
Abstract
Background
We screened out several hypermethylated solute carrier (SLC) family genes in acute myeloid leukemia by reduced representation bisulfite sequencing. SLC22A3 encodes an organic cation transport protein, which is critical for drug transportation and cellular detoxification. SLC22A3 is significantly downregulated and associated with tumor progression and worse prognosis in a variety of solid tumors. However, there are no data available regarding the role of SLC22 in AML. This study aimed to explore the regulatory mechanism of DNA methylation on SLC22A3 expression, as well as its clinical significance in AML prognosis.
Results
SLC22A3 was identified as the sole prognosis-associated gene among SLCs based on TCGA and Beat AML databases. Bone marrow mononuclear cells (BMMNCs) from AML, MDS patients, and healthy donors were enrolled in this study. SLC22A3 methylation was significantly increased in AML compared with controls and MDS patients; meanwhile, the expression level of SLC22A3 was decreased. SLC22A3 hypermethylation presented an obvious association with some specific clinical characteristics and affected the survival time of AML patients as an independent risk indicator. SLC22A3 expression changed regularly as the disease complete remissions and relapses. Demethylation drug 5-aza-2′-deoxycytidine (DAC) activated transcription and increased mRNA expression of SLC22A3 in leukemia cell lines and AML fresh BMMNCs. Knockdown of SLC22A3 in leukemia cells enhanced cell proliferation and suppressed cell apoptosis. Data from public programs were used for auxiliary screening of probable molecular mechanisms of SLC22A3 in the antileukemia effect.
Conclusions
Our results showed that increased methylation and decreased expression of SLC22A3 may be indicators of poor prognosis in AML. Methylation-silenced SLC22A3 expression may have potential guiding significance on antileukemia effect of DAC.
Similar content being viewed by others

Introduction
Recent decades have witnessed an increasing trend of leukemia incidence and mortality in china and worldwide [1,2,3]. AML is the most common adult acute leukemia that varies greatly in clinical features, immune phenotypes, morphology and genetics. Except for all-trans-retinoic acid (ATRA) for the treatment of acute promyelocytic leukemia, AML with conventional induction consolidation chemotherapy alone has a poor prognosis, low long-term survival rate and high recurrence rate. Hence, there is a high medical need to improve the outcome of AML patients [4,5,6]. Myelodysplastic syndrome (MDS) is a heterogeneous clonal myeloid neoplasm that is characterized by ineffective hematopoiesis and cytopenia in one or more of the myeloid lineages, as well as an apparent risk of progression to AML [7].
AML is commonly associated with a variety of different genetic abnormalities, such as chromosomal abnormalities, heterozygous deletions, gene mutations, and epigenetic abnormalities [8]. CpG island methylation is usually altered during malignant transformation, which plays an important role in transcriptional regulation and offers new ideas for AML and MDS surveillance and treatment [9]. SLCs are the largest family of transmembrane transport proteins. Dysregulation and mutations of SLC encoding gene have been associated with susceptibility to a variety of diseases, including metabolic disorders and many kinds of cancers [10,11,12]. The solute carrier family 22 (SLC22) members mainly contain the organic cation transporters OCT 1 (SLC22A1), OCT2 (SLC22A2), and OCT3 (SLC22A3), which are involved in many metabolic and detoxification processes as transcription factors. SLC22A3 has the widest tissue distribution among SLC22s [13]. Early studies focusing on metabolic functions and metabolic diseases found that SLC22A3 is critical for eliminating endogenous small organic cations and drugs [14, 15]. To date, many research works have indicated the association between SLC22A3 and cancers, including tumorigenesis, tumor invasion and metastasis, uptake and metabolism of antineoplastic drugs, and disease prognosis [16,17,18,19,20,21,22], but the significance of SLC22A3 in AML remains unclear so far. Therefore, we studied SLC22A3, one of several hypermethylated genes that we screened for in AML. Our aim was to investigate whether DNA methylation and mRNA expression of SLC22A3 are associated with AML and have potential value as a drug target for AML treatment.
Materials and method
Patients and samples
Three hundred and thirty-six adult AML patients in various clinical statuses [including 271 newly diagnosed cases, 66 complete remission (CR) cases, and 24 relapsed cases], 93 newly diagnosed MDS patients, and 45 healthy donors were enrolled in this study. BMMNCs were isolated by Ficoll density gradient centrifugation. Our research was approved by the Institutional Ethics Committee of the Affiliated People’s Hospital of Jiangsu University, and each individual provided signed informed consents for their participation. The diagnosis and classification of MDS and AML patients were based on the French–American–British (FAB) and the 2016 World Health Organization (WHO) criteria [7, 23].
Cytogenetic and molecular genetic analysis
A series of clinic-hematological profiles and auxiliary examination results of patients were enrolled in this study for clinical correlation research. Hematologic laboratory results, including morphologic identification of bone marrow aspirate, cytogenetic analysis, immune-phenotypic feature, and molecular testing were valuable for the diagnosis and prognosis of hematopoietic malignancies. These indexes were also applied to follow-up study after induction and consolidation chemotherapy [24, 25]. Cytogenetic characteristics were analyzed by R-banded standard karyotyping and/or fluorescence in situ hybridization at diagnosis. Twelve commonly gene mutations tested by high-resolution melting analysis and/or direct DNA sequencing were carried out on BMMNCs [4]. The prognosis of MDS patients can be grouped into four categories using the International Prognostic Scoring System (IPSS): low risk, INT‐1, INT‐2, and high risk [26, 27].
RNA isolation and real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated as per the TRIzol reagent instruction (Invitrogen, Carlsbad, CA, USA) from each BMMNCs sample and was transcriptionally reversed into cDNA using PrimeScript™ II 1st Strand cDNA Synthesis Kit (TaKaRa, Tokyo, Japan). RNA concentration and quality were assessed with a NanoDrop 2000 (NanoDrop Technologies, Wilmington, Delaware USA). SLC22A3 expression was detected by RT-qPCR using the SYBR Premix Ex Taq II (TaKaRa, Tokyo, Japan) and was calculated by relative expression level (2 − ΔΔCt) with ABL as internal reference [25]. Primers sequence: SLC22A3 (71 bp), 5′-CCACCATCGTCAGCGAGT-3′ (forward), 5′-CAGGATGGCTTGGGTGAG-3′ (reverse); internal reference ABL (118 bp), 5′-TCCTCCAGCTGTTATCTGGAAGA-3′ (forward), 5′-TCCAACGAGCGGCTTCAC-3′ (reverse). The appropriate primers and personalized RT-qPCR reaction temperature partly different from the manufacturer’s template were verified by gel electrophoresis and Sanger sequencing of PCR products, 60 °C for 30 s in annealing temperature and 82 °C for 30 s in collected fluorescence temperature.
DNA isolation and bisulfate modification
Genomic DNA was isolated from pretreated BMMNCs samples by Genomic DNA Purification Kit (Gentra, Minneapolis, MN, USA) and then modified using CpGenome DNA Modification Kit (Chemicon, Temecula, Canada). Methylation sites in genomic DNA were exposed after the bisulfite conversion and can be identified by targeted primers [28].
Targeted bisulfite sequencing assay and real-time quantitative methylation-specific PCR (RQ-MSP)
Based on the CpG sites of the genomic promoter region, we designed the primers and check them for feasibility and specificity. The bisulfite convert ratio and methylation levels of 27 CpG sites located at SLC22A3 promoter were validated by targeted bisulfite sequencing (methyl target) sequencing (Genesky Biotechnologies Inc., Shanghai, China), a multiple targeted CpG methylation analysis by next-generation sequencing [29]. The 20μL RQ-MSP reaction system was operated with 20 ng modified DNA, 0.8 μM primers, 10 μM SYBR Premix Ex Taq II Mix and 0.4 μM ROX Reference Dye II (TaKaRa, Tokyo, Japan). Primers sequence: methylated SLC22A3 (M-SLC22A3, 252 bp), 5′-GGGATTAAAAGGAGTTTCGC -3′ (forward), 5′-CACTCGCCCTAACGCTATAC -3′ (reverse); unmethylated SLC22A3 (U-SLC22A3, 252 bp), 5′-GTAGGGATTAAAAGGAGTTTTGT -3′ (forward), 5′-CCTCACTCACCCTAACACTATAC -3′ (reverse); internal reference ALU (110 bp), 5′-TTAGGTATAGTGGTTTATATTTGTAATTTTAGTA-3′ (forward), 5′-ATTAACTAAACTAATCTTAAACTCCTAACCTCA-3′ (reverse). The relative methylation level of SLC22A3 was calculated by the formula: NM/U-SLC22A3 = 2ΔCT M/U−SLC22A3(control−sample) ÷ 2ΔCT ALU(control−sample)(2−ΔΔCT). RQ-MSP reaction systems were personalized in annealing and collected fluorescence temperatures, 66 °C (M) or 63 °C (U) for 30 s and 80 °C for 30 s, respectively.
Human leukemia cell lines and AML fresh bone marrow mononuclear cells culture
The human leukemia cell lines K562 (FAB-M6), HL-60 (FAB-M2), THP-1 (FAB-M5), U937 (FAB-M5), SKM-1 (MDS-derived AML M5), and MV4-11 (FAB-M5) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and gifted from other laboratories. Cells were cultured in freshly prepared RPMI 1640 medium (WISENT, Nanjing, China) supplemented with 10% fetal calf serum (ExCell Bio, Shanghai, China) and 1% penicillin–streptomycin (Hyclone, Shanghai, China), surrounded by a 37◦C humidified atmosphere containing 5% CO2.
Fresh mononuclear cells from the bone marrow of four newly diagnosed untreated AML patients (P1-M2a, P2-M2b, P3-M4a, and P4-M5b) were separated through density gradient centrifugation using a PBMC separation tube (FcMACS, NanJing, China) and suspended in Dulbecco's phosphate-buffered saline (WISENT, Nanjing, China) for twice, according to the specification protocol. After cultured in the same medium and environment as cell lines for 48 h, all the suspension cells were collected for further culture, while mesenchymal stem cells which grew by static adherence were isolated.
RNA interference (RNAi)
Gene silencing induced by small interfering RNAs (siRNAs) was performed in HL60, K562, and AML fresh BMMNCs. siRNAs against SLC22A3 (siSLC22A3) and its related negative control (siNC) were designed and synthesized by GenePharma (Shanghai, China). Cell transfection was performed using EntransterTM-R4000 (Engreen Biosystem, Beijing, China).
Cell proliferation and apoptosis analysis
Cell lines (1.5 × 105 cells/ml) and AML fresh BMMNCs (1 × 105 cells/ml) were seeded onto a 6-well plate with normal substrate environment. After culturing for 0, 24, 48, and 72 h, cell proliferation status was counted on the counting board three times.
Cells (2 × 105 cells/ml) were seeded onto a 6-well plate in RPMI 1640 medium without fetal calf serum for 72 h. Cell apoptosis rate was detected by flow cytometry (Beckman Coulter, Miami, FL, USA) using Annexin V-PE/7-AAD apoptosis detection kits (BD Pharmingen, San Diego, CA, USA). Viable cells were counted using 0.4% trypan blue staining (Biosharp, Anhui, China). Each experiment was repeated three times.
Demethylation drug DAC sensitivity study
According to SLC22A3 expression and methylation pattern in above six leukemic cell lines, we selected HL60 and K562 cells for further 5-Aza-2′-deoxycytidine (DAC, Sigma-Aldrich, St. Louis, MO, USA) sensitivity experiment. Normal growing cells lines and AML fresh BMMNCs (1 × 105 cells/ml) were treated by DAC at a final concentration of 0 μM, 0.1 μM, 1 μM, and 10 μM for 72 h, respectively. The expression of SLC22A3 in treated cells at each concentration was determined by RT-qPCR. After treated by DAC at a final concentration of 1 μM for 24 h, cell apoptosis of K562 siNC/siSLC22A3 was detected by flow cytometry.
Sequencing and bioinformatics analyses
Our laboratory detected many differential methylation genes within four normal donors and four pairs of MDS-sAML (secondary AML) patients through RRBS [30]. The raw data have been submitted to NCBI SRA databases, whose accession number was PRJNA670308.
Gene expression (RNA Seq V2 RSEM), methylation (HM450), and clinical information of 200 adults with de novo AML (NEJM 2013) from the Cancer Genome Atlas (TCGA) dataset were downloaded via cBioPortal (http://www.cbioportal.org) and DiseaseMeth version 2.0 (http://bio-bigdata.hrbmu.edu.cn/diseasemeth/index.html) [31]. Genome-wide DNA methylation profiling of 15 AML and 5 normal bone marrow specimens from GSE63409 (http://www.ncbi.nlm.nih.gov/geo/; GSE63409) were used for gene screening. Genomic sequencing and clinical data of 558 cases from Beat AML program were downloaded via cBioPortal (http://www.cbioportal.org) [32]. Differential gene expression and enrichment analyses were calculated as reported previously [33].
Statistical analyses
SPSS 22.0 software package and GraphPad Prism 5 were applied to statistical analyses. Student's t test (Mann–Whitney’s U test and Paired T test) were performed to compare the differences of continuous variables. The Pearson chi-square analysis/Fisher exact test was conducted to analyze the diversities of categorical variables, while Spearman correlation test was used to evaluate the correlation between genes expression and methylation. The ROC curve and area under the ROC curve (AUC) were used for assessing discriminative capacity of SLC22A3 methylation between patients and controls. Kaplan–Meier and Cox regression (univariate and multivariate) analyses were carried out to evaluate the impact of SLC22A3 on survival. Statistical significance was set at P < 0.05, and each test was two-sided.
Results
Screening of methylation-related candidate SLCs involved in AML prognosis by public database
Combined with our previous genome-wide methylation pattern research by RRBS [30] and the public methylation array GSE63409 data, we identified numerous abnormally hypermethylated genes including a group of SLC gene family members in AML, which have arose our attention (Additional file 1). Then, we screened genes that associated with AML prognosis among these SLCs based on TCGA database (Fig. 1A–E). Prognostic significance of each gene was evaluated between two groups divided by the median level of their expression. Kaplan–Meier analysis showed that high expression of SLC22A3 predicted longer overall survival (OS) and disease-free survival (DFS) in both non-acute promyelocytic leukemia (APL) AML (non-APL AML) (P = 0.008 and 0.002) and cytogenetically normal AML (CN-AML) patients (P = 0.006 and 0.005; Fig. 1E). Independent assessments of paired SLCs methylation and expression sequencing data from TCGA AML project revealed a negative correlation between those of SLC22A3 (r = − 0.376, P < 0.001, n = 155, Fig. 1F), but not of other four genes (SLC5A8, SLC6A11, SLC7A14, and SLC34A2) (Additional file 2). Moreover, Kaplan–Meier analysis based on Beat AML patients also revealed this clinical significance between SLC22A3 high-expression and longer OS in both whole-cohort AML and CN-AML (P = 0.033 and 0.032; Fig. 1G). We also observed SLC22A3 up-regulation in MLLT3-KMT2A rearranged AML and FLT3-ITD negative mutations according to Beat AML cohort (Additional file 3).

Identification of SLCs expression associated with prognosis in AML by public database. A–E SLC5A8, SLC6A11, SLC7A14, SLC22A3, SLC34A2. The impact of SLCs expression on OS and DFS was detected among non-APL AML and CN-AML patients from TCGA databases. AML patients were divided into two groups by the median mRNA expression level of each gene, respectively. F Correlation between DNA methylation and mRNA expression of SLC22A3 in AML from TCGA database. G The impact of SLC22A3 expression on OS was detected among total-cohort AML and CN-AML patients from beat AML database. AML patients were divided into two groups by the median SLC22A3 expression level
Identification of aberrant SLC22A3 methylation by targeted bisulfite sequencing in MDS and AML patients
IN view of the significance of SLC22A3 methylation and expression as mentioned above, we wanted to identify aberrant SLC22A3 methylation involved in new diagnosed MDS and AML patients. We detected methylation pattern of CpG sites located at SLC22A3 promoter region in 30 MDS, 100 AML patients and 25 controls using MethylTarget assay (targeted bisulfite sequencing). The mean bait coverage attached 1694 × , and Q30 was 75.56% [33]. The methylation level of SLC22A3 was significantly increased in MDS and AML patients compared with controls (P < 0.001 and < 0.001), as well as in AML compared with MDS (P < 0.001; Fig. 2A).

Confirmation of SLC22A3 hypermethylation in AML. A SLC22A3 methylation density in controls, MDS and AML patients detected by targeted bisulfite sequencing. B The correlation of SLC22A3 methylation between two detection methods (RQ-MSP and targeted bisulfite sequencing). C SLC22A3 methylation level in larger cohort of controls, MDS and AML patients analyzed by RQ-MSP. D ROC curve analysis by SLC22A3 methylation for distinguishing AML patients from controls. **: P < 0.01; ***: P < 0.001
Confirmation of SLC22A3 hypermethylation in an expanded group of MDS and AML patients
To further confirm the pattern of SLC22A3 methylation, we enrolled a larger cohort of newly diagnosed MDS (n = 61) and AML (n = 153) samples by RQ-MSP, a more convenient method. Primers for RQ-MSP and targeted sequencing were designed most overlapped in ampliconic sequence. In addition, RQ-MSP results showed a high positive correlation with that in the targeted bisulfite sequencing as confirmed (r = 0.717, P < 0.001, n = 54, Fig. 2B). As shown in Fig. 2C, SLC22A3 methylation pattern was markedly higher in whole-cohort AML samples than in control and MDS groups (P < 0.001 and = 0.002), as well as in non-APL AML (n = 107) and CN AML (n = 58) vs. controls, respectively (P < 0.001 and = 0.002). However, we did not find a distinct variation between controls and MDS specimens.
Clinical properties and genetic features of MDS and AML with high SLC22A3 methylation
According to ROC curve analysis, SLC22A3 methylation could be helpful to distinguish AML from controls (Fig. 2D). To investigate the clinical correlation of SLC22A3 methylation in pathogenesis and prognosis of MDS/AML, patients were divided into two distinct groups (SLC22A3 hypermethylation and SLC22A3 hypomethylation) based on the cutoff value of 0.042 (according to Youden index of ROC curve), which can also put almost all healthy donors in hypomethylation set. SLC22A3 hypermethylation was associated with lower platelets and higher CEBPA mutation rate in non-APL AML patients (P = 0.020 and 0.006), as well as higher mutation rate of N/K-RAS in CN-AML (P = 0.080; Table 1). Besides, SLC22A3 methylation pattern was irregularly distributed among leukemia subtypes (FAB) (Table 1). However, we observed no statistical differences between clinical data of two MDS groups (Table 2).
Correlation of SLC22A3 hypermethylation with prognosis in MDS and AML patients
Based on follow-up investigation, we would like to analyze whether SLC22A3 methylation affect the life span expectancy of MDS and AML patients. We screened AML patients treated with 3 days of an anthracycline and 7 days of cytarabine (“3 + 7” regimens). Kaplan–Meier analysis indicated tendencies of shorter OS and leukemia-free survival (LFS) in CN-AML patients with SLC22A3 hypermethylation (P = 0.109 and 0.057; Fig. 3A). SLC22A3 hypermethylation group showed conspicuous shorter OS and LFS in non-APL AML patients (P = 0.043 and 0.035; Fig. 3B). However, SLC22A3 methylation pattern had no influence on MDS prognosis, no matter which method methylation data were measured (P > 0.050; Fig. 3C, D). Next, we enrolled variables that have statistically differences in univariate cox regression analysis (P < 0.200) and/or clinically recognized related to AML into multivariate analysis. In non-APL AML cohort, SLC22A3 hypermethylation was an independent risk indicator for shorter OS and LFS, respectively (P = 0.001 and < 0.001; Table 3).

Prognostic value of SLC22A3 methylation in AML and MDS patients. A, B The impact of SLC22A3 methylation on OS and LFS of CN-AML, and non-APL AML patients, respectively. AML patients were treated by “3 + 7” induction regimens. C, D The impact of SLC22A3 methylation on OS among MDS patients. SLC22A3 methylation was measured using RQ-MSP (C) and MethylTarget assay (D)
Transcriptional regulatory effects of SLC22A3 methylation on mRNA expression
To verify the regulatory role of SLC22A3 methylation in AML pathogenesis, specimens of newly diagnosed MDS (n = 20) and AML (n = 89) were used to assess SLC22A3 expression level by RT-qPCR. Our results showed that SLC22A3 expression was downregulated in AML compared with controls and MDS (P = 0.001 and 0.002; Fig. 4A). Besides, we observed a credibly negative correlation between SLC22A3 methylation and expression (r = − 0.550, P < 0.001, n = 46, Fig. 4B).

Regulation effect of DNA methylation on SLC22A3 expression. A SLC22A3 expression level detected by RT-qPCR in controls, MDS and AML patients. B Correlation between SLC22A3 methylation (RQ-MSP) and expression (RT-qPCR). C, D SLC22A3 methylation and expression pattern in six leukemic cell lines. E, F SLC22A3 expression on HL60 and K562 changed with the concentration of DAC treatment (0, 0.1, 1, 10 μmol/L). G–J SLC22A3 expression changed with the concentration of DAC treatment on AML fresh CMMNCs isolated from four newly diagnosed AML patients (Patient1-M2a, Patient2-M2b, Patient3-M4a, and Patient4-M5b). *: P < 0.05
DNA methylation and mRNA expression levels of SLC22A3 were performed on six leukemia cell lines (Fig. 4C, D). Next we selected HL60 and K562 cells with the lowest and highest methylation levels, respectively, and treated them with the demethylation drug DAC. A dose–response relationship can be observed between DAC treatment and SLC22A3 expression over a range of drug concentrations (Fig. 4E, F). Furthermore, we treated 4 AML fresh BMMNCs with graduate increased dosage of DAC, and found that DAC also promoted mRNA expression of SLC22A3 in the same dose range (Fig. 4G–J), which further verify our thesis that DNA methylation regulates SLC22A3 mRNA expression in AML.
Assessment of SLC22A3 expression for AML prognosis as a surveillance biomarker
Since SLC22A3 methylation level is associated with prognosis of AML, we attempted to further evaluate whether SLC22A3 expression level reflects the course of AML disease. We collected AML specimens from different clinical stages including 66 patients who achieved CR after induction therapy and 24 relapsed patients. RT-qPCR results revealed that SLC22A3 expression level was significantly improved in CR patients compared to newly diagnosed time, and subsequently fell back in the relapse phase (P = 0.002 and 0.009; Fig. 5A). As presented in Fig. 5B, SLC22A3 expression exhibited obviously dynamic changes with the clinical phases in a follow-up study of eight patients (P = 0.012).

SLC22A3 expression in the surveillance of AML. A SLC22A3 expression in different clinical stages (new diagnosis, complete remission, and relapsed time) of AML patients. B Dynamic change of SLC22A3 expression in eight patients during different clinical stages
Biological function of SLC22A3 on leukemia cells
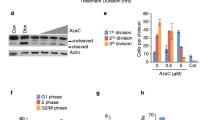
TO explore the biological role of SLC22A3 expression in AML, we created conditions of SLC22A3 silencing in HL60 and K562 cells, as well as fresh BMMNCs from two AML patients. We transfected siSLC22A3 and its related siNC into these tool cells, and obtained remarkable silencing effect (Fig. 6A). With the knock-down of SLC22A3 transcript level, proliferation ability of HL60, K562, and AML fresh BMMNCS were significantly climbing among 72 h (Fig. 6B–E). Compared with siNC controls, in addition, HL60 siSLC22A3 and K562 siSLC22A3 exhibited lesser apoptosis after culturing in starvation station for 72 h (Fig. 6F, G). In addition, pro-apoptotic effect of DAC was weakened in K562 siSLC22A3 after treated for 24 h, compared with siNC (20.24% and 65.63%, Additional file 4).

Biological role of SLC22A3 on leukemic cell lines and AML fresh BMMNCs. A Transcript level of SLC22A3 in HL60, K562 and 2 AML fresh BMMNCs (patient1 and patient4) after transfected with small interfering (si)RNA against SLC22A3 and negative control. B–E The effect of SLC22A3 under-expression on cell proliferation. SLC22A3 under-expression significantly increased cell proliferation ability in HL60, K562, and fresh BMMNCs from Patient1 and Patient4. F, G The effect of SLC22A3 under-expression on cell apoptosis. SLC22A3 under-expression significantly decreased cell apoptosis ratio in HL60 and K562 cells
Molecular exploration of SLC22A3 in AML
To further understand the biological insight of SLC22A3 in AML, we analyzed the transcriptome differences to be associated with SLC22A3 expression from Beat AML and TCGA cohorts. AML patients were divided by the median level of SLC22A3 expression into low- and high-expression groups. We identified 124 differentially expressed genes in Beat AML (Fig. 7A, B; Additional file 5) and 1015 in TCGA (Fig. 7C, D; Additional file 6). Combined with the results of the above two cohorts, a total of 66 positively correlated genes were singled out, including VCAM1, SOX9, ID4, and ITIH5 which have been clearly reported for antileukemia effects [34,35,36,37]. Moreover, these genes were concentrated in the extracellular matrix and involved in signaling receptor binding, cell adhesion, and ECM-receptor interaction according to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (Fig. 7E–H). Besides, the differentially expressed analysis based on microRNA data of TCGA screened out 43 microRNAs including 16 negatively associated with SLC22A3 (Fig. 7I; Additional file 7). These negatively correlated microRNAs such as hsa-let-7b, hsa-mir-19b, and hsa-mir-196a were involved in antileukemia effects as previously reported [38,39,40].

Molecular signatures associated with SLC22A3 in AML. A Heatmap of differentially expressed genes between patients with low- and high-expressed SLC22A3 among Beat AML datasets (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). B Volcano plot of up- and downregulated genes in lower expressed SLC22A3 group among Beat AML datasets (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). C Heatmap of differentially expressed genes between AML patients with low- and high-expressed SLC22A3 among TCGA datasets (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). D Volcano plot of up- and downregulated genes in lower expressed SLC22A3 group among TCGA datasets (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). E, F Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differentially expressed genes among Beat AML datasets by using online website of STRING (http://string-db.org). G, H GO and KEGG analysis of differentially expressed genes among TCGA datasets by using online website of STRING. I Heatmap of differentially expressed microRNAs between AML patients with low- and high-expressed SLC22A3 among TCGA datasets (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5)
Discussion
Hematopoietic malignancies are more susceptible to epigenetic interventions than solid malignancies. AML is a heterogeneous myeloid tumor that displays extensive variation in their clinical courses and in response to therapy. Epigenetics allows us to extend the exploration of the potential diversity among AML subsets [41]. In this research, we have found that SLC22A3 present general higher methylation and lower expression pattern in the whole-cohort de novo AML, non-APL AML and CN patients. Set a threshold for SLC22A3 methylation that can exclude almost all healthy donors, AML patients above this level tended to have lower OS and LFS. The similar poor prognosis tendencies have also been reflected in SLC22A3 under-expression groups among large AML samples from TCGA and OHSU database. Follow-up study and paired analysis provided a more intuitive view in dynamic changes of SLC22A3 expression with the clinical phases, which can promote its understanding and further application in AML surveillance. The genomes hypomethylation and aberrant hypermethylation in promoter are involved in many kinds of tumorigenesis, leading to activating of proto-oncogenes and inhibiting of tumor suppressor genes [42, 43]. Abnormal DNA methylation and expression levels of SLC22A3 have been demonstrated in several tumors. Higher methylation and lower expression of SLC22A1 and SLC22A3 was observed in hepatocellular carcinoma and prostate tumor compared with matched normal samples [44, 45]. SLC22A1 activity was reported to correlate with the sensitivity of imatinib, a tyrosine kinase inhibitor, in patients with chronic myeloid leukemia [46, 47]. In this study, our results based on clinical BM specimens and bioinformatics analysis reveal an association between SLC22A3-hypermethyl status and AML.
Our study showed that the DNA methylation level of SLC22A3 was also increased in MDS group compared with the control group, but lower than that of AML. MDS is a group of malignant clonal diseases with a high risk of transition to AML. However, MDS is a highly heterogeneous group of diseases, whose clinical course and outcome vary greatly, with no more than 30% actually transforming into AML [48, 49]. Overall, MDS and AML represent a disease continuum that undergoes genetic clonal evolution, but there are still differences in pathophysiology between AML and MDS.
The transcription factor CCAAT enhancer binding protein alpha (CEBPA) is a myeloid transcription factor. Ley et al. confirmed that mutations in CEBPA and other myeloid transcription factor genes, such as DNMT3A, NPM1, IDH1/2 and RUNX1, were common in AML and suggested that these mutations had functions related to the pathogenesis of AML [31]. Results of our clinical analysis showed that SLC22A3 DNA hypermethylation status was associated with CEBPA mutation in AML patients. However, we did not discuss two types (biallelic and single heterozygous) of CEBPA mutations separately because of the limited mutation cases. We observed no correlation between SLC22A3 expression and CEBPA biallelic mutations based on Beat AML databases. Considering of the prognosis differences between two types of CEBPA mutations, the causal relationship between SLC22A3 hypermethylation and CEBPA mutation and its clinical significance in AML remain to be clarified.
Promoter methylation plays an important role in SLC22A3 expression. Chen et al. confirmed that methylation in the SLC22A3 promoter region could explain the low expression level of SLC22A3 in high-Gleason grade prostate cancer, which may be related to the progression of prostate cancer. In SLC22A3-negative HCT116, SLC22A3 mRNA levels were significantly reactivated with increased dose of 5′ -AZADC (a demethylating agent) [44]. Xiong et al. observed that SLC22A3 methylation conferred susceptibility to esophageal squamous cell carcinoma [50].
Our demethylation studies with DAC also showed that SLC22A3 mRNA expression of two leukemic cell lines and AML bone marrow mononuclear cells increased with increasing drug dose within a certain concentration range. The SLC22A3 post-intervention experiments confirmed that SLC22A3 downregulation led to active proliferation and diminished apoptosis of leukemia cells. SLC22A3 is a widely expressed drug transporter. hSLC22A3-mediated oxaliplatin uptake in cancer is thought to be important for its cytotoxicity [16], but it is not clear whether SLC22A3 mediated the uptake of DAC. Our results showed that DNA hypermethylation may repress drug importer SLC22A3 located in membrane and resulted in enhanced drug resistance and diminished apoptosis. We considered that poor clinical outcomes of SLC22A3 downregulation/SLC22A3 hypermethylation patients could be associated with weakened drug pump function.
In summary, this current study showed that SLC22A3 DNA is aberrant hypermethylation in AML and different clinical status of disease display distinct patterns of DNA methylation. DNA methylation levels may be useful for AML prognosis. Just as Šestáková et al. [51] proposed, further validation of selected tumor markers is important especially for their clinical applications. Future studies are needed to investigate more cases to clarify the significance of SLC22A3 methylation level and expression in therapy of AML and to clarify whether SLC22A3 aberrant methylation facilitate or merely coexist with CEBPA mutation.
Conclusion
Our results showed that increased methylation and decreased expression of SLC22A3 may be indicators of poor prognosis in AML. Methylation-silenced SLC22A3 expression may have potential guiding significance on the antileukemia effect of DAC.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- AML:
-
Acute myeloid leukemia
- RRBS:
-
Reduced representation bisulfite sequencing
- BMMNCs:
-
Bone marrow mononuclear cells
- DAC:
-
5-Aza-2′-deoxycytidine
- ATRA:
-
All transretinoic acid
- MDS:
-
Myelodysplastic syndrome
- CR:
-
Complete remission
- FAB:
-
French–American–British
- WHO:
-
World Health Organization
- IPSS:
-
International Prognostic Scoring System
- PQ-MSP:
-
Real-time quantitative methylation-specific PCR
- RT-qPCR:
-
Real-time quantitative PCR
- TCGA:
-
The Cancer Genome Atlas
- AUC:
-
Area under the ROC curve
- DFS:
-
Disease-free survival
- LFS:
-
Leukemia-free survival
- OS:
-
Overall survival
- APL:
-
Acute promyelocytic leukemia
- CN-AML:
-
Cytogenetically normal AML
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32.
Zheng RS, Sun KX, Zhang SW, et al. Report of cancer epidemiology in China, 2015. Zhonghua Zhong Liu Za Zhi. 2019;41(1):19–28.
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Narayanan D, Weinberg OK. How I investigate acute myeloid leukemia. Int J Lab Hematol. 2020;42(1):3–15.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Song X, Peng Y, Wang X, et al. Incidence, survival, and risk factors for adults with acute myeloid leukemia not otherwise specified and acute myeloid leukemia with recurrent genetic abnormalities: analysis of the surveillance, epidemiology, and end results (SEER) database, 2001–2013. Acta Haematol. 2018;139(2):115–27.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Benetatos L, Vartholomatos G. Enhancer DNA methylation in acute myeloid leukemia and myelodysplastic syndromes. Cell Mol Life Sci. 2018;75(11):1999–2009.
Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52.
Sutherland R, Meeson A, Lowes S. Solute transporters and malignancy: establishing the role of uptake transporters in breast cancer and breast cancer metastasis. Cancer Metastasis Rev. 2020;39(3):919–32.
Cannizzaro M, Jarošová J, De Paepe B. Relevance of solute carrier family 5 transporter defects to inherited and acquired human disease. J Appl Genet. 2019;60(3–4):305–17.
Xie J, Zhu XY, Liu LM, et al. Solute carrier transporters: potential targets for digestive system neoplasms. Cancer Manag Res. 2018;10:153–66.
Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165(5):1260–87.
Nies AT, Koepsell H, Winter S, et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50(4):1227–40.
Qi Q, Workalemahu T, Zhang C, et al. Genetic variants, plasma lipoprotein(a) levels, and risk of cardiovascular morbidity and mortality among two prospective cohorts of type 2 diabetes. Eur Heart J. 2012;33(3):325–34.
Yokoo S, Masuda S, Yonezawa A, et al. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab Dispos. 2008;36(11):2299–306.
Shnitsar V, Eckardt R, Gupta S, et al. Expression of human organic cation transporter 3 in kidney carcinoma cell lines increases chemosensitivity to melphalan, irinotecan, and vincristine. Cancer Res. 2009;69(4):1494–501.
Mohelnikova-Duchonova B, Brynychova V, Hlavac V, Kocik M, et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother Pharmacol. 2013;72(3):669–82.
Li Fu, Qin Y-R, Ming X-Y, et al. RNA editing of SLC22A3 drives early tumor invasion and metastasis in familial esophageal cancer. Proc Natl Acad Sci U S A. 2017;114(23):E4631–40.
Grisanzio C, Werner L, Takeda D, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci U S A. 2012;109(28):11252–7.
Huang KM, Thomas MZ, Magdy T, et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc Natl Acad Sci USA. 2021;118(5):e2020168118.
Nishikura K. Oesophageal cancer: RNA editing of SLC22A3 mRNAs: Causative relevance to familial ESCC? Nat Rev Gastroenterol Hepatol. 2017;14(10):569–70.
Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620–5.
Lian X-Y, Zhang W, De-Hong Wu, et al. Methylation-independent ITGA2 overexpression is associated with poor prognosis in de novo acute myeloid leukemia. J Cell Physiol. 2018;233(12):9584–93.
Zhang T-J, Zhou J-D, Zhang W, Lin J, et al. H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin Epigenetics. 2018;10:47.
Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65.
Montalban-Bravo G, Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93(1):129–47.
Ramalho-Carvalho J, Henrique R, Jerónimo C. Methylation-specific PCR. Methods Mol Biol. 2018;1708:447–72.
Kachroo P, Szymczak S, Heinsen F-A, et al. NGS-based methylation profiling differentiates TCF3-HLF and TCF3-PBX1 positive B-cell acute lymphoblastic leukemia. Epigenomics. 2018;10(2):133–47.
Zhou J-D, Zhang T-J, Zi-Jun Xu, et al. Genome-wide methylation sequencing identifies progression-related epigenetic drivers in myelodysplastic syndromes. Cell Death Dis. 2020;11(11):997.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31.
Zhang TJ, Zi-jun Xu, Yu Gu, et al. Identification and validation of prognosis-related DLX5 methylation as an epigenetic driver in myeloid neoplasms. Clin Transl Med. 2020;10:e29.
Becker PS, Kopecky KJ, Wilks AN, et al. Very late antigen-4 function of myeloblasts correlates with improved overall survival for patients with acute myeloid leukemia. Blood. 2009;113(4):866–74.
Azadniv M, Myers JR, McMurray HR, et al. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia. 2020;34(2):391–403.
Zhou J-D, Zhang T-J, Li X-X, et al. Epigenetic dysregulation of ID4 predicts disease progression and treatment outcome in myeloid malignancies. J Cell Mol Med. 2017;21(8):1468–81.
Oing C, Jost E, Dahl E, et al. Aberrant DNA hypermethylation of the ITIH5 tumor suppressor gene in acute myeloid leukemia. Clin Epigenetics. 2011;2(2):419–23.
Dong L-H, Huang J-J, Peng Zu, et al. CircKDM4C upregulates P53 by sponging hsa-let-7b-5p to induce ferroptosis in acute myeloid leukemia. Environ Toxicol. 2021;36(7):1288–302.
Zhao T-F, Jia H-Z, Zhang Z-Z, et al. LncRNA H19 regulates ID2 expression through competitive binding to hsa-miR-19a/b in acute myelocytic leukemia. Mol Med Rep. 2017;16(3):3687–93.
Bamodu OA, Kuo K-T, Yuan L-P, et al. HDAC inhibitor suppresses proliferation and tumorigenicity of drug-resistant chronic myeloid leukemia stem cells through regulation of hsa-miR-196a targeting BCR/ABL1. Exp Cell Res. 2018;370(2):519–30.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–99.
Iacobuzio-Donahue CA. Epigenetic changes in cancer. Annu Rev Pathol. 2009;4:229–49.
Chen L, Hong C, Chen EC, et al. Genetic and epigenetic regulation of the organic cation transporter 3, SLC22A3. Pharmacogenomics J. 2013;13(2):110–20.
Schaeffeler E, Hellerbrand C, Nies AT, et al. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011;3(12):82.
Crossman LC, Druker BJ, Deininger MWN, et al. hOCT 1 and resistance to imatinib. Blood. 2005;106(3):1133–4.
White DL, Saunders VA, Dang P, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007;110(12):4064–72.
Granfeldt Østgård LS, Medeiros BC, Sengeløv H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33(31):3641–9.
Hulegårdh E, Nilsson C, Lazarevic V, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. 2015;90(3):208–14.
Xiong J-X, Wang Y-S, Sheng J, et al. Epigenetic alterations of a novel antioxidant gene SLC22A3 predispose susceptible individuals to increased risk of esophageal cancer. Int J Biol Sci. 2018;14(12):1658–68.
Šestáková Š, Cerovská E, Šálek C, et al. A validation study of potential prognostic DNA methylation biomarkers in patients with acute myeloid leukemia using a custom DNA methylation sequencing panel. Clin Epigenetics. 2022;14(1):22.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (81970118, 81900166), Medical Innovation Team of Jiangsu Province (CXTDB2017002), Research Innovation Program for College Graduates of Jiangsu Province (SJKY19_2584), Zhenjiang Clinical Research Center of Hematology (SS2018009), Social Development Foundation of Zhenjiang (SH2021052, SH2019065), Natural Science Foundation of Jiangsu Province (BK20221287), Research Project of Jiangsu Commission of Health (M2022123), and Scientific Research Project of The Fifth 169 Project of Zhenjiang (21).
Author information
Authors and Affiliations
Contributions
JQ and JL conceived and designed the experiments; YG and JZ performed the experiments; ZX and XW analyzed the data; YJ, QY, and PX collected the clinical data; YF and LY offered the technical support; YG and JZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This research was approved by Institutional Ethics Committee of the Affiliated People’s Hospital of Jiangsu University, and each individual provided signed informed consents for their participation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Identification of aberrantly hypermethylated SLCs in AML. A Heatmap of differentially methylated genes between normal and AML bone marrow specimens from GSE63409. B The Venn diagram of hypermethylated genes in AML. The intersection of hypermethylated genes in AML based on GSE63409 and the RRBS data that our lab has submitted to NCBI SRA databases previously (accession number PRJNA670308). C represents normal donors.
Additional file 2:
Correlation between DNA methylation and mRNA expression of SLCs in AML from TCGA database. A-D, SLC5A8, SLC6A11, SLC7A14, SLC34A2. The values of zero were excluded from log calculation.
Additional file 3
: SLC22A3 expression in genetics subsets of Beat AML cohort. A, SLC22A3 expression in subsets of recurrent genetic abnormalities. B, SLC22A3 expression in subsets of MLL rearranged AML. C-E, SLC22A3 expression in AML with CEBPA biallelic, NPM1, and FLT3-ITD mutations.
Additional file 4:
Apoptotic analysis of K562 siNC/siSLC22A3 treated by DAC. A, B, Comparison of cell apoptosis between K562 siSLC22A3 and siNC after DAC dosing.
Additional file 5:
Differently expressed mRNAs between lower and higher SLC22A3 expression groups from beat AML.
Additional file 6:
Differently expressed mRNAs between lower and higher SLC22A3 expression AML groups from TCGA.
Additional file 7:
Differently expressed microRNAs between lower and higher SLC22A3 expression AML groups from TCGA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, Y., Xu, Zj., Zhou, Jd. et al. SLC22A3 methylation-mediated gene silencing predicts adverse prognosis in acute myeloid leukemia. Clin Epigenet 14, 162 (2022). https://doi.org/10.1186/s13148-022-01373-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01373-w