
Abstract
The potential mechanism of myelodysplastic syndromes (MDS) progressing to acute myeloid leukemia (AML) remains poorly elucidated. It has been proved that epigenetic alterations play crucial roles in the pathogenesis of cancer progression including MDS. However, fewer studies explored the whole-genome methylation alterations during MDS progression. Reduced representation bisulfite sequencing was conducted in four paired MDS/secondary AML (MDS/sAML) patients and intended to explore the underlying methylation-associated epigenetic drivers in MDS progression. In four paired MDS/sAML patients, cases at sAML stage exhibited significantly increased methylation level as compared with the matched MDS stage. A total of 1090 differentially methylated fragments (DMFs) (441 hypermethylated and 649 hypomethylated) were identified involving in MDS pathogenesis, whereas 103 DMFs (96 hypermethylated and 7 hypomethylated) were involved in MDS progression. Targeted bisulfite sequencing further identified that aberrant GFRA1, IRX1, NPY, and ZNF300 methylation were frequent events in an additional group of de novo MDS and AML patients, of which only ZNF300 methylation was associated with ZNF300 expression. Subsequently, ZNF300 hypermethylation in larger cohorts of de novo MDS and AML patients was confirmed by real-time quantitative methylation-specific PCR. It was illustrated that ZNF300 methylation could act as a potential biomarker for the diagnosis and prognosis in MDS and AML patients. Functional experiments demonstrated the anti-proliferative and pro-apoptotic role of ZNF300 overexpression in MDS-derived AML cell-line SKM-1. Collectively, genome-wide DNA hypermethylation were frequent events during MDS progression. Among these changes, ZNF300 methylation, a regulator of ZNF300 expression, acted as an epigenetic driver in MDS progression. These findings provided a theoretical basis for the usage of demethylation drugs in MDS patients against disease progression.
Similar content being viewed by others


Introduction
Myelodysplastic syndromes (MDS) comprise a diverse group of clonal hematopoietic disorders characterized by peripheral blood cytopenias and ineffective production of blood cells1. Although the intensive treatments are carried out appropriately, ~30% of MDS patients will progress to acute myeloid leukemia (AML) with poor prognosis1,2. However, the potential mechanism of MDS progression remains poorly investigated. Accumulated pre-existing genetic/epigenetic abnormalities and newly emerging events play crucial roles in the progression of MDS. Thus, identification of specific molecular events could make better understanding of MDS pathogenesis and may guide clinical treatment against disease progression.
With a rapid advance in sequencing methodologies, whole-genome or whole-exome sequencing have been successively utilized to identify huge genetic changes in MDS3,4,5,6,7. Gene mutations in functional gene categories, including epigenetic modifiers (TET2 and IDH1/2), RNA splicing machinery (ASXL1), transcriptional factors (RUNX1), and signal transduction factors (ROBO1/2), have been considered to be partially responsible for progression in MDS3,4,5,6,7. These genetic drivers, however, could not generalize all the cases during MDS progression. The frequently mutant genes TET2 and IDH1/2 in MDS-derived secondary AML (sAML) are also methylation-related genes suggesting that aberrant epigenetic programming may play a crucial role in MDS progression8. Previously, we have already identified that single genes, including GPX3, ID4, and SOX30 methylation were associated with disease progression in MDS9,10,11. To gain new insights into the epigenetic mechanism underlying disease evolution in MDS, reduced representation bisulfite sequencing (RRBS) was conducted in bone marrow (BM) samples from four paired MDS/sAML patients and intended to discover methylation-associated epigenetic drivers in MDS progression.
In the current study, we investigated genome-wide methylation of four paired MDS/sAML patients by RRBS, and confirmed increased genomic hypermethylation as a frequent phenomenon during MDS progression. Secondly, several genes methylation patterns were screened and verified in MDS and AML patients using targeted bisulfite sequencing. Simultaneously, real-time quantitative methylation-specific PCR (RQ-MSP) was implemented to detect the identified gene methylation pattern in a larger group of clinical samples. Finally, the potential role and biological network of the targeted gene were further determined in MDS-derived AML cell-line SKM-112,13. The current work demonstrated a novel contribution to the epigenetic alterations profile of MDS during disease progression and could be potentially helpful to guide treatment decisions for MDS against disease progression.
Materials and methods
Patients and samples
Firstly, the BM samples from four paired MDS/sAML patients and four healthy donors were obtained in the Affiliated People’s Hospital of Jiangsu University and the First Affiliated Hospital of Soochow University. No significant differences were observed in age of controls (from 47 to 58 years old) and paired MDS/sAML patients (from 36 to 74 years old) (P = 0.828). All four paired MDS/sAML patients did not receive any demethylation treatment such as azacitidine and decitabine before. The detail information of four paired MDS/sAML patients was presented in Table S1. Secondly, another cohort of samples was enrolled for targeted bisulfite sequencing consisting of 35 de novo MDS, 111 de novo AML patients and 25 healthy donors treated at the Affiliated People’s Hospital of Jiangsu University. Furthermore, a total of 70 de novo MDS and 170 de novo AML patients together with 46 healthy donors treated at the Affiliated People’s Hospital of Jiangsu University were also included in validation assays. The diagnosis and classification of MDS and AML patients were established mainly according to the revised World Health Organization (WHO) criteria14. Common gene mutations of MDS and AML patients were detected as our previous report10. Treatment regimens for MDS and AML patients were reported previously as well10. BM was collected from all of the subjects after providing written informed consents. BM mononuclear cells (BMMNCs) were separated using Lymphocyte Separation Medium (Solarbio Science & Technology Co., Ltd., Beijing, China) based on density-gradient centrifugation, and RNA and DNA extraction were carried out subsequently15. This study was approved by the Ethics Committee of Affiliated People’s Hospital of Jiangsu University.
RRBS
RRBS was performed in Genesky Biotechnologies Inc. (Shanghai, China). Genomic DNA from BMMNCs was extracted and digested with MspI enzyme. After end-repair and A-tailing, methylated adaptors were ligated to MspI fragments. Adaptorligated fragments were size-selected (150–350 bp), bisulfite converted using EZ DNA Methylation kit and PCR amplified (12–15 cycles). After purification by TIANquick Midi Purification Kit, libraries were quantified and the quality was assessed. Single-ended RRBS libraries with 2 × 150 bp read length were sequenced on an Illumina HiSeq2500. The sequenced reads were mapped against the complete human reference genome GRCh37/h19 using the Bismark alignment tool.
Targeted bisulfite sequencing
DNA methylation of target genes was evaluated by Targeted Bisulfite Sequencing (MethylTarget) performed in Genesky Biotechnologies Inc. (Shanghai, China). The primers used for selected genes were listed in Table S2. The MethylTarget assay was described clearly as reported16,17.
Reverse transcription and RQ-PCR
Reverse transcription was carried out using random primers as reported15,16. Determination of selected gene expression was detected by real-time quantitative PCR (RQ-PCR) using AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Piscataway, NJ). ABL1 examined by 2×SYBR Green PCR Mix (Multisciences, Hangzhou, China) was applied to calculate the abundance of mRNA expression. The primers used for selected genes were given in Table S2. Relative mRNA expression was calculated by 2−∆∆CT method.
Bisulfite modification and RQ-MSP
Genomic DNA was bisulfite converted as our previous report15,16. The methylation level of selected genes was detected by RQ-MSP with primers listed in Table S2. ALU was utilized to calculate the abundance of gene methylation level. Relative gene methylation level was calculated using 2−∆∆CT method.
Cell lines and cell culture
The MDS-derived AML cell-line SKM-1 were cultured in RPMI 1640 medium containing 10% fetal calf serum (ExCell Bio, Shanghai, China) and grown at 37 °C in 5% CO2 humidified atmosphere12,13.
Demethylation studies
SKM-1 cells at a density of 5 × 105 cells/ml in 2 ml were treated with 5-aza-2′-deoxycytidine (5-aza-dC) with a final concentration of 0 μM, 1 μM, 2 μM, and 10 μM during 4 days.
Cell transfection
Human full-length ZNF300 CDS sequences were introduced into the BamHI/AgeI of GV569 (Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin) vector (GENECHEM, Shanghai, China), and were transfected with lentivirus (Lv).
Western blot analysis
Western blot was carried out as described10,18. The antibodies were rabbit anti-ZNF300 (Sigma-Aldrich, St. Louis, MO) and mouse anti-GAPDH (BOSTER, Wuhan, China).
Cell growth assays
The transfected cells were seeded in 96-well plates at a density of 5 × 103 cells per well in triplicate. After culture for 0 h, 24 h, 48 h, 72 h, and 96 h, CCK-8 was added to each well and incubated for 2 h. The absorbance at 450 nm was measured using a microplate reader. The rate of cell growth was calculated as OD value.
Cell apoptosis analysis
The transfected SKM-1 cells were cultured with serum-free medium for 48 h in 6-well plates (5 × 105 cells/well). Annexin V-APC Kit was used to analyze the apoptosis rate according to the manufacturer’s protocols and then analyzed via flow cytometry.
Cell cycle analysis
The transfected SKM-1 cells were harvested after 48 h incubation with a density of 5 × 105 cells per well in 6-well plates in triplicate. After fixed in 70% ice-cold ethanol in PBS, the cells were washed twice and added with propidium iodide to analyze the cell cycle distribution according to the manufacturer’s protocols via flow cytometry.
Statistical analysis
SPSS 22.0 and GraphPad Prism 5.0 were the main software package in statistical analysis. Independent T/Paired T/Mann–Whitney’s U-tests were applied for the comparison of continuous variables, whereas Pearson Chi-square/Fisher exact tests were used for the comparison of categorical variables. Spearman correlation test was performed to analysis the correlation between ZNF300 expression and methylation. The receiver operating characteristic (ROC) curve and area under the ROC curve (AUC) were applied to assess the discriminative capacity of ZNF300 methylation between AML patients and controls. The impact of ZNF300 methylation on complete remission (CR) was tested by univariate and multivariate logistic regression models. The prognostic effects of ZNF300 expression on leukemia-free survival (LFS) and overall survival (OS) were analyzed through Kaplan–Meier analysis (Log-rank test) and Cox regression analysis (univariate and multivariate proportional hazard regression). Two-tailed P-values < 0.05 in all analysis was considered as statistically significant differences.
Results
Genome-wide methylation analysis of paired MDS/sAML patients
To identify epigenetic alterations occurring in MDS progression, genome-wide methylation pattern was explored using RRBS in four newly diagnosed MDS patients and the matched patients at sAML stage. In addition, four healthy donors were also included as normal controls. The sequencing data were submitted to NCBI SRA databases with an accession number PRJNA670308. A mean of 2.2 × 107 total pair reads per sample were produced from RRBS libraries and mapped to the reference human genome (UCSC hg19) using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/). The mapping efficiency of sequenced reads was about 58% of each sample. A mean of 7.5 × 108 cytosines were analyzed per sample and about 17% (1.2 × 108) of these cytosines were located in CpG sites. RRBS data quality was shown in Table S3.
The mean global methylation in controls ranged from 45.4% to 51.0%, similar to the methylation patterns for MDS/sAML patients ranging from 36.8% to 53.1% (Table S3 and Fig. S1). However, in four paired MDS/sAML patients, cases at sAML stage exhibited significantly increased methylation level when compared with their matched MDS stage (Fig. 1). Hierarchical clustering of controls and MDS/sAML patients based on CpG site methylation separated MDS/sAML patients from controls (Fig. S1).

The percentage of methylated CpG sites in controls and MDS/sAML was demonstrated. P-values were calculated using the Paired T-test.
Differential methylation landscapes in paired MDS/sAML patients
Mspl fragments (40–220 bp) rather than individual CpG sites or a tiled window approach were regarded as the basic analysis units as described previously19,20,21. The fragments that passed statistical significance (paired/independent T test-P < 0.05, and also had >25% mean methylation difference) were considered as differentially methylated fragments (DMFs). We identified 1090 DMFs (441 hypermethylated and 649 hypomethylated) between MDS and controls (MDS vs. controls), which may be seen as molecular events contributing to MDS pathogenesis. Moreover, a total of 103 DMFs (96 hypermethylated and 7 hypomethylated) were identified between sAML and MDS (sAML vs. MDS), seen as molecular events contributing to MDS progression (Fig. 2). A detailed description of these DMFs was given in Table S4 and Table S5.

MDS pathogenesis indicated the difference compared between MDS and controls. MDS progression indicated the difference compared between sAML and MDS. The fragments/genes that passed statistical significance (paired/independent T test-P < 0.05, and also had >25% mean methylation difference) were considered as differentially methylated fragments/genes.
Next, CGI, promoter (±2000 bp from transcription start site), and gene body were also used as the unit of analysis, respectively. For CGI, we identified 74 differentially methylated genes (DMGs) (37 hypermethylated and 37 hypomethylated) in MDS vs. controls, which may be involved in MDS pathogenesis, and 32 DMGs (32 hypermethylated) in sAML vs. MDS, involved in MDS progression (Fig. 2). Secondly, for promoter, we discovered 54 DMGs (22 hypermethylated and 32 hypomethylated) in MDS vs. controls and 9 DMGs (9 hypermethylated) in sAML vs. MDS (Fig. 2). Furthermore, for gene body, the numbers were 1023 DMGs (144 hypermethylated and 879 hypomethylated in MDS vs. controls) and 26 DMGs (23 hypermethylated and 3 hypomethylated in sAML vs. MDS) respectively (Fig. 2). More detailed descriptions of these DMGs were illustrated in Tables S4 and S5.
Comparison of methylome of independent paired MDS/sAML patients
Since each of the four paired MDS/sAML patients contained distinct epigenomes, we further performed differential methylation analysis on each pairs independently. Using the promoter as the unit of analysis, we identified 5803, 3735, 4043, and 2748 DMGs (fragments annotated as genes with >25% mean methylation difference) between MDS and sAML stage in four individual paired patients (Fig. 3). A total of 4558 genes were abnormally methylated in at least two of the samples, while 2130 genes in at least three of the samples and 634 genes in all four samples (Fig. 4).

The circos plot represented methylation values in MDS/sAML patients. Outer ring indicated MDS sage, inner ring indicated sAML stage. The fragments/genes that passed statistical significance (paired/independent T test-P < 0.05, and also had >25% mean methylation difference) were considered as differentially methylated fragments/genes.

The circos plot represented methylation values in MDS/sAML patients. Outer ring indicated MDS sage, inner ring indicated sAML stage. Inside lines indicated genes that differentially methylated in at least two of the patients. The fragments/genes that passed statistical significance (paired/independent T test-P < 0.05, and also had >25% mean methylation difference) were considered as differentially methylated fragments/genes.
Identification and validation of DMFs by targeted bisulfite sequencing in MDS/AML patients
In order to identify and validate the candidate DMFs involved in MDS progression, we screened a series of DMFs (paired/independent T test-P < 0.05, and also had >10% mean methylation difference) shared in sAML vs. MDS and sAML vs. controls, and further annotated as DMGs (Fig. 5a and Table S6). A total of 486 genes were identified and five genes (GFRA1, IRX1, NPY, PRRT4, and ZNF300) were selected, of which may have biological functions in cancers analyzed by Coremine analysis (http://www.coremine.com/medical/#search), for further targeted bisulfite sequencing in additional 35 MDS, 111 AML patients and 25 controls using MethylTarget (GENESKY, Shanghai, China)17. The mean bait coverage attached 1694×, and Q30 was 75.56%17. As showed in Fig. 5b, the methylation level of GFRA1, IRX1, NPY, and ZNF300 were markedly increased in MDS and AML patients compared with controls (Fig. 5c). Moreover, GFRA1, NPY, and ZNF300 methylation level in AML patients also significantly higher than that in MDS patients (Fig. 5c).

a The flowchart of the differentially methylated genes screening. The fragments that passed statistical significance (paired/independent T test-P < 0.05, and also had >10% mean methylation difference) were considered as differentially methylated fragments/genes. b Heatmaps summarizing differentially methylated fragments/genes in MDS progression. c The methylation level of the candidate genes in additional samples of controls (n = 25), de novo MDS (n = 35) and AML patients (n = 111) analyzed by targeted bisulfite sequencing. P-values were calculated using the Mann–Whitney U-test. NS: no significance; *P < 0.05; **P < 0.01; ***P < 0.001.
Transcriptional regulatory effects on mRNA expression of the candidate genes methylation
To confirm the transcriptional regulatory effects on gene expression influenced by DNA methylation, we further evaluated the candidate gene expression in clinical samples and MDS-derived AML cell-line SKM-1 before and after the treatment with demethylation agent 5-aza-dC. Overall, IRX1, NPY, and ZNF300 expression were significantly downregulated in MDS and AML patients (Fig. 6a). Moreover, IRX1 and ZNF300 expression were negatively associated with gene methylation (Fig. 6b). In addition, a significantly increased expression of ZNF300 and NPY were observed after 5-aza-dC demethylation treatment (Fig. 6c).

a The expression level of the candidate genes in de novo MDS and AML patients by real-time quantitative PCR. P-values were calculated using the Mann–Whitney U-test. b The correlation between the candidate genes methylation and genes expression. The correlation was analyzed by Spearman correlation test. c The expression of the candidate genes in MDS-derived AML cell line SKM-1 before and after 5-aza-dC treatment. P-values were calculated using the independent T-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Further confirmation of ZNF300 methylation in a larger cohort of MDS/AML patients
To further investigate the value of ZNF300 methylation in clinical diagnosis and risk/treatment assessment, we expanded the clinical samples (70 MDS and 160 AML) to explore the clinical significance of ZNF300 methylation through a more rapid and inexpensive methodology—RQ-MSP. Primers for RQ-MSP were designed containing in the targeted sequencing primers (Fig. 7a). RQ-MSP results were highly correlated with that in the targeted sequencing (Fig. 7b). Patients with MDS and AML exhibited a markedly higher ZNF300 methylation level than controls, and ZNF300 methylation level in AML patients was even much higher than that in MDS patients (Fig. 7c). ROC curve analysis revealed that ZNF300 methylation could be used to segregate AML from controls with an AUC value of 0.832 (95% CI: 0.777–0.887, Fig. 7d). For the purpose of investigating the clinical relevance of ZNF300 methylation in MDS and AML patients, the patients were divided into two groups (ZNF300 hypermethylated and non-hypermethylated) based on the cutoff value of 0.408 (set as “mean + 2 SD” in controls). As presented in Tables 1 and 2, ZNF300 methylation tended to correlate with U2AF1 and SRSF2 mutations in MDS (P = 0.066 and 0.090, respectively), whereas its hypermethylation was associated with lower platelets and higher proportion of male patients/CEBPA mutations in AML (P = 0.002, 0.032, and 0.066, respectively).

a The genomic coordinates of ZNF300 promoter region CpG island and primer locations. The panel plots the GC content as a percentage of the total. Each vertical bar in the bottom panel represents the presence of a CpG dinucleotide. Black horizontal bars indicate regions amplified by targeted bisulfite sequencing primer pairs and RQ-MSP primer pairs. This figure was created using CpGplot (http://emboss.bioinformatics.nl/cgi-bin/emboss/cpgplot) and Methyl Primer Express v1.0 software. TSS: transcription start site; RQ-MSP: real-time quantitative methylation-specific PCR. b The correlation of the candidate gene methylation results between the targeted bisulfite sequencing and RQ-MSP. The correlation was analyzed by Spearman correlation test. c The methylation level of the ZNF300 in larger samples of controls (n = 46), de novo MDS (n = 70) and AML patients (n = 170) analyzed by targeted bisulfite sequencing. P-values were calculated using the Mann–Whitney U-test. d ROC curve analysis using ZNF300 methylation for discriminating AML patients from controls. (e): The impact of ZNF300 methylation on leukemia-free survival and overall survival of MDS and AML patients. Survival was analyzed through Kaplan–Meier analysis using Log-rank test.
Prognostic effect of ZNF300 methylation in MDS/AML patients
In MDS patients, Kaplan–Meier analyses indicated that ZNF300 hypermethylated cases exhibited significantly shorter OS and LFS time than ZNF300 non-hypermethylated cases (P = 0.022 and 0.010, respectively) (Fig. 7e). Moreover, Cox regression analyses also confirmed the independent prognostic effect of ZNF300 methylation on OS and LFS (P = 0.037 and 0.038, respectively) (Table S7). In AML patients, we first evaluated the association between ZNF300 methylation and CR. Cases with ZNF300 hypermethylation had lower CR rate as compared with cases with ZNF300 non-hypermethylation among total and non-M3 AML as well as cytogenetically normal AML (CN-AML) subgroups (P = 0.054, 0.073, and 0.049, respectively) (Table 2). Due to the significant associations of ZNF300 methylation with CR among CN-AML, Logistic regression analyses were further performed to confirm the effect of ZNF300 methylation on CR. After adjusting for the known factors, ZNF300 hypermethylation was a negative independent risk factor affecting CR in CN-AML patients (P = 0.015) (Table S8). Next, Kaplan–Meier analyses showed that ZNF300 hypermethylated cases exhibited shorter OS and LFS time compared with ZNF300 non-hypermethylated cases in both non-M3 AML (P = 0.039 and 0.054, respectively) and CN-AML (P = 0.005 and 0.035, respectively) (Fig. 7e). Additionally, Cox regression analysis revealed that ZNF300 hypermethylation was an independent prognostic factor affecting OS in both non-M3 AML and CN-AML patients (P = 0.013 and 0.018, respectively) (Table S9).
Biological role of ZNF300 in MDS-derived AML cell-line SKM-1
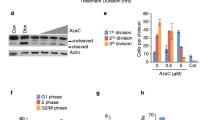
To investigate the underlying role of ZNF300 during MDS progression, we carried out gain-of-function experiments in MDS-derived AML cell-line SKM-1 in vitro. Firstly, we successfully established SKM-1 cells stably overexpressing ZNF300 by Lv-ZNF300 infection, which was determined by fluorescence, RQ-PCR, and western blot (Fig. 8a–c). Notably, SKM-1 cells overexpressing ZNF300 exhibited significantly lower growth rate than those without ZNF300 overexpression (Fig. 8d), and could cause G0/G1 arrest as well (Fig. 8e). Conversely, the apoptosis of SKM-1 cells were significantly promoted after ZNF300 overexpression (Fig. 8f).

a Fluorescence detection after LV-NC and LV-ZNF300 transfection in SKM-1. b Confirmation of ZNF300 mRNA level in SKM-1 after transfection by real-time quantitative PCR. c Confirmation of ZNF300 overexpression in SKM-1 after transfection by western blot. d The proliferation ability in SKM-1 affected by ZNF300 overexpression. e The cell cycle in SKM-1 affected by ZNF300 overexpression. f The apoptosis ability in SKM-1 affected by ZNF300 overexpression. P-values were calculated using the independent T-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Recent studies have made great progresses in understanding of the mechanism underlying MDS progression. Cytogenetic abnormalities, such as -7/7q-, +8, 6q-, 11q-, i(7q), 11q-, t(7;9), i(9q), and complex karyotypes play crucial roles in MDS evolution22,23. With the rapid advances in sequencing technologies, somatic mutations were also identified to contribute to disease progression in MDS, such as genes encoding transcription factors (ETV6 and TP53), epigenetic regulators [DNMT3A (methylation), TET2 and IDH1/2 (hydroxymethylation), EZH2 and ASXL1 (histones modifications)], and splicing factors (U2AF1)22,23. Recently, Xu et al. by whole-exome and targeted sequencing revealed that ROBO1/2 mutations acted as progression-related drivers in MDS7. In addition to genetic alterations, epigenetic modifications, especially for DNA methylation were also reported to be participated in cancer progression including MDS24,25,26. Previous studies almost focused on the single gene change during MDS progression, such as CDKN2B, SOCS1, NR4A2, ABAT, ID4, GPX3, SOX30, and so on9,10,11,27,28,29,30. However, few studies demonstrated the whole-genome DNA methylation alterations during MDS progression.
In this study, we for the first time used RRBS in four paired MDS/sAML patients to identify the methylation-associated epigenetic drivers underlying MDS progression. We identified and verified that whole-genome DNA hypermethylation was a frequent phenomenon during MDS progression. Moreover, as analyzed in details, the number of hypermethylated fragments was larger than hypomethylated fragments during MDS progression, but not presented during MDS incidence. Previously studies also exhibited that aberrant methylation existed in every MDS sample, on average affecting 91 of 1505 CpG loci in early MDS and 179 of 1505 loci after blast transformation26. Figueroa et al. reported that abnormal methylation in MDS and sAML tended to affect particular chromosomal regions, occurred more frequently in Alupoor genes, and included prominent involvement of genes involved in the WNT and MAPK signaling pathways25. In the current study, although some of these genes methylation (such as S0X17 and RAP1GAP) were previously reported in MDS31,32, the majority of them were unknown so far. Notably, several genes were reported to be methylated in other hematologic malignancies or solid tumors, such as HOXD11, GBX2, CRMP1, RBM47, NHLRC1, WNT2, TUSC3, NRG1, TSPYL5, CNTFR, NR4A3, PHOX2A, KCNA5, PTGDR, HS3ST2, CLDN7, CPT1C, and NKX2-433,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48. Based on these findings, we further selected the unreported hypermethylated genes with potential roles involving in MDS progression for further validation. The targeted bisulfite sequencing in additional expanded de novo MDS and AML samples identified GFRA1, IRX1, NPY, and ZNF300 were frequently hypermethylated in these patients. Moreover, GFRA1, NPY, and ZNF300 methylation were more frequently happened in AML patients than in MDS patients, suggesting that the aberrant hypermethylation of these genes played vital roles in MDS transformation.
Assignment of the DMFs to genomic regions demonstrated that DMFs were mostly located in or around CpG islands, suggesting that these DMFs might play a role in the regulation of transcriptional gene expression as methylation of CpG islands located in promoter were strongly associated with gene silencing. Nevertheless, it was a pity that we did not obtain enough RNA samples corresponding to these paired MDS/sAML patients for genome-wide gene expression sequencing in our study. We only detected the expression level of the selected genes (GFRA1, IRX1, NPY, and ZNF300) in de novo MDS and AML samples by conducting RQ-PCR. IRX1, NPY and ZNF300 expression were significantly reduced in de novo MDS and AML patients, and only IRX1 and ZNF300 expression were associated with their gene methylation analyzed by targeted bisulfite sequencing. Additionally, epigenetic studies further confirmed that ZNF300 expression was significantly associated with ZNF300 methylation in MDS. These results, in general, suggested that ZNF300 methylation functioned in MDS progression mainly via downregulating ZNF300 expression.
The direct role and clinical implication of ZNF300 in MDS and AML remain poorly investigated. Cai et al. demonstrated that ZNF300 knockdown inhibited forced megakaryocytic differentiation by phorbol and erythrocytic differentiation by arabinofuranosyl cytidine in chronic myeloid leukemia cell line K56249. Our study further exhibited the direct role of ZNF300 in MDS-derived AML cell-line SKM-1 with potential anti-proliferative and pro-apoptotic effects. In addition to hematologic malignancies, ZNF300 gene overexpression enhanced growth and metastasis of cancer cells through activating NF-κB pathway in cervical cancer50. Moreover, we for the first time determined clinical implication of ZNF300 methylation in de novo MDS and AML patients, and found that ZNF300 methylation was a potential biomarker helpful for diagnosis in AML. Furthermore, ZNF300 methylation could act as an independent prognostic biomarker affecting LFS, OS, and CR in MDS and/or AML. Taken all the results together, it is inferred that ZNF300 may act as a potential therapeutic target in MDS and AML against disease progression, and using ZNF300-based targeted therapy could improve the clinical outcome for MDS and AML patients. Since this was the first report regarding ZNF300 in MDS and AML, prospective studies and integrative analysis are needed before the promising biomarkers can be routinely used for risk stratification and planning therapy in MDS and AML.
In summary, by using next generation sequencing, it was apparently revealed that genome-wide DNA hypermethylation were frequent events during MDS progression. Among these changes, ZNF300 methylation through regulating ZNF300 expression acted as an epigenetic driver in MDS progression. These findings provided a theoretical basis for the usage of demethylation drugs in MDS patients against disease progression and opened up new insights for targeted therapy in MDS.
References
Adès, L., Itzykson, R. & Fenaux, P. Myelodysplastic syndromes. Lancet 383, 2239–2252 (2014).
Garcia-Manero, G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am. J. Hematol. 90, 831–841 (2015).
Ogawa, S. Genetics of MDS. Blood 133, 1049–1059 (2019).
Ganguly, B. B. & Kadam, N. N. Mutations of myelodysplastic syndromes (MDS): an update. Mutat. Res. Rev. Mutat. Res. 769, 47–62 (2016).
Haferlach, T. et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247 (2014).
Papaemmanuil, E. et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627 (2013).
Xu, F. et al. Whole-exome and targeted sequencing identify ROBO1 and ROBO2 mutations as progression-related drivers in myelodysplastic syndromes. Nat. Commun. 6, 8806 (2015).
Heuser, M., Yun, H. & Thol, F. Epigenetics in myelodysplastic syndromes. Semin. Cancer Biol. 51, 170–179 (2018).
Zhou, J. D. et al. GPX3 methylation in bone marrow predicts adverse prognosis and leukemia transformation in myelodysplastic syndrome. Cancer Med. 6, 267–274 (2017).
Zhou, J. D. et al. Epigenetic dysregulation of ID4 predicts disease progression and treatment outcome in myeloid malignancies. J. Cell. Mol. Med. 21, 1468–1481 (2017).
Zhou, J. D. et al. Identification and validation of SRY-box containing gene family member SOX30 methylation as a prognostic and predictive biomarker in myeloid malignancies. Clin. Epigenetics 10, 92 (2018).
Nakagawa, T. & Matozaki, S. The SKM-1 leukemic cell line established from a patient with progression to myelomonocytic leukemia in myelodysplastic syndrome (MDS)-contribution to better understanding of MDS. Leuk. Lymphoma 17, 335–339 (1995).
Nakagawa, T. et al. Establishment of a leukaemic cell line from a patient with acquisition of chromosomal abnormalities during disease progression in myelodysplastic syndrome. Br. J. Haematol. 85, 469–476 (1993).
Vardiman, J. W. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951 (2009).
Zhang, T. J. et al. H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin. Epigenetics 10, 47 (2018).
Xu, Z. J. et al. Reduced protocadherin17 expression in leukemia stem cells: the clinical and biological effect in acute myeloid leukemia. J. Transl. Med. 17, 102 (2019).
Zhang, T. J. et al. Identification and validation of prognosis-related DLX5 methylation as an epigenetic driver in myeloid neoplasms. Clin. Transl. Med. 10, e29 (2020).
Lin, J. et al. Arresting of miR-186 and releasing of H19 by DDX43 facilitate tumorigenesis and CML progression. Oncogene 37, 2432–2443 (2018).
Chatterjee, A. et al. Placental hypomethylation is more pronounced in genomic loci devoid of retroelements. G3. 6, 1911–1121 (2016).
Stockwell, P. A., Chatterjee, A., Rodger, E. J. & Morison, I. M. DMAP: differential methylation analysis package for RRBS and WGBS data. Bioinformatics 30, 1814–1822 (2014).
Chatterjee, A., Stockwell, P. A., Rodger, E. J. & Morison, I. M. Genome-scale DNA methylome and transcriptome profiling of human neutrophils. Sci. Data 3, 160019 (2016).
Dan, C., Chi, J. & Wang, L. Molecular mechanisms of the progression of myelodysplastic syndrome to secondary acute myeloid leukaemia and implication for therapy. Ann. Med. 47, 209–217 (2015).
Bravo, G. M., Lee, E., Merchan, B., Kantarjian, H. M. & García-Manero, G. Integrating genetics and epigenetics in myelodysplastic syndromes: advances in pathogenesis and disease evolution. Br. J. Haematol. 166, 646–659 (2014).
Qin, T. et al. Risk of disease progression in low-risk MDS is linked to distinct epigenetic subtypes. Leukemia 33, 2753–2757 (2019).
Figueroa, M. E. et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114, 3448–3458 (2009).
Jiang, Y. et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113, 1315–1325 (2009).
Tien, H. F. et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br. J. Haematol. 112, 148–154 (2001).
Wu, S. J. et al. Clinical implications of SOCS1 methylation in myelodysplastic syndrome. Br. J. Haematol. 135, 317–323 (2006).
Wong, Y. F. et al. Longitudinal analysis of DNA methylation in CD34+ hematopoietic progenitors in myelodysplastic syndrome. Stem Cells Transl. Med. 3, 1188–1198 (2014).
Zhao, G. et al. High methylation of the 4-aminobutyrate aminotransferase gene predicts a poor prognosis in patients with myelodysplastic syndrome. Int. J. Oncol. 54, 491–504 (2019).
Fan, R. et al. Abnormal methylation of the sex-determining region Y-box 17 (SOX17) promoter predicts poor prognosis in myelodysplastic syndrome. Clin. Lab. 60, 1465–1474 (2014).
Ding, W. J. et al. Methylation level of Rap1GAP and the clinical significance in MDS. Oncol. Lett. 16, 7287–7294 (2018).
Furuta, J. et al. Silencing of peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res 66, 6080–6086 (2006).
Jeyapala, R. et al. GBX2 Methylation is a novel prognostic biomarker and improves prediction of biochemical recurrence among patients with prostate cancer negative for intraductal carcinoma and cribriform architecture. Eur. Urol. Oncol. 2, 231–238 (2019).
Ammerpohl, O. et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int. J. Cancer 130, 1319–1328 (2012).
Tirosh, A. et al. Distinct genome-wide methylation patterns in sporadic and hereditary nonfunctioning pancreatic neuroendocrine tumors. Cancer 125, 1247–1257 (2019).
Li, Y. et al. Novel methylation-driven genes identified as prognostic indicators for lung squamous cell carcinoma. Am. J. Transl. Res. 11, 1997–2012 (2019).
Farkas, S. A., Vymetalkova, V., Vodickova, L., Vodicka, P. & Nilsson, T. K. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes. Epigenomics 6, 179–191 (2014).
Burgermeister, E. et al. Epigenetic silencing of tumor suppressor candidate 3 confers adverse prognosis in early colorectal cancer. Oncotarget 8, 84714–84728 (2017).
Chua, Y. L. et al. The NRG1 gene is frequently silenced by methylation in breast cancers and is a strong candidate for the 8p tumour suppressor gene. Oncogene 28, 4041–4052 (2009).
Kumar, S. R., Bryan, J. N., Esebua, M., Amos-Landgraf, J. & May, T. J. Testis specific Y-like 5: gene expression, methylation and implications for drug sensitivity in prostate carcinoma. BMC Cancer 17, 158 (2017).
Kober, P., Bujko, M., Olędzki, J., Tysarowski, A. & Siedlecki, J. A. Methyl-CpG binding column-based identification of nine genes hypermethylated in colorectal cancer. Mol. Carcinog. 50, 846–856 (2011).
Shimizu, R. et al. Possible role of intragenic DNA hypermethylation in gene silencing of the tumor suppressor gene NR4A3 in acute myeloid leukemia. Leuk. Res. 50, 85–94 (2016).
Ryland, K. E. et al. Promoter methylation analysis reveals that KCNA5 ion channel silencing supports Ewing sarcoma cell proliferation. Mol. Cancer Res. 14, 26–34 (2016).
Kim, H. J. et al. Whole genome MBD-seq and RRBS analyses reveal that hypermethylation of gastrointestinal hormone receptors is associated with gastric carcinogenesis. Exp. Mol. Med. 50, 156 (2018).
Huang, R. L. et al. Epigenetic loss of heparan sulfate 3-O-sulfation sensitizes ovarian carcinoma to oncogenic signals and predicts prognosis. Int. J. Cancer 143, 1943–1953 (2018).
Li, Y. et al. Downregulation of CLDN7 due to promoter hypermethylation is associated with human clear cell renal cell carcinoma progression and poor prognosis. J. Exp. Clin. Cancer Res. 37, 276 (2018).
Bell, A., Bell, D., Weber, R. S. & El-Naggar, A. K. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer 117, 2898–2909 (2011).
Cai, J. et al. ZNF300 knockdown inhibits forced megakaryocytic differentiation by phorbol and erythrocytic differentiation by arabinofuranosyl cytidine in K562 cells. PLoS ONE 9, e114768 (2014).
Wang, T. et al. Overexpression of the human ZNF300 gene enhances growth and metastasis of cancer cells through activating NF-kB pathway. J. Cell. Mol. Med. 16, 1134–1145 (2012).
Acknowledgements
The work was supported by National Natural Science Foundation of China (81900166, 81900163, 81970118, 81970156), Medical Innovation Team of Jiangsu Province (CXTDB2017002), Natural Science Foundation of Jiangsu Province for Youths (BK20180280), Zhenjiang Clinical Research Center of Hematology (SS2018009), Social Development Foundation of Zhenjiang (SH2017040, SH2018044, SH2019065, SH2019067, SH2020055), Scientific Research Project of The Fifth 169 Project of Zhenjiang (21), Youth Medical Talents Project of “Ke Jiao Qiang Wei” Project of Jiangsu Province (QNRC2016450), Medical Field of Zhenjiang “Jin Shan Ying Cai” Project, Scientific Research Foundation of Affiliated People’s Hospital of Jiangsu University for PhD (KFB202002). We are also very grateful for the technical support by Genesky Biotechnologies Inc., Shanghai, 201315.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Amelio
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, Jd., Zhang, Tj., Xu, Zj. et al. Genome-wide methylation sequencing identifies progression-related epigenetic drivers in myelodysplastic syndromes. Cell Death Dis 11, 997 (2020). https://doi.org/10.1038/s41419-020-03213-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03213-2
- Springer Nature Limited
This article is cited by
-
State-transition modeling of blood transcriptome predicts disease evolution and treatment response in chronic myeloid leukemia
Leukemia (2024)
-
Genome-wide analysis toward the epigenetic aetiology of myelodysplastic syndrome disease progression and pharmacoepigenomic basis of hypomethylating agents drug treatment response
Human Genomics (2023)
-
The validation and clinical significance of LPCAT1 down-regulation in acute myeloid leukemia
Molecular Biology Reports (2023)
-
All-trans retinoic acid enhances the cytotoxic effect of decitabine on myelodysplastic syndromes and acute myeloid leukaemia by activating the RARα-Nrf2 complex
British Journal of Cancer (2023)
-
SLIT2 promoter hypermethylation predicts disease progression in chronic myeloid leukemia
European Journal of Medical Research (2022)