm is associated with glycemic control in type 1 diabetes. In DCCT/EDIC, prior mean HbA1c was associated with DNAm at several CpGs and methylation at those sites was also prospectively associated with microvascular complications [6]. Given the differences in study designs between EDC and DCCT/EDIC, we are not able to perform similar analyses using prior mean HbA1c but have instead adjusted for concurrent HbA1c. Nonetheless, the results of the current analyses are consistent with our prior data showing differences in clinical risk factors for CVD by level of glycemic exposure [13] and suggest that epigenetic regulation of factors related to CVD pathogenesis may differ by glycemic exposure as well. A more comprehensive examination of the role of glycemia in the association between methylation and CVD is warranted and will be the subject of future analyses in the EDC cohort.
Our results overlap somewhat with prior large EWAS in general population studies. The Cohorts for Heart and Aging Genetic Epidemiology (CHARGE) Consortium has performed the largest prospective EWAS of coronary heart disease to date and reported associations with DNAm in PTPRN2 and MAD1L1 [8]. Multi-Ethnic Study of Atherosclerosis (MESA) similarly demonstrated PTPRN2 expression is associated with coronary artery calcification and carotid plaque score [28]. Our observation that methylation of cg02768721 in PTPRN2 has a suggestive association with CVD only in EDC participants with lower HbA1c corresponds to those associations between methylation of PTPRN2 and CVD in the general population, which has low glycemic exposure on average.
In addition to findings discussed above, we identified associations between DNAm and CVD at several CpGs that were driven by a small number of participants with extreme methylation probe β values (data shown in the Additional file 1). Upon examination of their clinical characteristics, all had either HbA1c > 9% or advanced kidney disease at baseline and three of the eight individuals had a greater proportion of extreme probe β values than the overall cohort. It has been shown that methylation outlier burden likely reflects biological age and is unlikely to be a result of technical artifacts [29]. Thus, the biological plausibility of the corresponding loci, including FBXO27, which is involved in autophagy [30] and highly expressed in cardiomyocytes [31]; CARD9, which encodes a protein that regulates cell apoptosis and NF-kappaB activation [32]; and NLN, which is involved in the renin–angiotensin–aldosterone system [33], suggest they may warrant further study in external cohorts.
Strengths and limitations
Our study has several strengths, including the use of data from a well-characterized, exclusively type 1 diabetes cohort with long-term follow-up to ascertain CVD incidence. The EDC cohort is epidemiologically representative of the childhood-onset type 1 diabetes population of Allegheny County, Pennsylvania [34]. The prospective study design avoids the possibility of reverse causation. Another strength is that CVD events were verified using death certificates and medical records by physician reviewers masked to risk factor status. A further strength is the availability of traditional cardiometabolic risk factor data, facilitating examination of cardiometabolic phenotypes for the CVD-associated DNAm. We also assessed DNAm-CVD associations separately by level of glycemic exposure, an important potential effect modifier in type 1 diabetes.
Limitations of our study include use of whole blood for methylation measurement and a lack of tissue-specific data. However, DNAm in whole blood is commonly examined in epidemiologic studies such as ours, due to ease of specimen collection and because it facilitates detection of multiple physiologic pathways that lead to complex phenotypes like CVD. We used a standard method to account for blood cell type composition in our analyses. Another limitation is the lack of long-term glycemic exposure data prior to DNA collection, so we are unable to assess the temporal association between glycemic exposure and differential methylation. The sample size of our study is relatively small with limited power for the secondary outcomes MACE and major CAD in particular, restricting our ability to identify strong associations. Thus the results should be validated in other cohorts as data become available. Additionally, due to the small sample size, we applied a more liberal cut-off of FDR ≤ 0.20 for including CpGs in the post hoc functional analyses to facilitate our goal of identifying potentially important pathways. This approach is similar to other prior epidemiological studies of DNAm which have used the approach of applying a more liberal FDR cut point for inclusion of CpGs in enrichment analyses [35, 36]. Finally, the EDC cohort is 98% white/European ancestry, primarily due to the demographics of Allegheny County, Pennsylvania, USA (< 15% black/African American) and historically lower incidence of type 1 diabetes among black individuals [37], so our results may not apply to more diverse populations.
Conclusions
We present novel evidence that DNAm at several CpGs may be associated with future CVD risk in type 1 diabetes and contribute to risk independently of traditional cardiometabolic risk factors. Importantly, our findings suggest epigenetic modification of GPM6A and CHRNA9, both involved in calcium channel activity and development, may play a role in the pathophysiology of CVD under conditions of higher glycemic exposure in type 1 diabetes. While experimental data and validation in external populations are needed, our observation of such glycemia-dependent associations supports the existence of heterogeneous pathways to CVD development in type 1 diabetes and may provide insight into new targets for intervention to prevent or delay CVD in this high-risk population.
Methods
Study population
The Pittsburgh Epidemiology of Diabetes Complications (EDC) study is a prospective cohort study of childhood-onset (< 17 years old) T1D. All participants in the parent cohort (n = 658) were diagnosed with T1D, or seen within one year of diagnosis, at Children’s Hospital of Pittsburgh between 1950 and 1980. The cohort has been described in detail [38] and a study timeline is shown in Fig. 1. Participants have been followed since 1986–1988, with biennial examinations and questionnaires for the first ten years and thereafter with biennial questionnaires and further examinations in 2004–2006, 2011–2013, and 2016–2018. DNA was collected at study visits between 1988 and 1998: 86% of the DNA specimens were collected at the 1988–1990 visit, 9% at the 1990–1992 visit, and the remaining 5% between 1992 and 1998. The date of each participant’s DNA specimen defines their baseline for these analyses.
A diagram detailing derivation of the final analytic sample is in Fig. 1. Of 496 participants consenting to blood leukocyte DNA collection, 436 European ancestry participants had suitable quality DNA available for methylation arrays remaining in 2020. After excluding participants not passing methylation quality control (n = 11), one randomly selected individual from each of 10 first-degree relative pairs, two outliers from genetic principal components analysis, and one randomly selected individual from each of 2 technical duplicate specimen pairs the final sample comprised 411 participants.
DNA methylation arrays, quality control, and data processing
High molecular weight DNA was isolated from whole blood-derived leukocytes. DNAm was assayed using Illumina Infinium MethylationEPIC BeadChip arrays (Illumina, San Diego, CA, USA) [39] at the University of Virginia Center for Public Health Genomics, Genomic Sciences Laboratory using the standard Illumina methylation array protocol. Quality control methods were implemented in two packages in R (v4.1.0; R Core Team 2021), minfi v1.32.0 [40] and SeSAMe v1.8.10 [41]. The pipeline, including the specific functions used, is summarized in Additional file 1: Fig. S1. The EPIC output Intensity Data (IDAT) file contained raw probe intensity data, as well as the control probe information needed for quality control. Using minfi, the IDAT files were loaded and converted into an RGSet object and thence to a GMSet object based on the Illumina 1.0 B5 hg38 annotation release. The minfiQC function was used to check for outlier samples of potentially poorer quality based on: (1) median intensity of methylated (M) versus unmethylated (U) probes (minfiQC() function, outlier cut-off = 3 stdev); (2) median X, Y chromosome probe signals for karyotype prediction (getSex() function cut-off = − 2); (3) sample probe detection rate (detectionP() function, p value < 0.01). The third step was repeated after dropping 72,868 low quality probes (probe detection rate < 95% as described below) and a previously published curated list of 95,923 probes recommended for exclusion as detailed by Zhou et al. [42]. Specifically, the list curated by Zhou et al. excludes probes with: inconsistent mapping; SNPs in the extension base that cause a color channel switch from the official annotation; non-unique 30bp3’ subsequence; 5bp3’ subsequence overlap with any of the SNPs with global minor allele frequency > 1%; and extension base inconsistent with the specified color channel or CpG.
To confirm and expand the minfi QC, a second pipeline of QC checks used functions in the SeSAMe package [41]. First, the RGset object was converted to a SigSet object using the RGSetChannelSetToSigSet() function. Then, the pipeline checked: (1) mean sample probe intensity distribution by sample, mean(M + U) (sesameQC() function); (2) sample bisulfite GCT score for completeness of bisulfite conversion (sesameQC() function); (3) inferred sex karyotype based on chromosome X and Y probes (inferredSexKaryotypes() function); (4) detection of duplicate samples using SNP genotype data extracted from a subset of the probes. Finally, probe quality checks were performed based on individual probe detection p values. In this check, any probe that was not detected in at least 95% of samples was dropped (72,868 unique probes excluded). Cryptic sample duplicates were inferred using KING [43] applied to the SNP genotype data extracted from the Illumina ‘spiked-in’ SNP probes (n = 44) and additional probes that manifest a reproducible pattern of genotype-stratified methylation signals driven by a flanking SNP (n = 865 SNPs in total). After the QC described above and further restricting analysis to probes mapped to autosomal chromosomes, 683,597 of the original 865,918 probes on the EPIC array were analyzed.
The final methylation fraction β values for analysis were generated using the following pipeline steps in SeSAMe [41]: (1) background subtraction based on normal-exponential deconvolution using out-of-band probes (noob); (2) dye bias correction in the two-color channels through nonlinear scaling (dyeBiasCorrTypeINorm); (3) probe quality masking based on the curated exclusion list (qualityMask); (4) detection masking at individual probe/sample level using pooBAH p value out-of-band array hybridization (detectionMask); (5) beta calculation, M/M + U (getBetas); (6) drop samples and probes identified during quality control pre-processing. Finally, for each probe we excluded β values > ± 3 standard deviations from the mean to remove extreme outliers for the primary analysis. As DNA was isolated from whole blood-derived leukocytes, variable cell type composition and differential methylation states in a sample could confound association tests. Thus, cell type compositions variables were estimated using the estimateCellCounts2 function from the R package FlowSorted.Blood.EPIC [44], using an IDOL-optimized probe set [45] derived from the original Houseman method [46] which has been shown to be an optimal algorithm for cell-type deconvolution in EWAS using whole blood specimens [47].
Ascertainment of cardiovascular outcomes
Follow-up time for each participant was calculated from the study visit during which their DNA was isolated (baseline) until CVD incidence or censoring (31 December 2018 or last follow-up). CVD was defined as the first instance of CVD death, nonfatal myocardial infarction (MI, including clinical events and subclinical myocardial infarction on ECG, i.e., Minnesota code 1.1 or 1.2), nonfatal stroke, coronary revascularization procedure, blockage ≥ 50%, ischemic EGC at EDC study visit (Minnesota codes 1.3, 4.1–4.3, 5.1–5.3, 7.1), or EDC physician-diagnosed angina. Two secondary outcomes were also examined: major adverse cardiovascular events (MACE, first instance of CVD death, nonfatal MI or nonfatal stroke) and major coronary artery disease (CAD, first instance of CAD death or nonfatal MI). Fatal events were ascertained using medical records, death certificates, autopsy reports, and/or interview with next of kin and classified according to the Diabetes Epidemiology Research International (DERI) system [48]. Nonfatal events were confirmed with medical records.
CVD EWAS
Participants with prevalent total CVD (n = 43), MACE (n = 14), or major CAD (n = 11) at the time of DNA collection were excluded from each respective analysis. We performed a time-to-event EWAS for total CVD incidence with CpGs on chromosomes 1–22 using Cox regression. Each CpG probe β-value was modeled as the main independent variable and adjusted for type 1 diabetes duration, sex, pack-years of smoking, cell type composition variables, plate/run number, GCT bisulfite score, DNA extraction method, and well position. As one of our aims was to identify genetic variants associated with DNAm, we also included the first two ancestry principal components based on GWAS data [49] as covariates in the EWAS. EDC is an exclusively childhood-onset (< 17 years) type 1 diabetes cohort; thus, age and diabetes duration are highly correlated (r = 0.86, p < 0.0001). As type 1 diabetes duration is the exposure of greater interest in the current analysis, the results we present were adjusted for diabetes duration only. However, results remained the same in alternative models adjusting for age instead of diabetes duration. Separate EWAS of the secondary outcomes MACE and major CAD were also performed using the same methods described above. As DNAm outliers could be associated with extreme phenotypes, we also performed alternative exploratory analyses including extreme probe β values (> ± 3 standard deviations from the mean). Given the sample size of the cohort, potentially important associations with small effect sizes would be disregarded if we only applied a conservative statistical significance cut-off. Thus, a Benjamini–Hochberg false discovery rate (FDR) ≤ 0.05 was considered significant and FDR ≤ 0.10 was considered nominally significant. Additionally, to avoid overlooking potentially important functional relationships, a more liberal FDR ≤ 0.20 was considered suggestive and warranting inclusion in the secondary post hoc functional analyses. As the traditional genomic inflation factor λ has been shown to overestimate test statistic inflation in EWAS [50], we used the bacon method (λ.bacon), a Bayesian method developed by Iterson et al. specifically for EWAS [50], to estimate inflation if traditional λ > 1.10.
HbA1c-stratified EWAS and cardiometabolic risk factor analyses
We repeated the EWAS for total CVD stratified by HbA1c to determine whether CVD-associated DNAm differed by level of glycemic control. Because there is no known optimal HbA1c cut point at which the CVD-DNAm association is modified, median baseline HbA1c (8.9% [74 mmol/mol]) was used as the cut point for stratification, ensuring approximately equal sample size in each stratum. We then assessed associations between DNAm at CpGs considered at least nominally suggestive (FDR ≤ 0.10) and levels of baseline cardiometabolic risk factors using linear regression separately by median HbA1c (8.9%). Cardiometabolic risk factors analyzed were HbA1c, body mass index (BMI), estimated glucose disposal rate (eGDR), HDL cholesterol, non-HDL cholesterol, triglycerides, systolic and diastolic blood pressures (SBP and DBP), pulse rate, albumin excretion rate (AER), and estimated glomerular filtration rate (eGFR). Details regarding ascertainment of cardiometabolic risk factors have been published previously [51] and are described in the Additional file 1. In separate minimally adjusted linear models, each cardiometabolic risk factor was the dependent variable and the methylation β value at each CVD-associated CpG was the independent variable, adjusting for type 1 diabetes duration, sex, pack-years of smoking, and cell type composition variables. An adjusted p value ≤ 0.0005 (0.05/99 comparisons, i.e., 9 CpGs × 11 risk factors) was considered conservatively statistically significant, while p ≤ 0.05 was nominally significant. Finally, for each CpG assessed, we re-fit Cox models for time-to-CVD, adjusting for cardiometabolic risk factors, selected using backward selection, to obtain cardiometabolic risk factor-independent estimates of the DNAm-CVD associations. Risk factors were entered into the models if they were univariately associated with the CpG at p ≤ 0.10 and were retained in the Cox model if p ≤ 0.05.
Post hoc functional analyses
For CpG associated with CVD at FDR ≤ 0.20 in EWAS, we identified methylation quantitative trait loci (meQTLs) via GWAS using existing imputed genotyping array data in the EDC cohort [49]. Genotyping was performed in n = 453 EDC participants using the Infinium HumanCore Exome-24 BeadChip (Illumina, San Diego, CA, USA), following the manufacturer’s protocol. After quality control and exclusions, quality genotyping data were available for n = 422. Determination of the analytic sample has been described previously [52]. Minimac3/ Minimac3-omp version 1.0.14 was used for imputation with the 1000 Genomes (1KG) Phase 3 version 5 reference panel (updated Oct 20, 2015). In the n = 411 EDC participants included in the DNAm analysis, we performed GWAS of the CVD-associated CpGs with FDR ≤ 0.20. DNAm probe β-values were modeled as the dependent variable and SNP dosage (additive coding) as independent variables in separate linear models, adjusting for sex, T1D duration, pack-years of smoking, estimated cell type composition, and the first three ancestry principal components. Genome-wide significant SNPs at p ≤ 5 × 10–8 were identified as meQTLs for the corresponding CpG. Results were compared to the Genetics of DNA Methylation Consortium (GoDMC) database of meQTLs [53] for external validation. Finally, we looked for evidence of the meQTLs’ effects on gene expression by determining whether the meQTLs were annotated as whole blood eQTLs in the Genotype-Tissue Expression (GTEx) Project database using a significant threshold of p < 0.0005 (p = 0.05/93 meQTLs assessed). The data used for the analyses described here were obtained from the GTEx Portal on 6 September 2022.
Gene information was obtained from Illumina annotation (March 2020, reference genome GRCh38/hg38). We performed gene set enrichment analysis (GSEA) using the GOmeth function in the missMethyl package for R with the GO.db and KEGG.db annotation packages [54] to identify enrichment of terms associated with ≥ 2 loci with CVD-associated DNAm. For all loci associated with CpGs with FDR ≤ 0.20, we identified a Reactome Functional Interaction (FI) network [16] using Cytoscape [55]. Modules within the network were identified using a spectral algorithm for clustering [56]; we performed Reactome pathway analysis on the resulting modules with ≥ 3 total nodes including ≥ 2 candidate loci.
Availability of data and materials
Data, code, and associated documentation are available from the corresponding author on reasonable request. In accordance with University of Pittsburgh and Institutional Review Board policies and regulations, a data-sharing agreement is required.
References
Saeed M, Stene LC, Ariansen I, Tell GS, Tapia G, Joner G, et al. Nine-fold higher risk of acute myocardial infarction in subjects with type 1 diabetes compared to controls in Norway 1973–2017. Cardiovasc Diabetol. 2022;21(1):59.
Ma RC. Genetics of cardiovascular and renal complications in diabetes. J Diabetes Investig. 2016;7(2):139–54.
Orchard TJ, Costacou T. Cardiovascular complications of type 1 diabetes: update on the renal link. Acta Diabetol. 2017;54(4):325–34.
Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016;118(1):119–31.
Agha G, Mendelson MM, Ward-Caviness CK, Joehanes R, Huan TX, Gondalia R, et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. 2019;140(8):645–57.
Chen Z, Miao F, Braffett BH, Lachin JM, Zhang L, Wu X, et al. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat Metab. 2020;2(8):744–62.
Miller RG, Mychaleckyj JC, Onengut-Gumuscu S, Orchard TJ, Costacou T. TXNIP DNA methylation is associated with glycemic control over 28 years in type 1 diabetes: findings from the Pittsburgh Epidemiology of Diabetes Complications (EDC) study. BMJ Open Diabetes Res Care. 2023;11(1):e003068.
Agardh E, Lundstig A, Perfilyev A, Volkov P, Freiburghaus T, Lindholm E, et al. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015;13(1):182.
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genom. 2010;3(1):33.
Gu T, Falhammar H, Gu HF, Brismar K. Epigenetic analyses of the insulin-like growth factor binding protein 1 gene in type 1 diabetes and diabetic nephropathy. Clin Epigenetics. 2014;6(1):10.
Swan EJ, Maxwell AP, McKnight AJ. Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet Med. 2015;32(8):1110–5.
Smyth LJ, Patterson CC, Swan EJ, Maxwell AP, McKnight AJ. DNA methylation associated with diabetic kidney disease in blood-derived DNA. Front Cell Dev Biol. 2020;8:561907.
Miller RG, Orchard TJ, Costacou T. 30-Year cardiovascular disease in type 1 diabetes: risk and risk factors differ by long-term patterns of glycemic control. Diabetes Care. 2022;45(1):142–50.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Reserch. 2000;28(1):27–30.
Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50(D1):D687–92.
Cribbs LL. Vascular smooth muscle calcium channels could “T” be a target? Circ Res. 2001;89(7):560–2.
Choi KM, Zhong Y, Hoit BD, Grupp IL, Hahn H, Dilly KW, et al. Defective intracellular Ca2+ signaling contributes to cardiomyopathy in type 1 diabetic rats. Am J Physiol Heart Circ Physiol. 2002;283:H1398–408.
Bochmann L, Sarathchandra P, Mori F, Lara-Pezzi E, Lazzaro D, Rosenthal N. Revealing new mouse epicardial cell markers through transcriptomics. PLoS ONE. 2010;5(6):e11429.
Chamberlain AA, Lin M, Lister RL, Maslov AA, Wang Y, Suzuki M, et al. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J Am Heart Assoc. 2014;3(3):e000976.
Wu J, Li C, Lei Z, Cai H, Hu Y, Zhu Y, et al. Comprehensive analysis of circRNA-miRNA-mRNA regulatory network and novel potential biomarkers in acute myocardial infarction. Front Cardiovasc Med. 2022;7:9.
Sandanger TM, Nøst TH, Guida F, Rylander C, Campanella G, Muller DC, et al. DNA methylation and associated gene expression in blood prior to lung cancer diagnosis in the Norwegian Women and Cancer cohort. Sci Rep. 2018;8(1):1–10.
Tian G, Huang Y, Rommelaere H, Vandekerckhove J, Ampe C, Cowan NJ. Pathway leading to correctly folded β-tubulin. Cell. 1996;86(2):287–96.
Warner EF, Li Y, Li X. Targeting microtubules for the treatment of heart disease. Circ Res. 2022;130(11):1723–41.
Feltham R, Jamal K, Tenev T, Liccardi G, Jaco I, Domingues CM, et al. Mind bomb regulates cell death during TNF signaling by suppressing RIPK1’s cytotoxic potential. Cell Rep. 2018;23(2):470–84.
Aavik E, Babu M, Ylä-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. 2019;1(281):168–79.
Monaco CMF, Gingrich MA, Hawke TJ. Considering type 1 diabetes as a form of accelerated muscle aging. Exerc Sport Sci Rev. 2019;47(2):98–107.
Liu Y, Reynolds LM, Ding J, et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat Commun. 2017;8:393. https://doi.org/10.1038/s41467-017-00517-4.
Seeboth A, McCartney DL, Wang Y, Hillary RF, Stevenson AJ, Walker RM, et al. DNA methylation outlier burden, health, and ageing in Generation Scotland and the Lothian Birth Cohorts of 1921 and 1936. Clin Epigenetics. 2020;12(1):1–13.
Glenn KA, Nelson RF, Wen HM, Mallinger AJ, Paulson HL. Diversity in tissue expression, substrate binding, and SCF complex formation for a lectin family of ubiquitin ligases. J Biol Chem. 2008;283(19):12717–29.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (1979). 2015;347(6220):1260419.
Ji C, Yang Z, Zhong X, Xia J. The role and mechanism of CARD9 gene polymorphism in diseases. Biomed J. 2021;44(5):560–6.
Wangler NJ, Santos KL, Schadock I, Hagen FK, Escher E, Bader M, et al. Identification of membrane-bound variant of metalloendopeptidase neurolysin (EC 3.4.24.16) as the non-angiotensin type 1 (non-AT 1), non-AT 2 angiotensin binding site. J Biol Chem. 2012;287(1):114–22.
Wagener DK, Sacks JM, LaPorte RE, Macgregor JM. The Pittsburgh study of insulin-dependent diabetes mellitus. Risk for diabetes among relatives of IDDM. Diabetes. 1982;31(2):136–44.
Brunst KJ, Tignor N, Just A, Liu Z, Lin X, Hacker MR, et al. Cumulative lifetime maternal stress and epigenome-wide placental DNA methylation in the PRISM cohort. Epigenetics. 2018;13(6):665–81.
Ward-Caviness CK, Agha G, Chen BH, Pfeiffer L, Wilson R, Wolf P, et al. Analysis of repeated leukocyte DNA methylation assessments reveals persistent epigenetic alterations after an incident myocardial infarction. Clin Epigenetics. 2018;10(1):1–13.
Laporte RE, Tajima N, Dorman JS, Cruickshanks KJ, Eberhardt MS, Rabin BS, et al. Differences between blacks and whites in the epidemiology of insulin-dependent diabetes mellitus in allegheny county, Pennsylvania. Am J Epidemiol. 1986;123(4):592–603.
Orchard TJ, Dorman JS, Maser RE, Becker DJ, Drash AL, Ellis D, et al. Prevalence of complications in IDDM by sex and duration. Pittsburgh epidemiology of diabetes complications study II. Diabetes. 1990;39(9):1116–24.
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8(3):389–99.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Zhou W, Triche TJ, Laird PW, Shen H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;46(20):e123.
Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017;45(4):e22.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–73.
Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018;19(1):1–14.
Koestler DC, Jones MJ, Usset J, Christensen BC, Butler RA, Kobor MS, et al. Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL). BMC Bioinform. 2016;17(1):1–21.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13(1):86.
Teschendorff AE, Zheng SC. Cell-type deconvolution in epigenome-wide association studies: a review and recommendations. Epigenomics. 2017;9(5):757–68.
Diabetes Epidemiology Research International Mortality Study Group. International evaluation of cause-specific mortality and IDDM. Diabetes Epidemiology Research International Mortality Study Group. Diabetes Care. 1991;14(1):55–60.
Salem RM, Todd JN, Sandholm N, Cole JB, Chen WM, Andrews D, et al. Genome-Wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J Am Soc Nephrol. 2019;30(10):2000–16.
van Iterson M, van Zwet EW, Heijmans BT, ‘t Hoen PAC, van Meurs J, Jansen R, et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18(1):1–13.
Miller RG, Orchard TJ, Costacou T. Risk factors differ by first manifestation of cardiovascular disease in type 1 diabetes. Diabetes Res Clin Pract. 2020;163:108141.
Miller RG, Orchard TJ, Onengut-Gumuscu S, Chen WM, Rich SS, Costacou T. Heterogeneous long-term trajectories of glycaemic control in type 1 diabetes. Diabet Med. 2021;38(8):e14545.
Min JL, Hemani G, Hannon E, Dekkers KF, Castillo-Fernandez J, Luijk R, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53(9):1311–21.
Phipson B, Maksimovic J, Oshlack A. MissMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–8.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Newman MEJ. Modularity and community structure in networks. Proc Natl Acad Sci U S A. 2006;103(23):8577–82.
Acknowledgements
We thank the Epidemiology of Diabetes Complications study participants and staff for their invaluable contributions and dedication to this research for over 30 years.
Funding
The parent EDC study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (Grant # R01-DK034818) and the Rossi Memorial Fund. The EDC DNA methylation sub-study was supported by the American Diabetes Association (Grant # 1-19-JDF-109). The funding agencies were not involved in study design, data collection, data analysis, interpretation, or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
RGM, EF, and TJO designed the study. RGM, TJO, and TC contributed to acquisition of funding and data. SO-G oversaw methylation array processing. JCM performed the methylation data processing and quality control. RGM conducted the statistical analyses. RGM, JCM, SO-G, EF, TJO, and TC interpreted the data. RGM drafted the manuscript. All authors edited and critically reviewed the manuscript for intellectual content and approved the final version. RGM is the guarantor of this work and as such ad full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Research protocols were approved by the University of Pittsburgh institutional review board (approval #19040065). All participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Supplementary Methods:
Cardiometabolic risk factors. Supplementary Results: Extreme probe β values. Table S1. All CpGs with DNA methylation associated with time to major adverse cardiovascular event (MACE) and major coronary artery disease (CAD) at FDR <0.20 included in post hoc functional analyses. Table S2. Top 5 CpGs with DNA methylation associated with time to total CVD, MACE, and major CAD incidence in the EDC, including extreme probe β outliers. Table S3. Cardiometabolic risk factors and methylation at CVD associated differentially methylated positions (FDR ≤ 0.10) – HbA1c <8.9%. Table S4. Cardiometabolic risk factors and methylation at CVD-associated differentially methylated positions (FDR ≤ 0.10) – HbA1c ≥8.9%. Table S5. Associations between DNA methylation and total CVD after adjustment for traditional cardiometabolic risk factors by median HbA1c at baseline. Table S6. Genome-wide significant (p<5.0E-8) methylation quantitative trait loci (meQTLs) for CVD-associated methylation identified in the EDC cohort. Table S7. Gene ontology (GO) terms with p<0.05 for total CVD in EDC participants with HbA1c ≥8.9%. Table S8. Loci included in Reactome Functional Interaction network analysis (alphabetical). Table S9. Significantly enriched Reactome pathways identified in functional interaction network analysis of loci containing CpGs with DNA methylation associated with CVD incidence at FDR<0.20. Figure S1. DNA methylation quality control (QC) pipeline used in the EDC study. Figure S2. Kaplan-Meier curves of proportion free of each CVD outcome. Figure S3. Manhattan plot (A) for the epigenome-wide association of DNAm with time-to-CVD event after excluding probe beta outliers and gene track plots for CpGs with FDR<0.20 (B-D) in the EDC study. Figure S4. QQ plot for the epigenome-wide association of DNAme with time-to-CVD event. Figure S5. QQ plot for the epigenome-wide association of DNAme with time-to-CVD event, all observations included.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miller, R.G., Mychaleckyj, J.C., Onengut-Gumuscu, S. et al. DNA methylation and 28-year cardiovascular disease risk in type 1 diabetes: the Epidemiology of Diabetes Complications (EDC) cohort study. Clin Epigenet 15, 122 (2023). https://doi.org/10.1186/s13148-023-01539-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01539-0