
Abstract
Background
Obesity and diabetes mellitus are directly implicated in many adverse health consequences in adults as well as in the offspring of obese and diabetic mothers. Hispanic Americans are particularly at risk for obesity, diabetes, and end-stage renal disease. Maternal obesity and/or diabetes through prenatal programming may alter the fetal epigenome increasing the risk of metabolic disease in their offspring. The aims of this study were to determine if maternal obesity or diabetes mellitus during pregnancy results in a change in infant methylation of CpG islands adjacent to targeted genes specific for obesity or diabetes disease pathways in a largely Hispanic population.
Methods
Methylation levels in the cord blood of 69 newborns were determined using the Illumina Infinium MethylationEPIC BeadChip. Over 850,000 different probe sites were analyzed to determine whether maternal obesity and/or diabetes mellitus directly attributed to differential methylation; epigenome-wide and regional analyses were performed for significant CpG sites.
Results
Following quality control, agranular leukocyte samples from 69 newborns (23 normal term (NT), 14 diabetes (DM), 23 obese (OB), 9 DM/OB) were analyzed for over 850,000 different probe sites. Contrasts between the NT, DM, OB, and DM/OB were considered. After correction for multiple testing, 15 CpGs showed differential methylation from the NT, associated with 10 differentially methylated genes between the diabetic and non-diabetic subgroups, CCDC110, KALRN, PAG1, GNRH1, SLC2A9, CSRP2BP, HIVEP1, RALGDS, DHX37, and SCNN1D. The effects of diabetes were partly mediated by the altered methylation of HOOK2, LCE3C, and TMEM63B. The effects of obesity were partly mediated by the differential methylation of LTF and DUSP22.
Conclusions
The presented data highlights the associated altered methylation patterns potentially mediated by maternal diabetes and/or obesity. Larger studies are warranted to investigate the role of both the identified differentially methylated loci and the effects on newborn body composition and future health risk factors for metabolic disease. Additional future consideration should be targeted to the role of Hispanic inheritance. Potential future targeting of transgenerational propagation and developmental programming may reduce population obesity and diabetes risk.
Similar content being viewed by others
Background
Childhood obesity and diabetes mellitus are an increasing epidemic in the USA [1]. In 2015, an estimated 30.3 million people in the USA had diabetes mellitus (DM). Approximately 12.7 million children and adolescents ages 2 to 19 are obese, and it is estimated that > 25% of children will be classified as overweight or obese by kindergarten [2]. Hispanic Americans are particularly at risk for obesity, diabetes, and end-stage renal disease [3]. The risk of obesity is 35% higher in the Hispanic population, with obese Hispanic and non-Hispanic black adolescent females among those at highest risk of developing type II diabetes [1, 4]. Concomitant obesity and diabetes during pregnancy are also associated with increased risk of metabolic syndrome in the offspring [5]. In South Texas alone, 29% of mothers have a pre-pregnancy BMI of 30 or above, and 4.8% of mothers go on to develop gestational diabetes (GDM) [6]. This study seeks to investigate whether previously identified and unidentified associations occur between maternal diabetes, obesity, and altered newborn methylation in an already high-risk Hispanic population of South Texas.
The combination of obesity and gestational diabetes mellitus is estimated to complicate up to 9.2% of pregnancies, with the highest risks for gestational diabetes affecting ethnic and racial minority women [7]. Exposure to a diabetic intrauterine environment during pregnancy is associated with an increase in dyslipidemia, subclinical vascular inflammation, and endothelial dysfunction processes in the offspring, all of which are linked with development of cardiovascular disease later in life [8]. Maternal obesity and gestational diabetes have additionally been linked to increased risk of asthma, poorer cognitive performance, mental health disorders, neurodevelopmental disorders including cerebral palsy, and immune and infectious disease-related outcomes [9].
Increasing evidence has shown that transgenerational non-genetic inheritance can occur through in utero exposure of the developing fetus to the maternal environment or through either the male or female germline [10]. The concept of “gestational programming” is associated with alterations to the epigenome (non-genomic) as opposed to alteration in the genomic DNA sequence [11,12,13]. Significant hypermethylation of DNA may also occur globally in the placenta of mothers with GDM as well as the cord and neonatal blood of infants born to mothers with GDM, particularly genes associated with metabolic disease [14,15,16,17]. This hypermethylation may repress transcription leading to dysregulation of metabolic pathways. Epigenetic mechanisms may contribute to altered beta cell mass and beta cell failure, similarly as observed in diabetes [18]. Pregnancy complications with fetal exposure to glucocorticoids, either from maternal stress or synthetic glucocorticoids, can also lead to prolonged alteration of hypothalamic-pituitary-adrenal function [19, 20].
While pre-pregnancy maternal obesity is associated with adverse offspring outcomes at birth and later in life, the role of pre-pregnancy BMI is less clear [21]. The Pregnancy and Childhood Epigenetics (PACE) Consortium found a causal intrauterine effect of maternal BMI on newborn methylation at just 8/86 sites in a recent meta-analysis, attributing the identified robust associations between maternal adiposity and variations in DNA methylation to genetic or lifestyle factors [22]. Additionally, abnormal maternal nutrition, diet, folic acid, and vitamin deficiency can induce epigenetic alterations including DNA methylation, histone modifications, chromatin remodeling, and/or regulatory feedback by microRNAs, all of which have the ability to modulate gene expression and promote a metabolic syndrome phenotype [23,24,25,26,27,28].
We conducted epigenetic analysis via epigenome-wide association studies (EWAS) and regional analysis targeting genes associated with infant insulin signaling, glucose metabolism, and free fatty acid pathways in term infants delivered to mothers with normal weight, obesity, and DM in a highly Hispanic population. There are clear disparities in the risk of gestational diabetes by race and ethnicity, and small population studies in high-risk ethnicities are lacking. Our primary analyses focused on identifying areas of significant methylation differences between diabetic and non-diabetic Hispanic populations and obese and non-obese Hispanic populations. Additional analyses were also performed to determine whether the observed mediation effects were related to diabetes alone, obesity alone, or in combination.
Methods
Sample description
The study protocol was approved by the Institutional Review Board at The University of Texas Health Science Center, San Antonio (IRB# HSC 2016 0097H). Infants were prospectively enrolled between the years of 2016–2018. Limited neonatal data were available for extrapolation and determination of an ideal sample size. Complementary enrollment in a body composition analysis with the same inclusion/exclusion criteria occurred during study completion. A priori power analysis was completed for body composition analysis enrolling competitive patient enrollment in this cohort. Initial intention was prospective enrollment of 10 patients from each targeted population [29] but was expanded to 23 per arm (69) given to variability within targeted populations as well as to maximize efficiency of array utilization.
Population
All babies ≥ 37 weeks gestational age and ≤ 48 h old who were admitted to the newborn nursery at the University Hospital in San Antonio, TX, and who were eligible for inclusion were screened for recruitment. Inclusion criteria for enrollment included infants with and without a maternal history of diabetes mellitus (DM) and obesity (OB). Maternal diabetes mellitus was defined as abnormal glucose tolerance test during current pregnancy +/− medical therapy to include insulin or other antidiabetic agents or diagnosis of type I or type II diabetes prior to current pregnancy. Maternal obesity was defined as BMI ≥ 30. Exclusion criteria included infants with congenital anomalies, complex congenital heart disease, and severe central nervous system disease (grade 4 intraventricular hemorrhage; malformations). Infants requiring supplemental oxygen or admission to the neonatal intensive care unit for any reason were also excluded. Infants of mothers with DM and/or OB were identified through the electronic medical record. Eligible participants were actively recruited after screening for inclusion/exclusion criteria and confirmed willingness to participate. Written informed consent was obtained from all participating mothers prior to enrollment. Potential subjects were contacted within 24 h of delivery with the cord blood obtained upon delivery.
Blood collection, DNA extraction, and genome-wide methylation assay
Whole cord blood samples of 3–5 mL were collected immediately after birth. Blood samples were processed via centrifugation with 4 mL CPT BD Vacutainer tubes within 24 h to isolate and enriched for agranular leukocytes. DNA extraction was performed using the DNeasy Blood and Tissue Kit DNA kit (Qiagen). Isolated DNA was treated with a bisulfite conversion and run on Illumina MethylationEPIC BeadChip 850K array (UT Health SA Genomics Core).
Statistical analysis
We calculated means and standard deviations (SD) for all maternal and newborn characteristics to describe the study population overall. We additionally examined maternal pre-pregnancy BMI, age at enrollment, gestational diabetes status, and mode of delivery as potential confounders. Maternal covariates were added to all final models if they were associated with any of the log-transformed outcomes in linear regression models at p < 0.05.
Applied software
Quality control (QC) and all statistical analyses were performed using the R version 3.5.2 statistical analysis software, and the R-packages SWAN, missmethyl, minfi, limma, IlluminaHumanMethylation450kanno.ilmn12.hg19, IlluminaHumanMethylation450kmanifest, IlluminaHumanMethylationEPICmanifest, IlluminaHumanMethylationEPICanno.ilm10b2.hg19, bumphunter, RColorBrewer, matrixStats, minfiData, Gviz, DMRcate, and stringr.
Data preprocessing, QC, and filtering
Detection p values were calculated for all samples with removal of samples below 0.05 from the data set. getQC and plotQC were used to estimate quality of samples as well. Subset-quantile within array normalization (SWAN) was used to normalize the data taking into account the fact that the array (EPIC methylation bead chip) contained two different types of probes. Additional quality controls were completed after normalization: (1) Detection p values were again used to remove any probes which failed in one or more samples (7169 probes). (2) Additional probes on sex chromosomes were removed to reduce sex-linked variation in methylation between the samples (18,975 probes). (3) Probes associated with single-nucleotide polymorphisms (SnP) were removed using dropLociWithSnPs from the minfi package (28,179 probes). (4) Probes known to be cross-reactive within the methylation probe set were removed (38,756 probes). After technical QC, a total of 773,012 sites and 69 individuals were in principle available for analysis. All images and data were validated by GenomeStudio as a quality control measure.
Data transformation
After normalization, the data were processed to calculate beta values (methylated probe intensity at that site, over the total probe intensity of both the methylated and unmethylated probes) and M values (log2 of the methylated probe intensity over the unmethylated probe intensity). When performing statistical tests, M values were utilized as beta values tend to have heteroscedasticity; beta values were used for visualization in the figures and plots [30].
Epigenome-wide association study
To find differentially methylated positions or probes, several methods were employed. Differentially methylated probes were identified using lmfit and ebayes in R [29]. Descriptors of either diabetic or non-diabetic, or obese or non-obese, were used under lmfit to design a linear fit to model the data. ebayes was then used to determine significant methylation values between the groups. Statistical significance for genome-wide associations was adjusted for multiple comparisons using a false discovery rate (Benjamini-Hochberg correction method) q < 0.05.
Regional analyses
We examined the association of diabetes and/or obesity with differentially methylated regions (DMRs) in the cord blood using the R Bioconductor package bumphunter. Instead of employing a probe-wise approach, clusters of probes in the array are identified using the function clusterMaker, and the bumphunter function is used to fit a linear model accounting for the given sample variables to each identified cluster of probes in the given length (1000 bp) to determine if a region has a significant change in methylation compared with the control group. For our analysis, 1000 permutations were performed [31].
Results
Descriptive statistics of the study population are shown overall and stratified by maternal comorbidities in Table 1. Mothers had a mean age of 29.3 (SD = 5.6) at enrollment and a mean BMI of 29.5 (SD = 4.2). An overall Hispanic predominance in patient ethnicity was seen across all groups. There were significant increases in age and BMI for both the diabetic (DM) and obese-diabetic (OB/DM) groups compared with the non-diabetic healthy weight (NT). There was an increased rate of cesarean section delivery for the obese (OB) and OB/DM mothers. Birth weight, length, and occipital-frontal circumference were similar between all groups.
Epigenome-wide association study
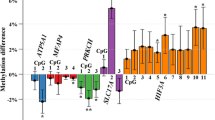
Unadjusted as well as following correction for maternal age, pre-pregnancy BMI, and method of delivery, a total of 15 CpG sites showed significant differential methylation in the diabetic subgroup (Fig. 1) with the top 10 CpG mapped genes of CCDC110, KALRN, PAG1, GNRH1, SLC2A9, CSRP2BP, HIVEP1, RALGDS, DHX37, and SCNN1D highlighted in Table 2. An additional six significant (adj. p value < 0.05) CpG sites were identified, but lacked an associated gene (cg08242354, cg23184039, cg00866179, cg17162208, cg24798727).

Scatter plots showing the beta value of the top 10 significantly methylated probes found by comparison between diabetic and non-diabetic groups using linear regression and empirical Bayes statistics for differential expression
A similar EWAS on obesity revealed no further epigenome-wide associated CpG sites (data not shown) using a linear modeling approach as well as including covariates (data not shown).
Regional analysis using bumphunter
In regional analyses, we identified three regions that met p value < 0.05 for diabetes and three regions for obesity (Table 3). A region of four CpG sites (cg06417478, cg04657146, cg11738485, and cg23899408) is associated with HOOK2, or Hook Microtubule Tethering Protein 2, and was significantly hypomethylated compared with NT in the diabetic subgroup. Additionally, LCE3C (cg09972436; Late Cornified Envelope 3C) and TMEM63B (cg25069157; Transmembrane Protein 63B) were also hypomethylated compared with NT. LTF (cg21787089, cg01427108; Lactotransferrin) and DUSP22 (cg01516881, cg26668828 (body); cg18110333, cg05064044 (1stExon; 5′UTR); Dual Specificity Phosphatase 22) were also differentially methylated with LTF hypermethylated and DUSP22 hypomethylated respectively compared with NT in the obese subgroup.
Correlation analysis with infant outcome
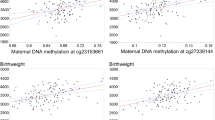
CpG sites identified during EWAS were directly compared with infantile demographic and body composition markers for potential phenotypic association (Table 4). No genes or cg IDs correlated with infantile birth weight. cg23184039 was associated with changes in birth length, frontal-occipital circumference, and gestational age; however, this CpG site is not associated with a gene identified. SLC2A9 and CSRP2BP correlated with gestational age.
Discussion
In this study, we sought to characterize how the fetal epigenome could be altered by the maternal environment, potentially predisposing the infant to long-term comorbidities of metabolic syndrome.
We identified multiple genes of interest through EWAS or regional analysis with significant differential methylation potentially caused by the presence of maternal diabetes or obesity in a largely Hispanic population. The genes identified from our population have not been previously reported in existing literature as differentially methylated in regard to maternal comorbidities during pregnancy. Genes identified are often hypomethylated when compared with the non-diabetic groups indicating potential elevated expression patterns in the newborns born to diabetic mothers, although we did not confirm gene expression profiles.
Epigenome-wide association study
Epigenome-wide association study found 15 (p adj. < 0.05) significant probes in a comparison between the diabetic and non-diabetic samples (Additional file 1); all 15 probes were hypomethylated compared with the non-diabetic group. No significant probes were found in a comparison between the NT and OB groups. Of the probes found in the DM comparison, ten are known to be associated with genes; the other five have no currently known gene association. A review of the genes associated with the significant probes found that several of the genes were associated with diabetes and/or obesity.
The SLC2A9 gene produces GLUT-9, a transport protein which facilitates the transport of glucose, fructose, and other sugars, and expression is specifically localized to insulin-containing β cells regulating glucose-stimulated insulin secretion [32]. In a study that looked at the levels of glucose transporter expression in placental tissue from mothers with diabetes mellitus, they found there was a significant increase in the expression of GLUT-9 in diabetic mothers controlled by insulin, as well as pregestational diabetes [33].
KALRN, or Kalirin RhoGEF Kinase, is a guanine exchange factor which acts on several Rho GTPases. Potential diseases associated with this gene include Huntington’s disease, coronary heart disease, various cardiovascular disorders, and ischemic stroke possibly related to nitric oxide signaling pathways [34].
GNRH1 encodes the precursor to gonadotropin-releasing hormone-1 (GnRH1). The effect of DM on GnRH expression is associated with the regulation of B and T cell response in pregnancy. One study found that antibodies against GnRH1, LH, and other related hormones were present more often in patients with diabetes mellitus [35].
RALGDS encodes a guanylyl-nucleotide exchange factor (GEFs) specifically involved in signal transduction pathways regulating cell growth and cancer/tumorigenesis in humans [36]. In addition, RalGDS activates Akt kinase whose abnormal expression is implicated in diabetes mellitus pathology [37, 38]. Akt interacts with insulin receptor substrate 1, PI3K, and GLUT4 translocation during insulin stimulation, as well as inactivates glycogen-synthase kinase-3, promoting glycogen synthesis [38].
Regional association study (DM)
Regional association study between DM and non-DM found three regions that were significant between the two comparisons. Of the most relevant to this study, four probes in a region associated with the gene HOOK2 were found to be hypomethylated in the DM group compared with the non-DM group. HOOK2 belongs to the HOOK family of proteins, which are responsible for trafficking and anchoring of organelles in the cell through the binding and directing of microtubules [36]. A study of DNA methylation in adipose tissue from subjects with type II diabetes and obesity, using the 450K Illumina beadchip, identified HOOK2 as significantly differentially methylated from the healthy group, although their findings indicated that HOOK2 was hypermethylated while our results show the genes being hypomethylated [39]. In an additional DNA methylation study on mothers with gestational diabetes, HOOK2 was found to be commonly differentially methylated in the maternal blood, placenta, and umbilical cord [40].
Regional association study (OB)
Three regions were identified as significantly differentially methylated between OB and non-OB groups using a regional association study. Two of the probes identified are associated with the gene LTF, which encodes lactotransferrin (Lf). Lf is a member of the iron-binding protein transferrin family and is involved in the regulation of iron homeostasis, anti-inflammatory response, cell growth regulation, differentiation, innate immune response, and antimicrobial activity [36]. Lf has also been shown to be positively correlated in individuals diagnosed with insulin resistance and type II diabetes and negatively correlated with body adiposity [41]. Moreno-Navarrete et al. confirmed these results, finding a decrease of Lf in hyperglycemic and obese individuals and an increase in insulin sensitive adults [42].
Four of the probes identified are associated with the gene DUSP22, encoding dual specificity phosphatase 22, or JNK pathway-associated phosphatase, and implicated in insulin receptor phosphorylation [36]. DUSP22 also represses the activation of T cells by phosphorylation of Lck, a Src-family tyrosine kinase involved in activation of T cell receptors during adaptive immune response. DUSP22 knockout mice were found to have depressed immune response, and later in life increased autoantibodies [43].
Correlation analysis with infant outcome
There were no identified CpG sites associated with birth weight changes. SLC2A9, or the GLUT-9 transport protein and CSRP2BP, a Cysteine-rich protein 2-binding protein correlated with gestational age. It is unclear the phenotypic significance of these associations as no changes were noted in the infant body composition anthropometrics. Interestingly, cg23184039, which has not been associated with a gene ID, was associated with increased birth length, frontal-occipital circumference, and gestational age in our population. This could potentially represent a future target for larger studies including expression and body composition beyond infancy.
Limitations
It is important to note that we have not shown experimentally that the methylation status of these significant genes has a phenotypic effect, only that there is a correlation between the conditions of obesity and diabetes and the differential methylation identified in the 69 samples. While we acknowledge the small sample size evaluated, the population of this study was almost exclusively comprised of mothers of Hispanic ethnicity, as such it should be noted that our results may reflect a more narrowed analysis of the epigenetic effects of maternal diabetes and obesity which is specific to populations of Hispanic ethnicity. Additionally, the identified genes detected through probe wise and peak detection and their differential methylation may or may not be associated with altered protein expression, which was not determined during this project. Without evaluation of protein expression or additional biomarkers, we are unable to potentially link these effects on phenotype or potential development of obesity or diabetes in the offspring beyond neonatal body composition.
Conclusion
Differential DNA methylation in the fetal epigenome is associated with exposure to maternal obesity and diabetes mellitus in a highly Hispanic population. DNA methylation of genes identified such as SLC2A9, HOOK2, LTF, and DUSP22 all have direct or indirect links to diabetes or obesity including immune or inflammatory regulatory pathways, signaling pathways, and clinical disorders related to diabetes and obesity. Future prospective studies are needed to assess the effects of maternal obesity and diabetes and its differential methylation effects on protein expression and offspring phenotypic effect including body composition and clinical risk of acquired disease in this high-risk population.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to legal restrictions but are available from the corresponding author on reasonable request.
Abbreviations
- DM:
-
Diabetes mellitus
- DNA:
-
Deoxyribonucleic acid
- EWAS:
-
Epigenome-wide association study
- OB:
-
Obesity
- QC:
-
Quality control
- SD:
-
Standard deviation
- SNP:
-
Single-nucleotide polymorphism
References
Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010. Jama. 2012;307(5):483–90.
Cunningham SA, Kramer MR, Narayan KM. Incidence of childhood obesity in the United States. N Engl J Med. 2014;370(5):403–11.
Yracheta JM, Alfonso J, Lanaspa MA, Roncal-Jimenez C, Johnson SB, Sanchez-Lozada LG, et al. Hispanic Americans living in the United States and their risk for obesity, diabetes and kidney disease: genetic and environmental considerations. Postgrad Med. 2015;127(5):503–10.
Pan L, May AL, Wethington H, Dalenius K, Grummer-Strawn LM. Incidence of obesity among young U.S. children living in low-income families, 2008-2011. Pediatrics. 2013;132(6):1006–13.
Ruchat SM, Hivert MF, Bouchard L. Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev. 2013;71(Suppl 1):S88–94.
Berlanga J, Mangla A. Health profiles 2013 vital statistics reports. City of San Antonio: Metropolitan Health District; 2013.
DeSisto CL, Kim SY, Sharma AJ. Prevalence estimates of gestational diabetes mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007-2010. Prev Chronic Dis. 2014;11:E104.
Vrachnis N, Antonakopoulos N, Iliodromiti Z, Dafopoulos K, Siristatidis C, Pappa KI, et al. Impact of maternal diabetes on epigenetic modifications leading to diseases in the offspring. Exp Diabetes Res. 2012;2012:538474.
Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VW, Eriksson JG, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017;5(1):53–64.
Elshenawy S, Simmons R. Maternal obesity and prenatal programming. Mol Cell Endocrinol. 2016;435:2–6.
Szyf M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol Med. 2015;21(2):134–44.
Desai M, Jellyman JK, Ross MG. Epigenomics, gestational programming and risk of metabolic syndrome. Int J Obes. 2015;39(4):633–41.
Li S, Wong EM, Dugue PA, AF MR, Kim E, Joo JE, et al. Genome-wide average DNA methylation is determined in utero. Int J Epidemiol. 2018;47(3):908–16.
Chen D, Zhang A, Fang M, Fang R, Ge J, Jiang Y, et al. Increased methylation at differentially methylated region of GNAS in infants born to gestational diabetes. BMC Med Genet. 2014;15:108.
Reichetzeder C, Dwi Putra SE, Pfab T, Slowinski T, Neuber C, Kleuser B, et al. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin Epigenetics. 2016;8:82.
Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, et al. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24(11):3021–9.
Hjort L, Martino D, Grunnet LG, Naeem H, Maksimovic J, Olsson AH, et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight. 2018;3(17).
Chavey A, Ah Kioon MD, Bailbe D, Movassat J, Portha B. Maternal diabetes, programming of beta-cell disorders and intergenerational risk of type 2 diabetes. Diabetes Metab. 2014;40(5):323–30.
Anwar MA, Saleh AI, Al Olabi R, Al Shehabi TS, Eid AH. Glucocorticoid-induced fetal origins of adult hypertension: association with epigenetic events. Vasc Pharmacol. 2016;82:41–50.
Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: mechanisms. Nat Rev Endocrinol. 2014;10(7):403–11.
Day SE, Coletta RL, Kim JY, Garcia LA, Campbell LE, Benjamin TR, et al. Potential epigenetic biomarkers of obesity-related insulin resistance in human whole-blood. Epigenetics. 2017;12(4):254–63.
Sharp GC, Salas LA, Monnereau C, Allard C, Yousefi P, Everson TM, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26(20):4067–85.
Jaeger K, Saben JL, Moley KH. Transmission of metabolic dysfunction across generations. Physiology. 2017;32(1):51–9.
Kitsiou-Tzeli S, Tzetis M. Maternal epigenetics and fetal and neonatal growth. Curr Opin Endocrinol Diab Obes. 2017;24(1):43–6.
Chango A, Pogribny IP. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients. 2015;7(4):2748–70.
Fleming TP, Watkins AJ, Velazquez MA, Mathers JC, Prentice AM, Stephenson J, et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet. 2018;391(10132):1842–52.
Lillycrop K, Murray R, Cheong C, Teh AL, Clarke-Harris R, Barton S, et al. ANRIL promoter DNA methylation: a perinatal marker for later adiposity. EBioMedicine. 2017;19:60–72.
Vanhees K, Vonhogen IG, van Schooten FJ, Godschalk RW. You are what you eat, and so are your children: the impact of micronutrients on the epigenetic programming of offspring. Cell Mol Life Sci. 2014;71(2):271–85.
Maksimovic J, Phipson B, Oshlack A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res. 2016;5:1281.
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–9.
Evans SA, Doblado M, Chi MM, Corbett JA, Moley KH. Facilitative glucose transporter 9 expression affects glucose sensing in pancreatic beta-cells. Endocrinology. 2009;150(12):5302–10.
Stanirowski PJ, Szukiewicz D, Pyzlak M, Abdalla N, Sawicki W, Cendrowski K. Impact of pre-gestational and gestational diabetes mellitus on the expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 in human term placenta. Endocrine. 2017;55(3):799–808.
Penzes P, Remmers C. Kalirin signaling: implications for synaptic pathology. Mol Neurobiol. 2012;45(1):109–18.
Roth B, Berntorp K, Ohlsson B. The expression of serum antibodies against gonadotropin-releasing hormone (GnRH1), progonadoliberin-2, luteinizing hormone (LH), and related receptors in patients with gastrointestinal dysfunction or diabetes mellitus. Drug Target Insights. 2014;8:45–50.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54(1.30):1–1 3.
Hao Y, Wong R, Feig LA. RalGDS couples growth factor signaling to Akt activation. Mol Cell Biol. 2008;28(9):2851–9.
Zdychova J, Komers R. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol Res. 2005;54(1):1–16.
Rodriguez-Rodero S, Menendez-Torre E, Fernandez-Bayon G, Morales-Sanchez P, Sanz L, Turienzo E, et al. Altered intragenic DNA methylation of HOOK2 gene in adipose tissue from individuals with obesity and type 2 diabetes. PLoS One. 2017;12(12):e0189153.
Wu P, Farrell WE, Haworth KE, Emes RD, Kitchen MO, Glossop JR, et al. Maternal genome-wide DNA methylation profiling in gestational diabetes shows distinctive disease-associated changes relative to matched healthy pregnancies. Epigenetics. 2018;13(2):122–8.
Mayeur S, Veilleux A, Pouliot Y, Lamarche B, Beaulieu JF, Hould FS, et al. Plasma lactoferrin levels positively correlate with insulin resistance despite an inverse association with total adiposity in lean and severely obese patients. PLoS One. 2016;11(11):e0166138.
Moreno-Navarrete JM, Botas P, Valdes S, Ortega FJ, Delgado E, Vazquez-Martin A, et al. Val1483Ile in FASN gene is linked to central obesity and insulin sensitivity in adult white men. Obesity (Silver Spring). 2009;17(9):1755–61.
Li JP, Yang CY, Chuang HC, Lan JL, Chen DY, Chen YM, et al. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat Commun. 2014;5:3618.
Acknowledgements
The authors would like to acknowledge the assistance and dedication of the Trinity (Chelsea Cole, Sarah Fordin) and University Health (Diana Anzueto, Rachel Jacob, and Robin Tragus) research teams.
Financial disclosure
All authors have indicated they have no financial relationships relevant to this article to disclose.
The view(s) expressed herein are those of the author(s) and do not reflect the official policy or position of the Brooke Army Medical Center, the US Army Medical Department, the US Army Office of the Surgeon General, the Department of the Air Force, the Department of the Army or the Department of Defense, or the US Government.
Funding
This work was supported by a Collaborative Research Grant from the San Antonio Medical Foundation (CLB, JMK, NRC), Trinity University Biology Department, and Long School of Medicine Pilot Projects (CLB).
Author information
Authors and Affiliations
Contributions
JMK, NRC, and CLB, as senior authors, contributed to the conception and design of the experiments, collected and analyzed the data, and oversaw the drafting of the manuscript. ENE, HER, EL, and NBA collected and analyzed the data, with HER, MAH, RF, and JG contributing significantly to the execution of the statistical analysis plan. All authors were involved in writing the manuscript, and all authors approved the final submitted and published versions.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The mothers provided written informed consent at recruitment. All study protocols were reviewed by the Institutional Review Board at University of Texas Health Science Center, San Antonio.
Competing interests
The authors declare that they have no competing interests. The view(s) expressed herein are those of the author(s) and do not reflect the official policy or position of Brooke Army Medical Center, the US Army Medical Department, the US Army Office of the Surgeon General, the Department of the Air Force and Army, Department of Defense or the US Government.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
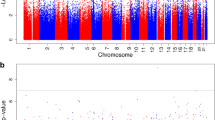
Additional file 1 Fig. S1.
Manhattan Plots of CpG sites for differentially methylated sites from either non-diabetic vs. diabetic (Panel A) or non-obese vs obese (Panel B) models. Unadjusted p-values models without correction for covariates is presented.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rizzo, H.E., Escaname, E.N., Alana, N.B. et al. Maternal diabetes and obesity influence the fetal epigenome in a largely Hispanic population. Clin Epigenet 12, 34 (2020). https://doi.org/10.1186/s13148-020-0824-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-0824-9