
Abstract
Background
DNA methylation biomarkers for early detection, risk stratification and treatment response in cancer have been of great interest over the past decades. Nevertheless, clinical implementation of these biomarkers is limited, as only < 1% of the identified biomarkers is translated into a clinical or commercial setting. Technical factors such as a suboptimal genomic location of the assay and inefficient primer or probe design have been emphasized as important pitfalls in biomarker research. Here, we use eleven diagnostic DNA methylation biomarkers for colorectal cancer (ALX4, APC, CDKN2A, MGMT, MLH1, NDRG4, SDC2, SFRP1, SFRP2, TFPI1 and VIM), previously described in a systematic literature search, to evaluate these pitfalls.
Results
To assess the genomic assay location, the optimal genomic locations according to TCGA data were extracted and compared to the genomic locations used in the published assays for all eleven biomarkers. In addition, all primers and probes were technically evaluated according to several criteria, based on literature and expert opinion. Both assay location and assay design quality varied widely among studies.
Conclusions
Large variation in both assay location and design hinders the development of future DNA methylation biomarkers as well as inter-study comparability.
Similar content being viewed by others

Background
DNA methylation biomarkers for early detection, risk stratification and treatment response have been of great interest in the clinical management of cancer. Over the past decades, the focus in DNA methylation biomarkers research has expanded from tissue to liquid biopsies. Since then, some of these biomarkers have been incorporated in commercially available diagnostic tests [1]. In a recent systematic literature review, 100 potentially published DNA methylation biomarkers for colorectal cancer (CRC) were identified in bodily fluids (Feng et al. unpublished data). Only three of these (NDRG4, BMP3 and SEPT9) have been translated into commercial tests currently available for the early detection of CRC [1]. Various reasons for this suboptimal clinical translation have been postulated [1,2,3]; many of these focus on issues such as a suboptimal study design, lack of validation and lack of clinical relevance. However, technical factors such as a suboptimal genomic location of the assay and inefficient primer or probe design have been emphasized as important pitfalls in biomarker research as well [1, 3,4,5]. The choice of which genomic location to study in the evaluation of DNA methylation biomarkers can influence the conclusion on the clinical value of this biomarker. Koch et al. previously described the importance of selecting the optimal genomic location, for example, by using publicly available data such as The Cancer Genome Atlas (TCGA) or whole-genome sequencing data [1, 6]. These data can be used to identify the genomic location with the largest methylation differences between sample groups, associated with the clinical outcome of interest. For example, we assume that the genomic locations with the largest difference in methylation between normal and tumor samples can be used to discriminate tumor tissue/patients from normal tissue/healthy individuals, as suggested in several of our previous publications [1, 7, 8].
In addition to the identification of these extracted locations with the largest difference between normal and tumor tissue, several technical assay design issues are crucial for optimal DNA methylation biomarker development and subsequent chances for successful clinical translation, including assay type and primer- and probe design. For DNA methylation analysis, the most widely used technique is (quantitative) methylation-specific PCR (MSP/qMSP), which requires primer and probe design on the bisulfite-converted sequence of the biomarker of interest [9]. Although MSP primer design tools (including Bisearch, Methprimer and PrimerSuite) are available [10], these tools do not incorporate publicly available genomic data, and therefore do not preselect the most optimal genomic region for assay design.
Here, we analyzed the diagnostic CRC methylation biomarkers identified in a previously conducted systematic literature search in order to provide an overview of the genomic locations. Moreover, we evaluated the quality of the described primers and probes and define recommendations that can guide assay design within the DNA methylation biomarker field.
Results and discussion
Dataset characteristics
Here, we provided an overview of the studied genomic locations, the extracted locations according to TCGA data, and the quality of used primers and probes for the 11 most studied diagnostic DNA methylation biomarkers in CRC (ALX4, APC, CDKN2A, MGMT, MLH1, NDRG4, SDC2, SFRP1, SFRP2, TFPI2, VIM). All genes were evaluated in a minimum of five (TFPI2) and a maximum of 12 (SFRP2) independent studies (Table 1). Markers had been studied in a variety of bodily fluids including stool, serum, plasma and urine. Diagnostic performance (sensitivity and specificity) showed considerable variation between individual studies evaluating the same marker, which might be attributed to sample type differences. MGMT showed the largest sensitivity range of 5.7–90.0% across sample types, with specificities varying from 93.8 to 100%. MLH1 showed the smallest range in sensitivity (30.0–45.1%); however, the specificity range was substantial (56.9–97.6%). Despite using identical assays, diagnostic performance of these studies varied widely; e.g., 20–80% sensitivity and 96.8–100% specificity for CDKN2A, 60–94.2% sensitivity and 54–100% specificity for SFRP2 and 32.6–81% sensitivity and 82–100% specificity for VIM (Fig. 1C, I, K). This might be attributed to sample type differences, as illustrated by the relatively low sensitivities of CDKN2A methylation in stool (20–40%), compared to serum (59–80%), plasma (61.1%) and peripheral blood (55.4%) using the same assay (Table 1; Fig. 1C). Similarly, the diagnostic performance of sFRP2 in stool varied more widely (sensitivity 60–94.2%, specificity 54–100%) compared to serum (sensitivity 66.9–86.8%, specificity 93.7%) using the same assay (Table 1; Fig. 1I). As stool contains PCR inhibitors like complex polysaccharides and bile salts, undigested debris, and an abundance of, e.g., bacterial DNA over human DNA, this can explain the lack of performance in these samples, compared to blood-based samples [11]. In addition, plasma seems to perform worse compared to serum using the same assay for most markers (Table 1; Fig. 1), which is in line with literature, suggesting that DNA is more abundant and stable in serum compared to plasma [12, 13]. On the other hand, the diagnostic performances within one gene using the same sample type but different also differs widely, as illustrated by the sensitivities of, e.g., sFRP1 in plasma (Rasmussen et al. 21.8%; Bedin et al. 62.9%), CDKN2A in plasma (Rasmussen et al. 9.3%; Frattini et al. 61.1%) and NDRG4 in stool (Lu et al. 28.6%; Melotte et al. 61.0%; Xiao et al. and Park et al. 68.8–76.2%). Therefore, it seems that the performance of these biomarkers is influenced by both sample type and assay.


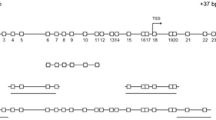
A–K Genomic locations, extracted CG’s (from TCGA) and diagnostic performances of the investigated assays per marker.
 : CpG islands,
: CpG islands,
 : CGs,
: CGs,
 : extracted CGs (obtained from TCGA),
: extracted CGs (obtained from TCGA),
 : primers,
: primers,
 : probes, TSS: transcription start site, Sens: sensitivity, Spec: specificity
: probes, TSS: transcription start site, Sens: sensitivity, Spec: specificity
Overview of genomic and extracted locations of selected assays
In the 11 most studied diagnostic DNA methylation biomarkers for CRC, multiple genomic locations were studied. An overview of all genomic locations in the individual studies is presented in Fig. 1. In addition, the extracted locations that we identified from TCGA data were compared to the locations used in all published assays.
For ALX4, APC, MGMT, sFRP1, sFRP2 TFPI2 and VIM, the least variation in genomic locations was observed among studies (three different genomic locations; Fig. 1A, B, D, H, I, J, K). For APC and VIM, most assays (4/5 and 5/7 respectively) included at least one of the extracted locations as identified in TCGA data (Fig. 1B, K). In contrast, none of the assays investigating ALX4, MGMT, sFRP1, sFRP2 and TFPI2 included an extracted CG (Fig. 1A, D, H, I, J). Although MLH1 and NDRG4 were studied in four and five genomic locations respectively, most assays did not contain an extracted CG (3/4 for MLH1, 3/5 for NDRG4; Fig. 1E, F). Largest variation in genomic location was observed for SDC2; however, none of these included an extracted CG (Fig. 1G).
These results show that there is a large variation in the investigated genomic locations among the different assays, whereas most studies do not specify a specific rationale for their used genomic location. Next to these variations in genomic location, we also observed a large variation in diagnostic performance even within the same genes. As previously postulated, the exact studied genomic location could influence the diagnostic performance of a biomarker, emphasizing the importance of considering genomic location of the assay upfront [1, 7, 8]. Currently, to our knowledge, no guidelines for identifying the optimal genomic location for diagnostic DNA methylation biomarkers are described. However, we previously recommended using TCGA data to identify the genomic location where the difference in methylation between normal and tumor tissue is largest. In theory, these locations might represent the most clinically relevant methylation sites for diagnostic purposes. Even though TCGA is a very accessible data source, it is limited in the amount of covered CGs. TCGA data is based on Infinium 450 K microarrays, of which the probes do not necessarily cover the most relevant CGs among the genome [1]. However, all genomic regions illustrated in this overview were covered by Illumina 450 K methylation array probes according to MEXPRESS. To assure full genomic coverage, sequencing prior to deciding on the genomic location covered in the methylation assay would be required. This has not always been feasible in the past, especially for small research groups with limited funding. The decreased sequencing costs and the availability of sequencing facilities (in both academic and commercial setting) combined with publicly available DNA methylation and gene expression data now provide opportunities to identify the most optimal genomic location for a DNA methylation marker [1]. Unfortunately, these sequencing data are rarely publicly available, which did not allow us to consider these in this manuscript.
Primer and probe assessment
Of the 47 assays used to measure the 11 included markers, 16 (34%) were MSP assays, 25 (53%) were qMSP assays with probe, and 6 (13%) were qMSP assays with SYBR (Fig. 2). As bisulfite-conversion changes unmethylated cytosines to uracil, while methylated cytosines remain unchanged, the CpG dinucleotides and non-CpG cytosines in the primers define the discriminative power of the primers to distinguish methylated from unmethylated DNA [9]. As an alternative to this damaging and fragmenting bisulfite conversion, an enzymatic modification kit to enable distinguishing methylated from unmethylated DNA has become available that is less damaging to the DNA in terms of fragmentation [56]. This novel enzymatic conversion could therefore impact assay design. However, as specific issues of, e.g., bisulfite conversion have been described in detail before [57], they are not evaluated in this manuscript.

Primer and probe quality assessment of all markers and studies in MSP (A), qMSP assays (probe/SYBR) (B). F: forward primer, R: reverse primer, P: probe, n/a: not applicable, *: Molecular beacon probe, additional bases included in evaluation, ˠ: Minor groove binder probe, X: probe sequence could not be mapped back to gene.
 : optimal design,
: optimal design,
 : suboptimal, but acceptable design,
: suboptimal, but acceptable design,
 : increased risk of technical problems with the primer or probe,
: increased risk of technical problems with the primer or probe,
 : not scorable because of molecular beacon probe
: not scorable because of molecular beacon probe
In the MSP assays, 1 forward and 1 reverse primer (6.25%) failed to include at least 2 CpG dinucleotides (Fig. 2A), whereas 2 forward primers and 1 reverse primer (4.8%) failed to include at least 2 CpG dinucleotides in the qMSP assays (Fig. 2B). All probes met this criterium (Fig. 2B). Additionally, in the MSP assays 2 forward primers and 1 reverse primer (9.4%), and 5 forward primers and 9 reverse primers (22.6%) in the qMSP assays failed to include at least 4 non-CpG cytosines (Fig. 2). Almost half of all probes (48%) in the qMSP assays with probe did not meet this criterium (Fig. 2B). Not meeting these criteria could lead to inefficient annealing and unspecific binding of the primers and probes, resulting in inconclusive findings. Inefficient annealing could result in false negatives due to the lack of amplification, even when the target sequence is available. Unspecific binding could result in false positives due to binding even when the target sequence is not fully complementary to the primer or probe [9, 58,59,60,61].
Next, 6 reverse primers (18.8%) in the MSP assays, and 6 forward and 14 reverse primers (32.2%) in the qMSP assays did not carry a CpG dinucleotide at the most 3’ end of the primer (Fig. 2). Not including a CpG dinucleotide at the most 3’ end of the primer might also result in inefficient or a lack of annealing, and unspecific binding, which could induce both false negative and false positive results [9, 58,59,60,61]. Optimal primer/probe length of 20–30 bases and 12–20 bases for minor groove binder (MGB) probes was met in 84.4% of the MSP primers, 69.4% of qMSP primers and 67% of the probes (Fig. 2). An additional 15.6% of the MSP primers, 29.2% of qMSP primers and 33% of probes were suboptimal in length (17–19 or 31–36 bases, < 12 or > 20 bases for MGB probes; Fig. 2). Among the probes suboptimal in length, 87.5% were molecular beacon probes. MGB probes generally allow a shorter probe sequence because of the increase in Tm by the MGB addition, which was accounted for in the results [62]. Molecular beacon probes carry an additional 5–7 bases complementary to each other at the start and end of the sequence, which means these probes are generally longer compared to Taqman probes [63]. In order to take these specific characteristics into account, the primer/probe length criterium was extended to 17–19 bases and to 31–36 bases. Nevertheless, 12.5% of these molecular beacon probes did not comply to the extended primer length criterium. This could potentially lead to inefficient or lack of annealing as well.
Eighty-one percent of MSP primer sets, and 55% of the qMSP primer sets had a similar melting temperature (Tm), meeting the criterium (i.e., Tm forward and reverse primers ≤ 2 °C difference). For the qMSP assays with Taqman probe, 75% of the probes met the criterium (i.e., Tm 5–10 °C higher than the corresponding primer set), and all of the MGB probes met the criterium (i.e., Tm 5–15 °C higher than the corresponding primer set; Fig. 2B). Not adhering to these Tm criteria could again lead to inefficient annealing of (one of) the primers or probe. For probes, an additional criterium was assessed (i.e., no G base at the most 5’ end of the probe), which was met in 95.8% of the included probes. A G base at the most 5’ end of the probe might prematurely quench the fluorophore, resulting in false negative results [64].
Last, optimal amplicon sizes of maximum 120 bp were used in 62.5% of the MSP assays, and 76.7% of the qMSP assays with probe or SYBR. An additional 25% of MSP assays and 13.3% of qMSP assays with probe or SYBR used suboptimal amplicon sizes (121–159 bp), and 12.5% of MSP assays, and 10% of qMSP assays exceeded the acceptable 160 bp amplicon size (Fig. 2). As DNA in liquid biopsies mostly originates from apoptotic and necrotic cells and in this case has to be bisulfite converted, it is highly fragmented with an estimated maximum of ~ 160 bp. However, depending on sample type, cell-free DNA in liquid biopsies can be as small as < 100 bp which should be taken into account when designing an assay [65,66,67,68,69].
Although assay design varied widely, the major criteria to distinguish methylated from unmethylated DNA were covered in most assays. However, several factors should receive additional consideration, such as primer length and Tm (Fig. 2). Probe design factors tend to score poorer compared to primer design factors, and generally, qMSP assays scored worse compared to MSP assays across all criteria (Fig. 2). In addition to the scored items, it is important to adhere to general primer/probe design criteria like a CG content of 30–80% and to ensure that no dimers or hairpin loops form [64, 70,71,72]. Further, it is important to consider genetic background and to make sure no prevalent single nucleotide polymorphisms (SNPs) appear at the 3’ end of the primer, to allow efficient annealing [73]. Moreover, assays including appropriate controls and a reference gene are most likely to generate reliable results [74].
To measure DNA methylation, several different techniques are currently available for research purposes, of which MSP and qMSP are most widely used. In general, qMSP assays with probe revealed more design flaws compared to both MSP and qMSP with SYBR assays. In the assays with a probe, especially the items regarding probe design showed low scores (Fig. 2). This emphasizes the difficulty of designing qMSP assays where the addition of a probe introduces another layer of complexity to the designing process. However, it can be questioned whether it is a necessity to fully optimize all separate subcomponents of primer and probe design, as assays with suboptimal scores for some criteria may also work. For example, if one of the primers in a set fails to meet the criterium of including ≥ 4–5 non-CpG cytosines per primer, the other primer could compensate, and the assay might work without any problems. This emphasizes that primers and probes should be designed as an assay, rather than single components.
Often, (nested) MSP assays are used in biomarker studies because they require substantially less DNA input compared to qMSP assays. Because of its quantitative nature and the specific binding properties of the utilized probe, qMSP with probe might be preferred over MSP assays for specific research questions. However, qMSP assays with SYBR are prone to false-positive results, as SYBR is an intercalating dye that binds to all double-stranded DNA [75].
After designing an assay, it is advised to perform an in silico analysis of this assay to check for dimers, hairpins and 3’-end primer stability, as extensively described by Davidović et al. [58]. In addition, assays should be optimized in terms of PCR conditions, such as PCR component concentrations and annealing temperature, using gradient PCRs. Bisulfite-converted fully methylated, fully unmethylated and no template controls, as well as non-converted DNA and a non-converted no template control should be used in the assay optimization process [76, 77]. Next, pilot studies using small sample sets of interest can evaluate the feasibility of an assay for cancer diagnosis, and minimize false positive and false negative results. Additionally, when analyzing quantitative data, it is important to select an appropriate cutoff value to determine whether a sample is methylated or unmethylated, and several methods to determine the optimal cutoff have previously been postulated [78, 79]. Different cutoffs among studies examining the same assays could, among others, explain the large variation in diagnostic performance, and could therefore hamper comparability of studies [78, 79].
Conclusions
In this study, using CRC markers as an example, we emphasized the importance of assay design for diagnostic DNA methylation biomarkers, indicating that a rational choice of genomic location and proper primer/probe design upfront are crucial when striving toward a clinically relevant and useful biomarker. This not only applies for diagnostic biomarkers, but for all DNA methylation markers intended to discriminate between two patient categories, such as prognostic and predictive biomarkers. However, only using the recommendations summarized in Box 1 does not guarantee a successful clinically relevant assay. Next to the factors discussed in this article, additional experimental factors can influence the diagnostic performance of DNA methylation biomarkers, such as sample type, quality and composition, assay amplicon size, and bisulfite conversion efficiency [11, 57, 80,81,82,83,84,85], as well as methodological factors such as sample size, using appropriate controls and statistical analyses. Nevertheless, considering both assay location and assay design upfront could greatly improve future DNA methylation biomarker development and inter-study comparability. To achieve this, future research should focus on linking the technical considerations discussed here to diagnostic parameters and clinical outcome. By optimizing these technical considerations in DNA methylation biomarker development, clinically relevant DNA methylation biomarkers are more likely to be developed.
Methods
Search strategy and study selection
A systematic search until December 2020 was performed in Pubmed, Embase, Cochrane library and Google Scholar, to identify all diagnostic DNA-methylation biomarker studies for CRC. Only original articles in the English language were considered; reviews, editorials and conference abstracts were excluded. Only articles studying DNA methylation through MSP (nested/direct) and qMSP (probe/SYBR), which provided the assay sequences in the article, and studied liquid biopsies (blood, serum, plasma, stool or urine) were included. Articles discussing hereditary cancer syndromes were excluded. From all diagnostic DNA methylation markers for CRC reported in the included studies, eleven were selected for further evaluation (ALX4, APC, CDKN2A, MGMT, MLH1, NDRG4, SDC2, SFRP1, SFRP2, TFPI1 and VIM) as they were described in at least five studies. Diagnostic performance (sensitivity and specificity) was extracted for all genes when available. Although it is one of the most commonly studied biomarkers for early CRC detection, SEPT9 was excluded due to the fact that most studies (62%) used one of the two commercial assays to measure SEPT9 methylation.
Identification optimal genomic location within TCGA data
In order to identify the genomic location where the methylation difference between normal and tumor tissue is the largest, the online available TCGA data visualization tool MEXPRESS [86, 87] was used. TCGA methylation data of the genes of interest in the CRC patient dataset (COAD) were assessed. MEXPRESS visualizes data for specific genes, and all Illumina 450 K methylation array CpGs that have been linked to that gene. All CpGs, irrespective of their location relative to the gene, were assessed. The three locations with the largest methylation difference between normal and tumor tissue (tumor hypermethylated compared to normal in all genes, except for MGMT) were extracted and will be referred to as ‘the extracted locations’ throughout this manuscript.
Primer and probe quality assessment
In order to assess primer and probe quality, two independent observers (M.M. & K.L.) scored all primers and probes according to criteria were constructed based on both literature and expert experience (Table 2). Although we are aware that designing the perfect primers and probes is challenging, and many different criteria have been postulated, we attempted to evaluate the optimal design criteria.
All criteria apply to (q)MSP primers and probes on the bisulfite-converted sequence of the gene of interest, and to the methylation-specific primer set in case of (q)MSP without probe. As bisulfite-conversion changes unmethylated cytosines to uracil, while methylated cytosines remain unchanged [9], primers and probes should be designed to distinguish methylated from unmethylated DNA and to anneal efficiently. Therefore, at least 2–3 CpG dinucleotides and 4–5 non-CpG cytosines should be included in the primer or probe [9, 58,59,60,61]. For optimal annealing, a CpG dinucleotide should be at the most 3’ end of each primer, and preferably the other CpGs are also at the 3’ end of the primer [9, 58,59,60,61]. Also, the ideal primer length is 20–30 bases [58], and preferably the forward and reverse primer should have a similar Tm (calculated using Gene Runner software). When using a probe, ideally it is 20–30 bases long, and the Tm is 5–10 °C above the primers’ Tm (calculated using Gene runner software); when using an MGB probe, its length is preferentially 12–20 bases long, and the Tm of MGB probes should preferably be 5–15 °C above the primers’ Tm (calculated using Primer Express software) [64, 72, 88,89,90]. Additionally, the most 5’ end of the probe cannot be a G, as this might quench the fluorophore [64]. Last, liquid biopsies mostly carry highly fragmented cell-free DNA of maximum ~ 160 bp (length of DNA wrapped around one nucleosome), and DNA is additionally fragmented by bisulfite conversion. Therefore, amplicon size was evaluated, with the preferred amplicon size being a maximum of 120 bp [65,66,67,68,69]. All primers and probes were scored according to these criteria (defined in Table 2). In case a nested approach was used, the inner assay was evaluated. For molecular beacon probes, one of our criteria might not be completely suitable, as (to our knowledge) no design tools exist to calculate the Tm of these probes. Therefore, we were unable to assess the Tm of these probes, and they were specifically marked within Fig. 2. Green dots represent optimal design, orange dots represent suboptimal, but acceptable design. Red dots do not necessarily mean a primer or probe does not work, but rather that there is an increased risk of technical problems with the primer or probe for that specific criterium (Table 2). Black dots mean that the criterium was not assessed for that probe, as it was a molecular beacon probe.
Availability of data and materials
All data are available in the main text. MEXPRESS is an open source visualization tool for DNA methylation-, expression- and clinical data extracted from TCGA (https://mexpress.be/).
Abbreviations
- CRC:
-
Colorectal cancer
- MGB:
-
Minor groove binder
- MSP:
-
Methylation-specific polymerase chain reaction
- PCR:
-
Polymerase chain reaction
- qMSP:
-
Quantitative methylation-specific polymerase chain reaction
- Sens:
-
Sensitivity
- Spec:
-
Specificity
- SNP:
-
Single-nucleotide polymorphism
- TCGA:
-
The Cancer Genome Atlas
- Tm:
-
Melting temperature
- TSS:
-
Transcription start site
References
Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15(7):459–66.
Van Vlodrop IJ, Niessen HE, Derks S, Baldewijns MM, Van Criekinge W, Herman JG, et al. Analysis of promoter CpG island hypermethylation in cancer: location, location, location! Clin Cancer Res. 2011;17(13):4225–31.
Poste G. Bring on the biomarkers. Nature. 2011;469(7329):156–7.
van Gool AJ, Bietrix F, Caldenhoven E, Zatloukal K, Scherer A, Litton J-E, et al. Bridging the translational innovation gap through good biomarker practice. Nat Rev Drug Discovery. 2017;16(9):587–8.
Lommen K, Vaes N, Aarts MJ, van Roermund JG, Schouten LJ, Oosterwijk E, et al. Diagnostic DNA methylation biomarkers for renal cell carcinoma: a systematic review. Eur Urol Oncol. 2021;4(2):215–26.
Zhao EY, Jones M, Jones SJM. Whole-Genome Sequencing in Cancer. Cold Spring Harb Perspect Med. 2019;9(3): a034579.
van Vlodrop IJ, Baldewijns MM, Smits KM, Schouten LJ, van Neste L, van Criekinge W, et al. Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am J Pathol. 2010;176(2):575–84.
Joosten SC, Odeh SNO, Koch A, Buekers N, Aarts MJB, Baldewijns M, et al. Development of a prognostic risk model for clear cell renal cell carcinoma by systematic evaluation of DNA methylation markers. Clin Epigenetics. 2021;13(1):103.
Derks S, Lentjes MH, Hellebrekers DM, de Bruïne AP, Herman JG, van Engeland M. Methylation-specific PCR unraveled. Cell Oncol. 2004;26(5–6):291–9.
Lu J, Johnston A, Berichon P, Ru KL, Korbie D, Trau M. PrimerSuite: a high-throughput web-based primer design program for multiplex bisulfite PCR. Sci Rep. 2017;7:41328.
Lommen K, Odeh S, Theije CC, Smits KM. Biobanking in molecular biomarker research for the early detection of cancer. Cancers (Basel). 2020;12(4):776.
Jung M, Klotzek S, Lewandowski M, Fleischhacker M, Jung K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin Chem. 2003;49(6):1028–9.
Lee TH, Montalvo L, Chrebtow V, Busch MP. Quantitation of genomic DNA in plasma and serum samples: higher concentrations of genomic DNA found in serum than in plasma. Transfusion. 2001;41:276–82.
Ebert MPA, Model F, Mooney S, Hale K, Lograsso J, Tonnes-Priddy L, et al. Aristaless-like homeobox-4 gene methylation is a potential marker for colorectal adenocarcinomas. Gastroenterology. 2006;131(5):1418–30.
Herbst A, Rahmig K, Stieber P, Philipp A, Jung A, Ofner A, et al. Methylation of NEUROG1 in serum is a sensitive marker for the detection of early colorectal cancer. Am J Gastroenterol. 2011;106(6):1110–8.
Salehi R, Atapour N, Vatandoust N, Farahani N, Ahangari F, Salehi AR. Methylation pattern of ALX4 gene promoter as a potential biomarker for blood-based early detection of colorectal cancer. Adv Biomed Res. 2015;4:252.
Rasmussen SL, Krarup HB, Sunesen KG, Johansen MB, Stender MT, Pedersen IS, et al. Hypermethylated DNA, a circulating biomarker for colorectal cancer detection. PLoS ONE. 2017;12(7): e0180809.
Tänzer M, Balluff B, Distler J, Hale K, Leodolter A, Röcken C, et al. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PLoS ONE. 2010;5(2): e9061.
Belshaw NJ, Elliott GO, Williams EA, Bradburn DM, Mills SJ, Mathers JC, et al. Use of DNA from human stools to detect aberrant CpG island methylation of genes implicated in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2004;13(9):1495–501.
Elliott KL, Catimel B, Church NL, Coates JL, Burgess AW, Layton MJ, et al. Immunopurification of adenomatous polyposis coli (APC) proteins. BMC Res Notes. 2013;6(1):429.
Leung WK, To K-F, Man EP, Chan MW, Bai AH, Hui AJ, et al. Quantitative detection of promoter hypermethylation in multiple genes in the serum of patients with colorectal cancer. Am J Gastroenterol. 2005;100(10):2274–9.
Nunes SP, Moreira-Barbosa C, Salta S, Palma de Sousa S, Pousa I, Oliveira J, et al. Cell-free DNA methylation of selected genes allows for early detection of the major cancers in women. Cancers (Basel). 2018;10(10):357.
Abbaszadegan MR, Moaven O, Sima HR, Ghafarzadegan K, A’Rabi A, Forghani MN, et al. p16 promoter hypermethylation: a useful serum marker for early detection of gastric cancer. World J Gastroenterol. 2008;14(13):2055–60.
Chang E, Park DI, Kim YJ, Kim BK, Park JH, Kim HJ, et al. Detection of colorectal neoplasm using promoter methylation of ITGA4, SFRP2, and p16 in stool samples: a preliminary report in Korean patients. Hepatogastroenterology. 2010;57(101):720.
Sakamoto J, Fujiya M, Okamoto K, Nata T, Inaba Y, Moriichi K, et al. Immunoprecipitation of nucleosomal DNA is a novel procedure to improve the sensitivity of serum screening for the p16 hypermethylation associated with colon cancer. Cancer Epidemiol. 2010;34(2):194–9.
Frattini M, Gallino G, Signoroni S, Balestra D, Lusa L, Battaglia L, et al. Quantitative and qualitative characterization of plasma DNA identifies primary and recurrent colorectal cancer. Cancer Lett. 2008;263(2):170–81.
Karam RA, Zidan HE, Abd Elrahman TM, Badr SA, Amer SA. Study of p16 promoter methylation in Egyptian colorectal cancer patients. J Cell Biochem. 2018.
Baek YH, Chang E, Kim YJ, Kim BK, Sohn JH, Park DI. Stool methylation-specific polymerase chain reaction assay for the detection of colorectal neoplasia in Korean patients. Dis Colon Rectum. 2009;52(8):1452–9 (discussion 9–63).
Huang Z-H, Li L-H, Yang F, Wang J-F. Detection of aberrant methylation in fecal DNA as a molecular screening tool for colorectal cancer and precancerous lesions. World J Gastroenterol. 2007;13(6):950.
Naini MA, Mokarram P, Kavousipour S, Zare N, Atapour A, Zarin MH, et al. Sensitive and noninvasive detection of aberrant SFRP2 and MGMT-B methylation in iranian patients with colon polyps. Asian Pac J Cancer Prev. 2016;17(4):2185–93.
Chen J, Sun H, Tang W, Zhou L, Xie X, Qu Z, et al. DNA methylation biomarkers in stool for early screening of colorectal cancer. J Cancer. 2019;10(21):5264–71.
Lu H, Huang S, Zhang X, Wang D, Zhang X, Yuan X, et al. DNA methylation analysis of SFRP2, GATA4/5, NDRG4 and VIM for the detection of colorectal cancer in fecal DNA. Oncol Lett. 2014;8(4):1751–6.
Melotte V, Lentjes MH, van den Bosch SM, Hellebrekers DM, de Hoon JP, Wouters KA, et al. N-Myc downstream-regulated gene 4 (NDRG4): a candidate tumor suppressor gene and potential biomarker for colorectal cancer. J Natl Cancer Inst. 2009;101(13):916–27.
Park SK, Baek HL, Yu J, Kim JY, Yang HJ, Jung YS, et al. Is methylation analysis of SFRP2, TFPI2, NDRG4, and BMP3 promoters suitable for colorectal cancer screening in the Korean population? Intest Res. 2017;15(4):495–501.
Xiao W, Zhao H, Dong W, Li Q, Zhu J, Li G, et al. Quantitative detection of methylated NDRG4 gene as a candidate biomarker for diagnosis of colorectal cancer. Oncol Lett. 2015;9(3):1383–7.
Yang C, Wu W, Yang Y, Yang X, Sun J, Zhang W, et al. Multitarget stool DNA test compared with fecal occult blood test for colorectal cancer screening. Oncol Lett. 2020;20(2):1193–200.
Melotte V, Yi JM, Lentjes MH, Smits KM, Van Neste L, Niessen HE, et al. Spectrin repeat containing nuclear envelope 1 and forkhead box protein E1 are promising markers for the detection of colorectal cancer in blood. Cancer Prev Res (Phila). 2015;8(2):157–64.
Niu F, Wen J, Fu X, Li C, Zhao R, Wu S, et al. Stool DNA test of methylated syndecan-2 for the early detection of colorectal neoplasia. Cancer Epidemiol Prev Biomarkers. 2017;26(9):1411–9.
Oh T, Kim N, Moon Y, Kim MS, Hoehn BD, Park CH, et al. Genome-wide identification and validation of a novel methylation biomarker, SDC2, for blood-based detection of colorectal cancer. J Mol Diagn. 2013;15(4):498–507.
Mitchell SM, Ho T, Brown GS, Baker RT, Thomas ML, McEvoy A, et al. Evaluation of methylation biomarkers for detection of circulating tumor DNA and application to colorectal cancer. Genes. 2016;7(12):125.
Zhang W, Bauer M, Croner RS, Pelz JO, Lodygin D, Hermeking H, et al. DNA stool test for colorectal cancer: hypermethylation of the secreted frizzled-related protein-1 gene. Dis Colon Rectum. 2007;50(10):1618–27.
Pasha HF, Radwan MI, Yehia AM, Toam MM. Circulating methylated RUNX3 and SFRP1 genes as a noninvasive panel for early detection of colorectal cancer. Eur J Gastroenterol Hepatol. 2019;31(11):1342–9.
Bedin C, Enzo MV, Del Bianco P, Pucciarelli S, Nitti D, Agostini M. Diagnostic and prognostic role of cell-free DNA testing for colorectal cancer patients. Int J Cancer. 2017;140(8):1888–98.
Huang Z, Li L, Wang J. Hypermethylation of SFRP2 as a potential marker for stool-based detection of colorectal cancer and precancerous lesions. Dig Dis Sci. 2007;52(9):2287–91.
Tang D, Liu J, Wang DR, Yu HF, Li YK, Zhang JQ. Diagnostic and prognostic value of the methylation status of secreted frizzled-related protein 2 in colorectal cancer. Clin Invest Med. 2011;34(2):E88-95.
Wang D-R, Tang D. Hypermethylated SFRP2 gene in fecal DNA is a high potential biomarker for colorectal cancer noninvasive screening. World J Gastroenterol: WJG. 2008;14(4):524.
Babaei H, Mohammadi M, Salehi R. DNA methylation analysis of secreted frizzled-related protein 2 gene for the early detection of colorectal cancer in fecal DNA. Niger Med J. 2016;57(4):242.
Kim Y-S, Kim S-H, Kim M-N, Sim U-R. SFRP2 promoter methylation analysis in tumor tissue, stool, and plasma DNA of patients with colorectal cancer. Arch Intern Med Res. 2019;2:034–9.
Zhang X, Song Y-F, Lu H-N, Wang D-P, Zhang X-S, Huang S-L, et al. Combined detection of plasma GATA5 and SFRP2 methylation is a valid noninvasive biomarker for colorectal cancer and adenomas. World J Gastroenterol. 2015;21(9):2629.
Glöckner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009;69(11):4691–9.
Zhang J, Yang S, Xie Y, Chen X, Zhao Y, He D, et al. Detection of methylated tissue factor pathway inhibitor 2 and human long DNA in fecal samples of patients with colorectal cancer in China. Cancer Epidemiol. 2012;36(1):73–7.
Chen W-D, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L, et al. Detection in fecal DNA of colon cancer–specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst. 2005;97(15):1124–32.
Itzkowitz SH, Jandorf L, Brand R, Rabeneck L, Schroy PC 3rd, Sontag S, et al. Improved fecal DNA test for colorectal cancer screening. Clin Gastroenterol Hepatol. 2007;5(1):111–7.
Pakbaz B, Jabinin R, Soltani N, Ayatollahi H, Farzanehfar MR. Quantitative study of vimentin gene methylation in stool samples for colorectal cancer screening. J Adv Pharm Technol Res. 2019;10(3):121.
Shirahata A, Hibi K. Serum vimentin methylation as a potential marker for colorectal cancer. Anticancer Res. 2014;34(8):4121–5.
Sun Z, Vaisvila R, Yan B, Baum C, Saleh L, Samaranayake M, et al. Non-destructive enzymatic deamination enables single molecule long read sequencing for the determination of 5-methylcytosine and 5-hydroxymethylcytosine at single base resolution. bioRxiv. 2019:2019.12.20.885061.
Brandes JC, Carraway H, Herman JG. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene. 2007;26(42):6229–37.
Davidović R, Božović A, Mandušić V, Krajnović M. Methylation-specific PCR: four steps in primer design. Open Life Sci. 2014;9(12):1127–39.
Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–31.
Patterson K, Molloy L, Qu W, Clark S. DNA methylation: bisulphite modification and analysis. J Vis Exp. 2011;(56).
Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes. 2008;1:54.
IDT. MGB probes 2021. https://eu.idtdna.com/pages/products/gmp-oem-and-integrations/gmp-products/mgb-eclipse-probes.
IDT. Molecular beacons 2021. https://eu.idtdna.com/pages/products/qpcr-and-pcr/custom-probes/molecular-beacons.
Snellenberg S, Strooper LMAD, Hesselink AT, Meijer CJLM, Snijders PJF, Heideman DAM, et al. Development of a multiplex methylation-specific PCR as candidate triage test for women with an HPV-positive cervical scrape. BMC Cancer. 2012;12(1):551.
Maass KK, Schad PS, Finster AME, Puranachot P, Rosing F, Wedig T, et al. From sampling to sequencing: a liquid biopsy pre-analytic workflow to maximize multi-layer genomic information from a single tube. Cancers. 2021;13(12):3002.
Martins I, Ribeiro IP, Jorge J, Gonçalves AC, Sarmento-Ribeiro AB, Melo JB, et al. Liquid biopsies: applications for cancer diagnosis and monitoring. Genes (Basel). 2021;12(3):349.
Salvi S, Gurioli G, De Giorgi U, Conteduca V, Tedaldi G, Calistri D, et al. Cell-free DNA as a diagnostic marker for cancer: current insights. Onco Targets Ther. 2016;9:6549–59.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14(9):531–48.
Udomruk S, Orrapin S, Pruksakorn D, Chaiyawat P. Size distribution of cell-free DNA in oncology. Crit Rev Oncol Hematol. 2021;166: 103455.
IDT DNA technologies. https://www.idtdna.com/pages/education/decoded/article/designing-pcr-primers-and-probes.
ThermoFisher. Real-time PCR 2021. https://www.thermofisher.com/nl/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/designing-taqman-gene-expression-assay.html.
IDT. Designing PCR primers and probes 2021. https://www.idtdna.com/pages/education/decoded/article/designing-pcr-primers-and-probes.
Boyle B, Dallaire N, MacKay J. Evaluation of the impact of single nucleotide polymorphisms and primer mismatches on quantitative PCR. BMC Biotechnol. 2009;9:75.
Chervoneva I, Li Y, Schulz S, Croker S, Wilson C, Waldman SA, et al. Selection of optimal reference genes for normalization in quantitative RT-PCR. BMC Bioinformatics. 2010;11(1):253.
Tajadini M, Panjehpour M, Javanmard SH. Comparison of SYBR Green and TaqMan methods in quantitative real-time polymerase chain reaction analysis of four adenosine receptor subtypes. Adv Biomed Res. 2014;3:85.
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–6.
Hernández HG, Tse MY, Pang SC, Arboleda H, Forero DA. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques. 2013;55(4):181–97.
Woo SY, Kim S. Determination of cutoff values for biomarkers in clinical studies. Precis Future Med. 2020;4(1):2–8.
Budczies J, Klauschen F, Sinn BV, Győrffy B, Schmitt WD, Darb-Esfahani S, et al. Cutoff finder: a comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS ONE. 2012;7(12): e51862.
Alcaide M, Cheung M, Hillman J, Rassekh SR, Deyell RJ, Batist G, et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci Rep. 2020;10(1):12564.
Ioannidis JPA, Bossuyt PMM. Waste, leaks, and failures in the biomarker pipeline. Clin Chem. 2017;63(5):963–72.
Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10(466):eaat4921.
Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE. 2011;6(9): e23418.
Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12(7): e1006162.
Wu TL, Zhang D, Chia JH, Tsao K, Sun CF, Wu JT. Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin Chim Acta. 2002;321(1–2):77–87.
Koch A, De Meyer T, Jeschke J, Van Criekinge W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genomics. 2015;16(1):636.
Koch A, Jeschke J, Van Criekinge W, van Engeland M, De Meyer T. MEXPRESS update 2019. Nucleic Acids Res. 2019;47(W1):W561–5.
Afonina IA, Reed MW, Lusby E, Shishkina IG, Belousov YS. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridization-triggered fluorescence. Biotechniques. 2002;32(4):940-4-946–9.
Biosystems A. Designing TaqMan MGB Probe and Primer Sets for Gene expression Using Primer Express Software Version 2.02002.
Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, et al. 3’-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 2000;28(2):655–61.
Acknowledgements
Not applicable.
Funding
This work was supported by KWF Kankerbestrijding [UM-2-13-6075] and by SU2C-DCS International Translational Cancer Research Dream Team Grant [Stand Up To Cancer (SU2C)-AACR- DT1415, MEDOCC].
Author information
Authors and Affiliations
Contributions
KS had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. MM, KL, KW, KS were involved in study concept and design. MM, KL, KW, KS helped in acquisition of data. MM, KL, KW, JV, WvC, JH, VM, LS, MvE, KS contributed to analysis and interpretation of data. MM, KL, KS helped in drafting of the manuscript. MM, KL, KW, JV, WvC, JH, VM, LS, MvE, KS were involved in critical revision of the manuscript for important intellectual content. VM, MvE helped in obtaining funding. VM, MvE, KS contributed to supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
MvE is cofounder and shareholder of Epify BV and MLA Diagnostics BV, and received grants from MDxHealth, outside the submitted work. In addition, MvE has a patent on DNA methylation markers for colorectal cancer detection.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Massen, M., Lommen, K., Wouters, K.A.D. et al. Technical considerations in PCR-based assay design for diagnostic DNA methylation cancer biomarkers. Clin Epigenet 14, 56 (2022). https://doi.org/10.1186/s13148-022-01273-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-022-01273-z