
Abstract
Background
In utero exposure to maternal hyperglycemia has been associated with an increased risk for the development of chronic diseases in later life. These predispositions may be programmed by fetal DNA methylation (DNAm) changes that persist postnatally. However, although some studies have associated fetal exposure to gestational hyperglycemia with DNAm variations at birth, and metabolic phenotypes in childhood, no study has yet examined how maternal hyperglycemia during pregnancy may be associated with offspring DNAm from birth to five years of age.
Hypothesis
Maternal hyperglycemia is associated with variation in offspring DNAm from birth to 5 years of age.
Methods
We estimated maternal hyperglycemia using the area under the curve for glucose (AUCglu) following an oral glucose tolerance test conducted at 24–30 weeks of pregnancy. We quantified DNAm levels in cord blood (n = 440) and peripheral blood at five years of age (n = 293) using the Infinium MethylationEPIC BeadChip (Illumina). Our total sample included 539 unique dyads (mother–child) with 194 dyads having DNAm at both time-points. We first regressed DNAm M-values against the cell types and child age for each time-point separately to account for the difference by time of measurement for these variables. We then used a random intercept model from the linear mixed model (LMM) framework to assess the longitudinal association between maternal AUCglu and the repeated measures of residuals of DNAm. We adjusted for the following covariates as fixed effects in the random intercept model: maternal age, gravidity, smoking status, child sex, maternal body mass index (BMI) (measured at first trimester of pregnancy), and a binary variable for time-point.
Results
In utero exposure to higher maternal AUCglu was associated with lower offspring blood DNAm levels at cg00967989 located in FSD1L gene (β = − 0.0267, P = 2.13 × 10–8) in adjusted linear regression mixed models. Our study also reports other CpG sites for which DNAm levels were suggestively associated (P < 1.0 × 10–5) with in utero exposure to gestational hyperglycemia. Two of these (cg12140144 and cg07946633) were found in the promotor region of PRDM16 gene (β: − 0.0251, P = 4.37 × 10–07 and β: − 0.0206, P = 2.24 × 10–06, respectively).
Conclusion
Maternal hyperglycemia is associated with offspring DNAm longitudinally assessed from birth to 5 years of age.
Similar content being viewed by others

Background
Gestational diabetes mellitus (GDM) is defined by hyperglycemia first occurring during pregnancy in an individual who did not previously have diabetes [1]. GDM affects approximately 14% of pregnancies worldwide [2] with variation in prevalence depending on ethnicity [3] and the diagnostic criteria applied [4]. Regardless of GDM diagnosis, maternal hyperglycemia along the whole spectrum has been linearly associated with adverse perinatal outcomes in both mother and offspring [5]. Exposure to maternal hyperglycemia in utero is also associated with an increased risk for the development of chronic diseases later in childhood, including obesity [6], metabolic [7] and cardiovascular diseases [8], asthma [9], autism [10], as well as other adverse neurodevelopmental outcomes [11]. The Developmental Origin of Health and Disease (DOHaD) hypothesis is central to the concept of fetal programming and based on lasting fetal metabolic adaptions to environmental/maternal stressors during the critical developmental period [12]. Epigenetic marks are highly malleable during in utero development and in early life but can be stable over time [13]. Epigenetics is one of the most likely molecular mechanisms as part of the DOHAD theory and fetal metabolic programming.
DNA methylation (DNAm) is the most studied epigenetic modification. It involves the addition of a methyl group to the fifth carbon of a cytosine when located just upstream of a guanine [14]. Growing evidence supports that DNAm may be the bridge between prenatal exposure to maternal hyperglycemia and the predisposition to chronic disease in later life. The most comprehensive study consists of a meta-analysis of an epigenome-wide association study (EWAS) investigating associations between maternal hyperglycemia exposure and cord blood DNAm variations. This study reported significant associations (FDR < 0.05) at two loci between cord blood DNAm and the area under the curve of glucose (AUCglu) following an oral glucose tolerance test (OGTT) [15]. Candidate gene studies also reported significant correlations between maternal hyperglycemia exposure during pregnancy and offspring DNAm in genes implicated in chronic disease pathways [16, 17].
However, although the association between maternal hyperglycemia exposure and DNAm measured at birth has been explored in a few studies, no study has yet examined how maternal hyperglycemia may be associated with offspring DNAm from birth to five years of age. To address this gap, we conducted an EWAS examining the association between maternal glycemic response to an OGTT performed during pregnancy, and offspring DNAm assessed longitudinally at birth (cord blood) and at age 5 years (peripheral blood), using modeling approaches that integrate DNAm at both time-points.
Methods
Study population
Our study sample consists of 539 mother–child dyads from the Genetics of Glucose Regulation in Gestation and Growth (Gen3G) prospective cohort. We recruited Gen3G participants in the Sherbrooke City area, Canada, to investigate environmental and genetic determinants of glucose regulation in pregnancy, and their impacts on fetal development and offspring health [18]. Between 2010 and 2013, we invited pregnant women to participate in the study when they attended a routine prenatal blood sampling visit during their first trimester of pregnancy at the Centre Hospitalier Universitaire de Sherbrooke (CHUS), if they planned to deliver at CHUS [18]. Eligible women were ≥ 18 years old, with a singleton pregnancy, and not reporting using medication that alters glucose. We excluded women with pre-existing diabetes or diabetes diagnosed in the first trimester based on laboratory screening (A1c > 6.5% or 1 h-glucose ≥ 10.3 mmol/L post-50 g glucose challenge test [GCT]). Details on Gen3G participants have been published previously [18]. All women provided signed consent, and the CHUS IRB approved all Gen3G protocols.
At enrollment, women provided information on age, ethnicity, smoking habits during pregnancy (never/former smoker vs current smoker) and gravidity (primigravid vs multiparous). During the first (6–14 weeks) and second (24–30 weeks) pregnancy visits, research staff measured weight (in kg) and height (in m) using standardized procedures, and we calculated body mass index (BMI) using the standard formula (kg/m2) [18]. At the first visit, most women completed a non-fasting 50 g-GCT. At the second visit, all women conducted a 75 g-OGTT in order to measure glucose tolerance. At delivery, we abstracted from the medical records information about gestational age at birth (in weeks), child sex, and birth weight (in g). Research staff collected umbilical cord blood samples rapidly after delivery (< 30 min).
We conducted in-person follow-up visits with mother–child pairs at the CHUS Research Center approximately 5 years after birth. During this visit, we collected updated medical history and anthropometric measures, in addition to child blood samples.
Maternal glucose measured during a 75 g-OGTT in the second trimester, and estimation of the area under the curve for glucose
We measured glucose levels (mmol/L) using the glucose hexokinase method (Roche Diagnostics, Indianapolis, US) at the CHUS biochemistry clinical laboratory rapidly after blood collection [18, 19]. We calculated the AUCglu based on the trapezoidal formula [20], to capture a global measure of maternal hyperglycemia in pregnancy that encompasses all three time-points of the OGTT since all of them have been associated with adverse pregnancy outcomes [5], and the AUCglu was the most informative phenotype in prior EWAS investigating dysregulation in child cord blood DNAm [15].
Genome-wide DNA methylation level assessment
Genomic DNA was purified from umbilical cord blood cells and from blood cells, respectively, collected at delivery and 5 years of age using the AllPrep DNA/RNA/Protein Mini kit (QIAGEN, Hilden, Germany). DNA was then bisulfite converted with the EZ-Methylation kit (Zymo Research, CA, US), and subsequently analyzed for DNAm at > 850,000 CpG sites at single-nucleotide resolution using the Infinium MethylationEPIC BeadChip (Illumina, San Diego, CA). We first performed a manual inspection of the quality of the methylation assay looking at probes call rate (> 99%), color balance, staining, extension, hybridization, specificity and bisulfite conversion (probe types I and II) using the BeadArray Controls Reporter software. We transferred raw methylation files into R (version 4.1.1) for further preprocessing using the minfi R package [21]. Our final dataset included 440 cord blood samples and 293 peripheral blood samples collected at the ~ 5-year visit after quality controls. In terms of participants, this represents 539 unique dyads (mother–child pairs), from which 194 dyads had both time-points (cord blood and peripheral blood) included in the analyses (see support Fig. 1).

Flowchart illustrating the participant selection from the Gen3G prospective cohort
We implemented functional normalization for background and dye bias correction [22] of the methylation data, and the regression on correlated probes (RCP) method [23] to adjust for probe-type bias, as previously described [24]. We filtered out probes with null methylation variance and detection P-value > 0.05 in at least 5% of the samples. We annotated CpG sites in the array using the R package IlluminaHumanMethylationEPICmanifest (version 1.0 B4), and we excluded probes located on sex chromosomes, non-CpG probes (“rs” or “ch”), single-nucleotide polymorphism (SNP)-associated probes at the CpG site, the single-base extension, or anywhere within the probe, if the SNP had a minor allele frequency (MAF) ≥ 0.05. In addition, we removed cross-reactive probes as previously described [25]. We used ComBat [26] to correct for technical variability and batch effects, while protecting our variables of interest. The model thus included the three glucose values (fasting glucose, 1 h-glucose, and 2 h-glucose post-OGTT), maternal age, BMI in the first trimester of pregnancy, gravidity, smoking status, and child sex. After quality control, we included up to 719,360 high-quality CpG sites in the analysis using DNAm from combined cord blood and childhood peripheral blood. We calculated the predicted cell counts in cord blood based on the method described by Gervin et al.[27], and the predicted cell counts in child blood based on the method from Salas et al.[28], implemented in the FlowSorted.Blood.EPIC R package.
Statistical analyses
We described baseline characteristics of the overall sample, and of samples at each time-point, using standard descriptive statistics. For each CpG site, we used methylation values measured in the β-value scale, as a range estimating between 0% (unmethylated) and 100% (completely methylated), to transform into M-values prior to analyses based on the statistical validity of this method to identify differential DNAm [29]. Due to differences in the cell types predicted in each time-point (i.e., cord blood = CD8 + T cells, CD4 + T cells, monocytes, natural killer, B cells, granulocytes and nucleated red blood cells (nRBC); peripheral blood at ~ 5 years = CD8 + T cells, CD4 + T cells, monocytes, natural killer, B cells and neutrophils) and different age measures (cord blood: gestational age in weeks; peripheral blood: child age in years), we regressed DNAm M-values against the cell types and age for each time-point separately, and retrieved residuals from this association to perform the longitudinal EWAS. For the linear mixed model (LMM), we considered a random intercept model using the nlme R package [30] to assess the longitudinal association between maternal AUCglu (primary exposure) and repeated measures of residuals of DNAm (in M-values adjusted for cell types and age, respectively, in each DNAm time-point) at ~ 719,360 CpG sites as outcomes. We conducted similar analyses for individual glucose components of maternal AUCglu: fasting glucose, 1 h-glucose, and 2 h-glucose post-OGTT as secondary exposures. We included the following covariates as fixed effects: maternal age, gravidity (primigravid vs multigravida), smoking in the first trimester of pregnancy (smoker versus non-smoker), child sex, maternal BMI in the first trimester of pregnancy, and a binary variable for time-point (i.e., birth = 0, early childhood = 1). We used a random intercept for the subject to account for the correlation of multiple measures on the same individual. We also conducted separate linear models for individual CpG at each of the two time-points (cord blood and 5y peripheral blood) using blood DNAm transformed in the same way (residuals on cell counts and age), for all four maternal glycemic exposures (maternal AUCglu, fasting glucose, 1 h post-OGTT and 2 h post-OGTT) using the same covariates (without the time-point variable). We also explored if we could find differentially methylated regions (DMRs) using the DMRff method [31] using the residuals that were used for the EWAS as the individual-level data. We choose this method as it provides decent power for detecting DMRs and without an inflated Type I error rate [32].
We reported coefficient estimates at the individual CpG site as the mean difference in DNAm residuals on cells and age, per unit increase in the AUCglu. We calculated the genomic inflation factor or lambda (λ) for each EWAS (Additional file 1). We adjusted associations for multiple testing using Bonferroni, deeming epigenome-wide significant associations as P < 6.9 × 10–8, while we considered “suggestive” associations identified with an arbitrary P ≤ 1.0 × 10–5. For each top CpG site (P ≤ 1.0 × 10–5 in at least one model (mixed model, or linear models in cord blood and 5y peripheral blood)), we visually inspected outliers and distribution by plotting the residuals (standardized or not) versus the fitted values, and by using quantile plots. Top CpG sites identified in the EWAS were annotated to their gene using the Illumina annotation (IlluminaHumanMethylationEPICanno.ilm10b2.hg19 R package [33]) and if not annotated, we completed using the closest gene using the UCSC Gene Browser. We visualized the results using Manhattan and quantile plots. All analyses were performed in R (version 4.1.1).
Results
Participants’ characteristics
The characteristics of the mothers and children are shown in Table 1. Briefly, mothers were 28.2 ± 4.2 years old (mean ± SD) and had a median [IQR] BMI of 23.9 [21.6; 27.9] kg/m2 at the first trimester of pregnancy (median 9.4 [8.1; 11.7] weeks). Forty-nine (9.1%) women reported smoking and 180 (33.4%) were primigravid. Median and IQR values of maternal glycemia during the 75 g OGTT (median gestational age; 26.3 [25.9; 27.1] weeks) are indicated in Table 1. The median AUCglu was 12.0 [10.5; 13.4] mmol/L*h and fifty (9.3%) women developed GDM. At delivery, the median gestational age was 39.7 [38.9; 40.4] weeks and 252 of these children were female (46.8%). At the 5-year visit, the children had a median age of 5.2 [5.1; 5.4] years old and 130 (44.4%) were female.
Maternal AUC glu is associated with DNAm variations in cord blood and in blood at 5 years of age
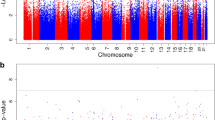
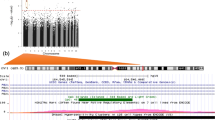
We first identified a total of 10 CpG sites (9 loci) at which offspring DNAm levels were associated with exposure to maternal AUCglu at a suggestive threshold of P-value ≤ 10–5 (Fig. 2). The strongest association reached the genome-wide significant threshold and was observed at the cg00967989 located within the proximal promotor of the FSD1L gene (β = − 0.0267, P = 2.13 × 10–8) (Fig. 3a). Among the other loci with suggestive associations, cg07946633 (β = − 0.0206, P = 2.24 × 10–6) and cg12140144 (β = − 0.0251, P = 4.37 × 10–7) were both located within the promotor region of the PRDM16 gene (Fig. 3b). The seven other CpG sites were located at the HES1, CCDC28A, INPP5F, CKB, GP1BB/SEPT5, SAR1B, and RGS3 gene loci. We presented in Table 2 the complete list of CpGs at which offspring blood DNAm levels were associated with exposure to high maternal AUCglu at a suggestive threshold (P ≤ 1.0 × 10–5), their associated genes, genomic location, regression β values and P-values from the main longitudinal EWAS (based on LMM). Using DMRff, we identified two DMRs, one containing the two CpG sites (cg07946633 and cg12140144; adj P = 0.03) previously reported at the PRDM16 gene locus and the second containing the one CpG site (cg00967989; adj P = 0.02), previously reported at the FSD1L gene locus (Additional file 2).

Epigenome-wide association plot (− log10 P-values) for maternal AUCglu. Manhattan plot for the EWAS of maternal AUCglu following a 75 g-OGTT with DNAm measured longitudinally at birth and five years of age (red line: Bonferroni threshold = P-value < 6.9 × 10–8; blue line: Suggestive threshold = P < 1.0 × 10–5)

FSD1L and PRDM16 gene regional plot association (− log10 P-values) for maternal AUCglu. Genomic regional plots for a FSD1L (on Chr 9) and b PRDM16 (on Chr 1) genes of maternal AUCglu following a 75 g-OGTT with DNAm measured longitudinally at birth and five years of age (Solid line: Bonferroni threshold = P-value < 6.9 × 10–8; dashed line: Suggestive threshold = P < 1.0 × 10–5)
We also tested associations between maternal AUCglu and DNAm measured at birth (cord blood) and at 5 years (peripheral blood), separately (Additional file 3). When looking at individual time-points for FSD1L, the signal appears to be primarily driven by its association in childhood, with only nominal association observed at birth. A similar pattern applies to the CpGs localized at the GP1BB/SEPT5 gene locus. In contrast, the signals identified for PRDM16 (cg12140144), SAR1B, CCDC28A, and INPP5F appear to be primarily driven by their associations reported at birth. Interestingly, the signal observed for PRDM16 (cg07946633) appears to be equally driven by its associations at birth and at age 5 years with a similar pattern in HES1, CKB, and RGS3 genes.
Most of the CpG sites associated with maternal AUC glu were associated with 1 h and 2 h post-OGTT glucose levels
Using LMM, we tested the association between offspring DNAm levels (cord blood and 5-year blood), and maternal fasting glucose, 1 h, and 2 h post-OGTT glucose levels as separate exposures (Additional files 4, 5, and 6). From the 10 CpG sites initially associated with maternal AUCglu (P-value ≤ 10–5), DNAm levels at cg00967989, located in the FSD1L gene, were associated with maternal 1 h-glucose (β = − 0.0347, P = 6.68 × 10–8) and suggestively with the 2 h-glucose (β = − 0.0357, P = 2.80 × 10–6) post-OGTT. DNAm at CpG sites within the PRDM16 gene associated with maternal AUCglu were suggestively associated with maternal 1 h-glucose (only cg12140144) and 2 h-glucose (both cg12140144 and cg07946633) post-OGTT. In addition, DNAm levels at cg17385945, cg19978242, cg03703356, and cg08694430 located within the CCDC28A, INPP5F, CKB, and RGS3 genes, respectively, were suggestively associated with the 1 h post-OGTT glucose levels (same direction of effect to what we had found in the analyses with maternal AUCglu). None of the CpG sites initially associated with maternal AUCglu or with the 1 h- and 2 h-glucose was found associated with fasting glucose levels. These results, along with all CpG sites we found significantly associated with fasting glucose, 1 h and 2 h post-OGTT, are summarized in a Venn diagram (Fig. 4) and presented in Additional files 7, 8, and 9.

Venn diagram showing common CpGs between maternal AUCglu, fasting, 1 h and 2 h post-OGTT glucose levels. All CpG sites shown in the Venn diagram were at least suggestively associated (P < 1.0 × 10–5) with maternal glucose measurements. Cg00967989 is located at the FSD1L gene locus; cg12140144 and cg07946633 are located at the PRDM16 gene locus
Discussion
In this study, we conducted a longitudinal EWAS based on blood DNAm from birth and 5 years of age in childhood in relation to maternal glycemic response after a 75 g-OGTT conducted during the second trimester of pregnancy. We found CpG sites at which offspring blood DNAm was associated with maternal AUCglu as well as other glycemic indices measured during pregnancy, including 1 h and 2 h post-OGTT glucose levels, but not fasting glucose levels. The different results we obtained with fasting glucose levels could be explained by the different metabolic characteristics observed in the impaired glucose tolerance (IGT) and impaired glucose fasting (IGF) conditions. For example, the insulin resistance state affects different tissues in the two conditions (peripheral in IGT vs. hepatic in IGF) and only IGT subjects show a defect in late-phase insulin secretion in a non-pregnant population [34]. In addition, the two conditions affect different populations that only partially overlap [35], suggesting likely variations in the impact of these complications on potential epigenetic adaptations. Of note, in our previous EWAS of maternal glycemic traits (PACE cohorts meta-analysis), we did not find cord blood DNAm variations that were robustly associated with maternal fasting glucose, while we identified a few CpG sites at which cord blood DNAm was associated with AUCglu and other glycemic indices measured during pregnancy [15]. We identified one CpG site (cg00967989), located in the promotor region of FSD1L, at which higher maternal AUCglu was significantly associated with lower values of DNAm at birth and in childhood (after Bonferroni correction for multiple testing). This association was also confirmed with maternal 1 h-glucose and the 2 h-glucose post-OGTT (with a less stringent P-value threshold). Our study also reported other CpG sites for which offspring blood DNAm levels were suggestively associated (P-value < 10–5) with maternal AUCglu. Interestingly, two of these were found in the promotor region of PRDM16 with the same direction of association. We also observed nominal associations between maternal AUCglu and offspring DNAm at individual time-points (cord blood DNAm and blood DNAm at 5 years of age).
The FSD1L gene is located on chromosome 9 and encodes for type 2 cystatins. Among other functions, cystatins regulate the activity of endogenous cysteine proteinases which are involved in tumor cell invasion and metastasis [36] potentially via angiogenic properties of FSD1L [37]. Genome-wide association studies (GWAS) also reported significant associations between SNPs near its location and age at menarche in females [38], as well as early reproductive traits contributing to fertility in female cattle [39]. However, the exact role of FSD1L in humans has not been clearly established yet. Thus, although our results might support its implication in metabolic health programming in the context of prenatal exposure to gestational hyperglycemia, further studies will be needed to better understand the exact role of FSD1L in this context.
Our results also suggest lower blood DNAm levels in PRDM16 promotor at birth and 5 years in the offspring as a response to elevated gestational glycemia. This result at PRDM16 is consistent in the direction of association with findings by Côté, Gagné-Ouellet et al. who also reported inverse correlations between lower PRDM16 DNAm levels in the placenta of newborns and higher maternal glycemia [40]. The PRDM16 gene is located on chromosome 1 and encodes for one of the 17 PRDM protein family members. These proteins contain a PR domain involved in the regulation of transcriptional activity mediated by chromatin histone modifications [41]. PRDM16 is a transcription factor primarily involved in brown adipose tissue (BAT) differentiation and the transition from white adipose tissue (WAT) to beige adipose tissue (iBAT) [42]. The BAT is enriched in mitochondria whose inner membrane expresses the uncoupling protein 1 (UCP1). UCP1 allows protons to leak across the mitochondrial membrane, which leads to an increase in heat production at the expense of oxidative phosphorylation and adenosine triphosphate (ATP) production [43]. Consistently, BAT and iBAT have been positively associated with leanness and metabolic health in human adults [44]. More specifically, PRDM16 induces the expression of peroxisome proliferator-activated receptor-y (PPAR-y) and PPAR-y coactivator-1α (PGC-1α) with which it forms a complex that regulates the expression of genes associated with lipid catabolism and thermogenesis [45]. Dysregulation of DNAm and gene expression of PGC-1α was observed in the placenta of GDM-exposed participants [46]. Studies involving different polymorphisms of PRDM16 have also shown significant correlations between genetic variants of the gene and the development of obesity in humans [47, 48]. Moreover, Lui et al. [49] demonstrated lower DNAm levels in the promotor region of PRDM16, measured in omental adipose tissue (OAT) in individuals with obesity compared with normal-weight subjects, suggesting a role for epigenetic regulation of PRDM16 in fat mass redistribution of peripheral iBAT to OAT in obesity. However, our findings are based solely on blood DNAm, which may reflect variations in DNAm in other tissues exposed similarly during in utero environmental factors, but we could not biopsy various tissues in healthy newborns for ethical reasons.
Even though computationally intensive, the use of multilevel modeling to analyze trends in DNAm variation is a better strategy to understand the biological relevance of methylation changes over time, versus analyzing their variation at each time-point separately [50]. In our analysis, we identified one signal at FS1DL with epigenome-wide significance in the longitudinal analysis and in the analysis of blood DNAm at 5 years. With few exceptions, most of the top signals detected in the longitudinal analysis showed a higher level of statistical significance in this versus the time-point specific analysis, with consistent association estimates observed across analyses. These findings support the advantage of using multilevel modeling over the single time-point analysis to capture CpGs with a time-varying association with the exposure, in this case, maternal hyperglycemia in mid-pregnancy. To our knowledge, this is the first study that has used a longitudinal approach to investigate changes in offspring DNAm associated with maternal hyperglycemia. Previous studies have looked at associations between cord blood DNAm in relation to elevated maternal AUCglu or individual glucose traits measured after an OGTT in pregnancy and have followed up these findings by investigating the cross-sectional association of important baseline markers with cardiometabolic phenotypes in infants and adults [15]. Similarly, the metabolic risk of infants exposed to GDM has been assessed in early childhood (3–10 years of age), identifying epigenetic age acceleration in those born to moms with GDM, which was further associated with cardiometabolic risk factors [51]. Future prospective studies including larger sample sizes and multiple time-points over the life course are warranted to investigate variation in offspring DNAm in response to maternal hyperglycemia in pregnancy and to evaluate the implication of these markers in the offspring’s future health.
Strengths and limitations
A strength of our study is the prospective cohort design, which allowed collection of samples to evaluate DNAm levels longitudinally from birth to mid-childhood, and their association with maternal glycemia objectively measured during a full 75 g-OGTT in pregnancy. In addition, we measured DNAm using the Illumina MethylationEPIC microarray which provides a coverage of more than 850,000 CpG sites across the human genome. Some limitations are also noteworthy to mention. One CpG site was significantly associated based on a predetermined statistical threshold after correction for multiple testing; however, other findings with suggestive or nominal significance should be interpreted with caution. We investigated DNAm using blood cells; other tissues (liver, beta-cells, adipose, and brain) might be of more relevance for studies of metabolic epigenetic programming related to exposure to maternal hyperglycemia but these tissues are not easily available in healthy children for obvious ethical reasons. We have excluded participants with pre-existing diabetes prior to pregnancy, but 9% of our included population did develop GDM, which may account partially for the observed associations; however, in the past, we have showed that associations between maternal glycemic traits and offspring DNAm markers were stronger in non-GDM participants [15]. Our sample size of participants who had DNAm measurements at both time-points (birth and 5 years) was relatively small (N = 194). For this reason, we chose a statistical model (LMM) that combines the two time of measurements, which improves power to detect associations. However, we cannot exclude that other associations with smaller effect sizes were not detected. Finally, our study only included participants of European descent, which limits the application of our results to other races and ethnicities.
Conclusions
We found associations between maternal hyperglycemia in pregnancy and offspring blood DNAm levels measured at birth and at 5 years. The association was significant after accounting for multiple testing at a CpG site near the FSD1L gene. Notably, our study also reports two CpG sites, located in the promotor region of PRMD16—a gene implicated in adipose tissue regulation, and that we had previously observed associations between maternal hyperglycemia and placenta DNAm. We also confirmed some of our suggestive associations with other maternal glycemic traits, predominantly with 1 h-glucose and 2 h-glucose post-OGTT. Overall, our study provides results supporting that maternal hyperglycemia might be implicated in offspring epigenetic programming which may last until 5 years after birth.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- ATP:
-
Adenosine triphosphate
- AUCglu :
-
Area under the curve for glucose
- BAT:
-
Brown adipose tissue
- BMI:
-
Body mass index
- CHUS:
-
Centre hospitalier universitaire de Sherbrooke
- CpG:
-
Cytosine–phosphate–guanine
- Chr:
-
Chromosome
- DNAm:
-
DNA methylation
- DOHaD:
-
Developmental origin of health and disease hypothesis
- EWAS:
-
Epigenome-wide association study
- FG:
-
Fasting glucose
- Gen3G:
-
Genetics of glucose regulation in gestation and growth cohort
- GCT:
-
50-G glucose challenge test
- GDM:
-
Gestational diabetes mellitus
- GWAS:
-
Genome-wide association study
- iBAT:
-
Beige adipose tissue
- LMM:
-
Linear mixed model
- MAF:
-
Minor allele frequency
- nRBC:
-
Nucleated red blood cells
- OGTT:
-
Oral glucose tolerance test
- OAT:
-
Omental adipose tissue
- PGC-1 α :
-
PPAR-y coactivator-1α
- PPAR-y :
-
Peroxisome proliferator-activated receptor-y
- QC:
-
Quality control
- RCP:
-
Regression on correlated probes
- SD:
-
Standard deviation
- SNP:
-
Single-nucleotide polymorphism
- UCP1:
-
Uncoupling protein 1
- WAT:
-
White adipose tissue
References
Quintanilla Rodriguez BS, Mahdy H. Gestational Diabetes. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 May 17]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK545196/
Wang H, Li N, Chivese T, Werfalli M, Sun H, Yuen L, et al. IDF diabetes atlas: estimation of global and regional gestational diabetes mellitus prevalence for 2021 by international association of diabetes in pregnancy study group’s criteria. Diabetes Res Clin Pract. 2022;183: 109050.
Pu J, Zhao B, Wang EJ, Nimbal V, Osmundson S, Kunz L, et al. Racial/ethnic differences in gestational diabetes prevalence and contribution of common risk factors: racial/ethnic differences in GDM risk factors. Paediatr Perinat Epidemiol. 2015;29(5):436–43.
Gupta Y, Kalra B. Screening and diagnosis of gestational diabetes mellitus. JPMA J Pak Med Assoc. 2016;66(9 Suppl 1):S19-21.
HAPO Study Cooperative Research Group, Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358(19):1991–2002.
On behalf of the HAPO Follow-up Study Cooperative Research Group, Lowe WL, Lowe LP, Kuang A, Catalano PM, Nodzenski M, et al. Maternal glucose levels during pregnancy and childhood adiposity in the hyperglycemia and adverse pregnancy outcome follow-up study. Diabetologia. 2019;62(4):598–610.
Vääräsmäki M, Pouta A, Elliot P, Tapanainen P, Sovio U, Ruokonen A, et al. Adolescent manifestations of metabolic syndrome among children born to women with gestational diabetes in a general-population birth cohort. Am J Epidemiol. 2009;169(10):1209–15.
Li Z, Wu Y, Du B, Yu X, Wang H, Niu Y, et al. Associations of maternal gestational diabetes mellitus with alterations in cardiovascular system in early childhood. Diabetes Metab Res Rev. 2022. https://doi.org/10.1002/dmrr.3551.
Adgent MA, Gebretsadik T, Reedus J, Graves C, Garrison E, Bush N, et al. Gestational diabetes and childhood asthma in a racially diverse US pregnancy cohort Genuneit J, editor. Pediatr Allergy Immunol. 2021;32(6):1190–6.
Xiang AH. Association of maternal diabetes with autism in offspring. JAMA. 2017;317(5):537–8.
Wang P, Xie J, Jiao XC, Ma SS, Liu Y, Yin WJ, et al. Maternal glycemia during pregnancy and early offspring development: a prospective birth cohort study. J Clin Endocrinol Metab. 2021;106(8):2279–90.
Heindel JJ, Vandenberg LN. Developmental origins of health and disease: a paradigm for understanding disease cause and prevention. Curr Opin Pediatr. 2015;27(2):248–53.
Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–6.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2013;38(1):23–38.
Tobi EW, Juvinao-Quintero DL, Ronkainen J, Ott R, Alfano R, Canouil M, et al. Maternal glycemic dysregulation during pregnancy and neonatal blood dna methylation: meta-analyses of epigenome-wide association studies. Diabetes Care. 2022;45(3):614–23.
Gagné-Ouellet V, Houde AA, Guay SP, Perron P, Gaudet D, Guérin R, et al. Placental lipoprotein lipase DNA methylation alterations are associated with gestational diabetes and body composition at 5 years of age. Epigenetics. 2017;12(8):616–25.
Allard C, Desgagné V, Patenaude J, Lacroix M, Guillemette L, Battista MC, et al. Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics. 2015;10(4):342–51.
Guillemette L, Allard C, Lacroix M, Patenaude J, Battista MC, Doyon M, et al. Genetics of glucose regulation in gestation and growth (Gen3G): a prospective prebirth cohort of mother–child pairs in Sherbrooke, Canada. BMJ Open. 2016;6(2): e010031.
Hivert MF, Cardenas A, Allard C, Doyon M, Powe CE, Catalano PM, et al. Interplay of placental DNA methylation and maternal insulin sensitivity in pregnancy. Diabetes. 2020;69(3):484–92.
Tai MM. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care. 1994;17(2):152–4.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinforma Oxf Engl. 2014;30(10):1363–9.
Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(12):503.
Niu L, Xu Z, Taylor JA. RCP: a novel probe design bias correction method for illumina methylation BeadChip. Bioinformatics. 2016;32(17):2659–63.
Juvinao-Quintero DL, Starling AP, Cardenas A, Powe CE, Perron P, Bouchard L, et al. Epigenome-wide association study of maternal hemoglobin A1c in pregnancy and cord blood DNA methylation. Epigenomics. 2021;13(3):203–18.
Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):208.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostat Oxf Engl. 2007;8(1):118–27.
Gervin K, Salas LA, Bakulski KM, van Zelm MC, Koestler DC, Wiencke JK, et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin Epigenet. 2019;11(1):125.
Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018;19(1):64.
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 2010;11:587.
J Pinheiro, Bates D, DebRoy S, Sarkar D, R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. [Internet]. R package version 3.1-155. 2022. Available from: https://CRAN.R-project.org/package=nlme.
Suderman M, Staley JR, French R, Arathimos R, Simpkin A, Tilling K. dmrff: identifying differentially methylated regions efficiently with power and control [Internet]. Bioinformatics. 2018. https://doi.org/10.1101/508556.
Lent S, Cardenas A, Rifas-Shiman SL, Perron P, Bouchard L, Liu CT, et al. Detecting differentially methylated regions with multiple distinct associations. Epigenomics. 2021;13(6):451–64.
Hansen KD. IlluminaHumanMethylationEPICanno.ilm10b2.hg19: Annotation for Illumina’s EPIC methylation arrays. [Internet]. R package version 0.6.0. 2016. Available from: https://bitbucket.com/kasperdanielhansen/Illumina_EPIC.
Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29(5):1130–9.
Qiao Q, Lindström J, Valle TT, Tuomilehto J. Progression to clinically diagnosed and treated diabetes from impaired glucose tolerance and impaired fasting glycaemia. Diabet Med J Br Diabet Assoc. 2003;20(12):1027–33.
Talluri R, Shete S. Evaluating methods for modeling epistasis networks with application to head and neck cancer. Cancer Inform. 2015;14(Suppl 2):17–23.
Gao Y, Liu J, Zhao D, Diao G. A novel prognostic model for identifying the risk of hepatocellular carcinoma based on angiogenesis factors. Front Genet. 2022;13: 857215.
Perry JRB, Stolk L, Franceschini N, Lunetta KL, Zhai G, McArdle PF, et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genet. 2009;41(6):648–50.
Tahir MS, Porto-Neto LR, Gondro C, Shittu OB, Wockner K, Tan AWL, et al. Meta-analysis of heifer traits identified reproductive pathways in Bos Indicus cattle. Genes. 2021;12(5):768.
Côté S, Gagné-Ouellet V, Guay SP, Allard C, Houde AA, Perron P, et al. PPARGC1α gene DNA methylation variations in human placenta mediate the link between maternal hyperglycemia and leptin levels in newborns. Clin Epigenet. 2016;8:72.
Chi J, Cohen P. The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol Metab TEM. 2016;27(1):11–23.
Becerril S, Gómez-Ambrosi J, Martín M, Moncada R, Sesma P, Burrell MA, et al. Role of PRDM16 in the activation of brown fat programming. Relevance to the development of obesity. Histol Histopathol. 2013;28(11):1411–25.
Ishibashi J, Seale P. Functions of Prdm16 in thermogenic fat cells. Temp Austin Tex. 2015;2(1):65–72.
Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360(15):1509–17.
Jiang N, Yang M, Han Y, Zhao H, Sun L. PRDM16 regulating adipocyte transformation and thermogenesis: a promising therapeutic target for obesity and diabetes. Front Pharmacol. 2022;13: 870250.
Wang L, Fan H, Zhou L, Wu Y, Lu H, Luo J. Altered expression of PGC-1 α and PDX1 and their methylation status are associated with fetal glucose metabolism in gestational diabetes mellitus. Biochem Biophys Res Commun. 2018;501(1):300–6.
Yue H, He JW, Ke YH, Zhang H, Wang C, Hu WW, et al. Association of single nucleotide polymorphism Rs2236518 in PRDM16 gene with BMI in Chinese males. Acta Pharmacol Sin. 2013;34(5):710–6.
AlAmrani A, AbdelKarim M, AlZoghaibi M. PRDM16 gene polymorphism is associated with obesity and blood lipids profiles in Saudi population. J Clin Med. 2018;7(6):141.
Liu L, Chen Y, Chen J, Lu M, Guo R, Han J, et al. The relationship between PRDM16 promoter methylation in abdominal subcutaneous and omental adipose tissue and obesity. Clin Nutr. 2021;40(4):2278–84.
Staley JR, Suderman M, Simpkin AJ, Gaunt TR, Heron J, Relton CL, et al. Longitudinal analysis strategies for modelling epigenetic trajectories. Int J Epidemiol. 2018;47(2):516–25.
Shiau S, Wang L, Liu H, Zheng Y, Drong A, Joyce BT, et al. Prenatal gestational diabetes mellitus exposure and accelerated offspring DNA methylation age in early childhood. Epigenetics. 2021;16(2):186–95.
Acknowledgements
We first and foremost thank the participants of the Gen3G cohort for their commitment and support over the last 12 years. We also wish to thank Chamaine Washington for editing the English language.
Funding
This work was supported by a Fonds de recherche du Québec - Santé (FRQS) operating grant (to M-FH, grant #20697); a Canadian Institute of Health Research (CIHR) operating grants (to M-FH grant #MOP 115071 and to LB #PJT-152989); AT was supported by Diabète Québec. KT was recipient of a doctoral research award from Fonds de la recherche du Québec en santé (FRQS). LB is a senior research scholar from the FRQS.
Author information
Authors and Affiliations
Contributions
M-FH, SML, DJQ, LB, and PP conceptualized the study; AT, KT, CA, DJQ, LB, and M-FH wrote the main manuscript text; CA prepared Figs. 2 and 3 and supplementary figures; CA performed the statistical analyses; LB and M-FH supervised the work; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of CIUSSS de l’Estrie-CHUS (protocol code #MP-04-2012-189 and September 2011). Informed consent was obtained from all subjects involved in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Genomic inflation factor or lambda for each LMM and linear regression models testing associations between maternal hyperglycemia outcomes and DNAm in cord blood and/or in blood at 5 years of age; Table presenting lambda for each LMM and linear regression models.
Additional file 2
: Differentially methylated regions based on results from LMM testing associations between maternal AUCglu and DNAm measured in cord blood and blood at 5 years of age, using the DMRff method; Table presenting the differentially methylated regions, including their chromosome number, genomic position, number of CpGin the region, estimate value, standard error, P-value and adjusted P-value, based on results from LMM testing the association between maternal AUCglu and DNAm measured in cord blood at 5 years of age.
Additional file 3
: CpG sites identified based on LMM or linear regression models testing associations between maternal AUCglu and DNAm in cord blood and/or in blood at 5 years of age; Table presenting CpG sites, including their chromosome number, genomic position, associated gene and regression beta and p-value, identified at suggestive P < 10−5 based on LMM or linear regression models testing association between maternal AUCglu and DNAm in cord blood and/or in blood at 5 years of age.
Additional file 4
: Epigenome-wide association plot for maternal fasting glucose levels; Figure of the epigenome-wide association plot presenting CpG sites from the LMM associated with maternal fasting glucose.
Additional file 5
: Epigenome-wide association plot for maternal 1h-post-OGTT glucose levels; Figure of epigenome-wide association plot presenting CpG sites from the LMM associated with maternal 1h-post-OGTT glucose levels.
Additional file 6
: Epigenome-wide association plot for maternal 2h-post-OGTT glucose levels; Figure of epigenome-wide association plot presenting CpG sites from the LMM associated with maternal 2h-post-OGTT glucose levels.
Additional file 7
: CpG sites identifiedin linear mixed models testing associations between maternal fasting glucose and DNAm measured in cord blood and blood at 5 years of age; Table presenting CpG sites, including their chromosome number, genomic position, and associated gene, identified at suggestive P < 10−5 in linear mixed models testing associations between maternal fasting glucose and DNAm measured in cord blood and blood at 5 years of age.
Additional file 8
: CpG sites identified in linear mixed models testing associations between maternal 1h-glucose post-OGTT and DNAm measured in cord blood and blood at 5 years of age; Table presenting CpG sites, including their chromosome number, genomic position, and associated gene, identified at suggestive P < 10−5 in linear mixed models testing associations between maternal 1h post-OGTT glucose levels and DNAm measured in cord blood and blood at 5 years of age.
Additional file 9
: CpG sites identified in linear mixed models testing associations between maternal 2h-glucose post-OGTT and DNAm measured in cord blood and blood at 5 years of age; Table presenting CpG sites, including their chromosome number, genomic position, and associated gene, identified at suggestive P < 10−5 in linear mixed models testing associations between maternal 2h post-OGTT glucose levels and DNAm measured in cord blood and blood at 5 years of age.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Taschereau, A., Thibeault, K., Allard, C. et al. Maternal glycemia in pregnancy is longitudinally associated with blood DNAm variation at the FSD1L gene from birth to 5 years of age. Clin Epigenet 15, 107 (2023). https://doi.org/10.1186/s13148-023-01524-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-023-01524-7