
Abstract
Background
High-risk human papillomavirus (HR-HPV) infection is the main cause of cervical cancer, but additional alterations are necessary for its development. Abnormal DNA methylation has an important role in the origin and dissemination of cervical cancer and other human tumors. In this work, we analyzed the methylation of eight genes (AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17) that participate in several biological processes for the maintenance of cell normality. We analyzed DNA methylation by methylation-specific PCR (MSP) and HPV infection using the INNO‑LiPA genotyping kit in 59 samples diagnostic of normal cervical tissue (non-SIL), 107 low-grade squamous intraepithelial lesions (LSILs), 29 high-grade squamous intraepithelial lesions (HSILs) and 51 cervical cancers (CCs).
Results
We found that all samples of LSIL, HSIL, and CC were HPV-positive, and the genotypes with higher frequencies were 16, 18, 51 and 56. In general, the genes analyzed displayed a significant tendency toward an increase in methylation levels according to increasing cervical lesion severity, except for the CDH13 gene. High CpG island methylator phenotype (CIMP) was associated with a 50.6-fold (95% CI 4.72–2267.3)-increased risk of HSIL and a 122-fold risk of CC (95% CI 10.04–5349.7).
Conclusions
We found that CIMP high was significantly associated with HSIL and CC risk. These results could indicate that CIMP together with HR-HPV infection and other factors participates in the development of HSIL and CC.
Similar content being viewed by others


Background
Cervical cancer remains a health problem in developing countries. Worldwide, there are approximately 529,800 new cases and 275,100 deaths every year [1]. The main cause of cervical cancer is persistent infection with HR-HPV [2]. This cancer evolves from the well-known precursor lesions LSIL and HSIL. Since not all cervical lesions with HR-HPV infections progress to cervical cancer, it is clear that additional events are necessary for progression [3, 4]. Epigenetic mechanisms are modifications that could have an important contribution to the progression of CC, particularly DNA methylation [5, 6].
DNA methylation frequently occurs at the cytosine-5-carbon adjacent to guanine (CpG), and CpG dinucleotides are abundant in regions denominated CpG islands [7]. DNA methylation plays important roles in genomic imprinting, embryonic development, chromatin structure and diseases such as cancer because it modulates the transcription of genes [8]. A fraction of the CpG islands is in gene promoters. In cancer, including cervical cancer, the hypermethylation of promoters of tumor suppressor and DNA repair genes is common [9,10,11]. Abnormal methylation participates in the initiation, transformation, and dissemination of cancer [8, 12]. Several studies have analyzed the abnormal methylation of genes that participate in cellular proliferation, apoptosis, differentiation, cell cycle, and cellular transformation during cervical carcinogenesis [13,14,15,16,17]. From these reports, it has been proposed that measurement of methylation could serve as a biomarker for early detection, diagnosis, and prognosis in cancer.
In this work, we analyzed the methylation of eight genes (AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17) that participate in many biological processes. These genes were selected because they are involved in essential functions in the maintenance of cell normality and have CpG islands. Methylation of CpG islands in promoters decreases gene expression, and there are reports of hypermethylation in multiple types of cancer. The methylation of these eight genes has been evaluated in cervical cancer but has been evaluated individually; though their methylation has not been related to HPV, only a small number of samples have been analyzed, rather than systematically in normal cervical tissue (non-SIL), LSIL, HSIL, and CC. Therefore, in this work, we evaluated the methylation of eight genes and HPV genotypes in 58 non-SIL, 107 LSIL, 29 HSIL and 51 CC samples.
Results
A total of 245 samples were included in this study: 58 non-SIL, 107 LSIL, 29 HSIL and 51 CC samples. Sociodemographic and sexual characteristics associated with the development of precancerous lesions and cervical cancer are shown in Table 1. The mean ages were 37.59 ± 10.97 years for non-SIL, 35.78 ± 11.94 years for LSIL, 39.37 ± 13.29 years for HSIL and 57.12 ± 13.18 for CC. The smoking status, alcohol consumption, parity, sexual age of onset and education years were statistically significant between non-SIL, LSIL, HSIL and CC.
High-risk HPVs are the cause of cervical cancer. We determined the prevalence of single, multiple, and mixed HPV infections in the samples included in this study (Table 2). The prevalence rates of HPV infection were 49.15% in non-SIL and 100% in LSIL, HSIL, and CC. Single infection with HR-HPV was detected in 23.73% of non-SILs, 41.12% of LSILs, 34.48% of HSILs and 60.78% of CCs, while multiple infections with HR-HPV were detected in 3.39% of non-SILs, 19.63% of LSILs, 24.14% of HSILs and 21.57% of CCs. Mixed HPV infections were more frequent, with HR and low-risk (LR) infections in 10.17% of non-SILs, HR and probably high-risk (PHR) infections in 21.5% of LSILs, HR and PHR infections in 13.79% of HSILs and HR and LR infections in 9.8% of CCs. Additionally, in Additional file 1: Table S1, we show the prevalence of all HPV genotypes found in this study. HPV16, 18, 51 and 56 were the genotypes more frequently detected.
At the present time, there is evidence that DNA methylation is involved in the genesis and progression of cervical cancer. In this work, we analyzed the DNA methylation status of eight genes (AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17) in 245 cervical tissue samples (Fig. 1). In general, the genes displayed a significant tendency toward an increase in methylation levels according to increasing cervical lesion severity, except for the CDH13 gene. Additionally, we analyzed the DNA methylation level and expression of these genes in cervical cancer samples (TCGA data; Additional file 3: Fig. S2). In this data set, we found methylation and lower expression in AJAP1, SOX17, CDH1, and RASSF1A genes. Furthermore, we analyzed the DNA methylation level and mRNA expression levels of these genes in four cervical cancer cell lines as a reference to the HaCaT cell line (Fig. 2 and Additional file 4: Fig. S3). AJAP1 methylation was significantly more prevalent in the SiHa, HeLa and C-33A cell lines. MYOD1 and CDH13 methylation was significantly more prevalent in CaSki, SiHa and HeLa cell lines. Only MGMT methylation was significantly more prevalent in the four cell lines, while CDH1 methylation was not significantly prevalent in any of the cell lines. SOX17 methylation was significantly more prevalent in the SiHa and HeLa cell lines, and RASSF1A and MAGI2 methylations were only observed in C-33A and SiHa cells, respectively (Fig. 2). In general, mRNA expression levels are lower in cervical cancer cells lines than in HaCaT cells, except for SOX17 and MYOD1 in SiHa cells, MGMT in C-33A cells and AJAP1, SOX17 and MYOD1 in HeLa cells (Additional file 4: Fig. S3).

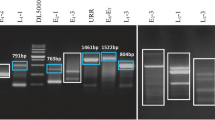
Analysis of the methylation levels of the AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17 genes in cervical tissue. Methylation was analyzed in 58 samples negative for squamous intraepithelial lesions (non-SILs), 107 low-grade squamous intraepithelial lesions (LSILs), 29 high-grade squamous intraepithelial lesions (HSILs) and 51 cervical cancers (CCs). The p values were calculated using the Kruskal–Wallis test

Analysis of the methylation levels of the AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17 genes in cervical cancer cell lines. The data are presented as fold changes in the cancer cell line relative to the HaCaT cell line. *p < 0.05
Using densitometric analysis, we semiquantified the methylation levels of all the samples by MSP. The methylation level of each gene was categorized into three groups, unmethylated, methylated and hypermethylated, and an arbitrary value was assigned to each category: 0, 1 and 2, respectively. Additional file 5: Fig. S4 and Additional file 6: Fig. S5 show the methylation levels of eight genes in non-SIL, LSIL, HSIL, CC samples and cell lines.
Previously, the presence of the CIMP has been reported in several types of human cancer. This phenotype is characterized by the existence of commonly methylated genes that can influence different aspects of cancer biology. For CIMP analysis, the arbitrary values of the three categories were used (unmethylated 0, methylated 1 and hypermethylated 2). Thus, the sum of arbitrary values of the eight genes by the patient was used to define CIMP categories: CIMP negative (0 to 1), CIMP low (2 to 5) and CIMP high (> 6). The CIMP analysis of non-SIL, LSIL, HSIL, and CC samples is shown in Fig. 3. CIMP high was found mainly in CC and HSIL, and CIMP negative was found in non-SIL and LSIL.

CpG island methylation phenotype (CIMP) frequencies in non-SIL, LSIL, HSIL and CC
The CIMP status represents the sum of abnormal methylation of the eight genes analyzed by sample. Therefore, we evaluated the CIMP status and the risk of LSIL, HSIL, and CC development (Table 3). CIMP high was associated with a 50.6-fold (95% CI 4.72–2267.3)-increased risk of HSIL and a 122-fold risk of CC (95% CI 10.04–5349.7). Given that the CIMP statuses between non-SIL and LSIL or HSIL and CC are very similar (Fig. 3), we analyzed the risk of this mode. CIMP high was associated with a 158.6-fold (95% CI 28.9–1471.41) increased risk of HSIL + CC. Additionally, Additional file 1: Tables S2 to S9 show the methylation status of each gene and the risk of LSIL, HSIL, and CC development.
Discussion
It is known that persistent infection with HR-HPV is the cause of cervical cancer [2]. However, additional alterations are necessary for the development of cervical lesions and cervical cancer [4]. Abnormal DNA methylation has an important role from genesis to the dissemination of cervical cancer and other human tumors [12, 18]. In this study, we evaluated the DNA methylation of eight genes, identified and genotyped DNA-HPV, and analyzed the CIMP status and the risk of LSIL, HSIL, and CC development.
CIMP-positive tumors are a class of cancers that have concurrent hypermethylation of multiple genes, and such genes are involved in cellular transformation, proliferation, migration, and invasion, which are key functions in the genesis of human cancer [19, 20]. In our work, CIMP high represents a subclass of patients with methylation or hypermethylation in most of the genes analyzed, and it is likely that such genes are decreased or not expressed. In fact, several studies have reported DNA methylation and decreases or a lack of expression of these genes in several types of human cancer [6, 13, 21]. Furthermore, in general, the mRNA levels of these genes were lower in cervical cancer than in normal cervical tissue (TCGA data; Additional file 7: Fig. S6). We found that CIMP high was significantly associated with an increased risk of HSIL (OR = 50.6) and CC (OR = 122). In agreement with our results, two studies have reported that CIMP high is significantly associated with HSIL and CC development [12, 21]. Biologically, this result can be explained by the individual contribution of each abnormally methylated gene in the molecular alterations that cause HSIL and CC development.
The analyzed genes in this work (SOX17, MYOD1, MAGI2, CDH1, AJAP1, MGMT, CDH13, and RASSF1A) participate in essential biological processes to maintain cell normality, and there is sufficient evidence for their protective role against the development of cancer. For example, SOX17 is a transcription factor involved in cellular reprogramming and has an antagonist role in WNT signaling [22]. There is evidence of its silencing by methylation in several types of human cancer [23,24,25]. MYOD1 is a master regulator of differentiation in muscle cells [26]. Abnormal promoter methylation is frequent in cervical cancer and other human cancers [27,28,29]. MAGI2 belongs to the membrane-associated guanylate kinase superfamily and is an important part of tight junction proteins in epithelial cells [30]. MAGI2, beta-catenin and other proteins participate in migration and proliferation in some cell types [21]. Abnormal expression and hypermethylation in this gene are common events in different types of cancer [31, 32]. In our work, the hypermethylation of the SOX17, MYOD1 and MAGI2 genes was significantly associated with CC development. The CDH1 gene encodes E-cadherin, which is a transmembrane glycoprotein involved in cell–cell adhesion and is key for controlling cell maturation and movement [33]. CDH1 is considered a tumor suppressor gene and is frequently silenced by the methylation of its promoter in cervical cancer and other human tumors [13, 33, 34]. AJAP1 is a transmembrane protein that is an important part of cell–cell and cell-extracellular matrix interactions [35]. This gene has been considered a tumor suppressor and is epigenetically silenced by methylation in cervical cancer, glioma, esophageal squamous cell carcinoma, lung cancer and endometrial carcinoma [21, 35,36,37,38]. We found that the hypermethylation of CDH1 and AJAP1 was significantly associated with HSIL and CC development. MGMT encodes a DNA repair protein that is removed by direct repair of oxygen 6-methylguanine lesions [39]. Lack of expression and promoter hypermethylation are frequent in human cancers, including cervical cancer [40, 41]. We found that the hypermethylation of MGMT was significantly associated with HSIL development. RASSF1A is a tumor suppressor gene that encodes scaffold proteins [42]. Additionally, hypermethylation and lack of expression of this gene are frequent in cancer [43, 44]. In our work, the methylation level of RASSF1A increased according to cervical lesion severity.
CDH13 is an atypical cadherin since it lacks the transmembrane and cytoplasmic domains to mediate its functions through its signaling properties [45]. This gene is considered a tumor suppressor, and its inactivation by hypermethylation is common in colorectal cancer and human lung adenocarcinoma cell lines [46,47,48]. Although CDH13 has a clear function in cancer, we did not find that its methylation was associated with HSIL and CC development. In agreement with our results, there is work that shows low-frequency or absence of methylation in this gene [13, 21, 49]. There is no evidence of CDH13 methylation in cervical cancer. We believe that this is because DNMT3B and 3A (de novo methylases) does not show an affinity for the promoter of this gene and that the decrease in its expression is explained by other components of epigenetic regulation [8, 10, 20].
Four of the genes (SOX17, MAGI2, AJAP1 and CDH1) that we analyzed and that were significantly associated with HSIL, and CC development play essential roles in the WNT/β-catenin signaling pathway [21, 50]. This signaling pathway is crucial in embryogenesis and cell differentiation, and abnormalities in this pathway are common in human cancers, including cervical cancer [51,52,53].
We do not show concrete evidence that this panel of genes is involved in the initiation, development and progression of HSIL or CC. We only show epidemiological evidence of the methylation role and the risk of development HSIL and cervical cancer. Another limitation of our study is that unfortunately, we did not analyze the expression of the 8 genes in the included samples. Instead, we analyzed the expression of these genes in cervical cancer cell lines and databases. Although there is concordance between the expression and methylation data of the cell lines and the analyzed samples, we must take these similarities with caution. The cell lines are homogeneous and do not reflect the complexity of cervical tumor tissue.
Conclusions
We found that CIMP high was significantly associated with HSIL and CC development. This result could indicate that CIMP in precancerous lesions and cervical cancer participates in its genesis together with HR-HPV infection. Additionally, the SOX17, MAGI2, AJAP1, MYOD1, MGMT, and CDH1 genes can be used as biomarkers in HSIL and CC development.
Methods
DNA samples
This study was conducted with 245 DNA samples from the Sample Bank of the Laboratory of Biomedicina Molecular, Universidad Autónoma de Guerrero, México. The population consisted of 58 samples diagnostic of non-SIL, 107 LSIL, 29 HSIL and 51 CC. The diagnosis of non-SIL and LSIL was performed by cytomorphological examination through the Papanicolaou test and diagnosis of HSIL and CC by histological diagnosis, according to the International Federation of Gynecology and Obstetrics. The Bioethics and Research Committee of the Institute approved the study (UAGro-IECan 04/18/2016), according to the ethical guidelines of the 2008 Helsinki Declaration.
Cell culture
Human cervical carcinoma (C-33A, SiHa, CaSki and HeLa) and human skin keratinocyte (HaCaT) cell lines were obtained from the American Type Culture Collection (ATCC, USA). Cells were grown at 37 °C in a 5% CO2 atmosphere with DMEM and F-12 1:1 (Sigma–Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin.
HPV detection and genotyping
Genomic DNA of clinical samples was extracted from cervical cells by the phenol chloroform method. Genomic DNA of cell lines was extracted using the Wizard® Genomic DNA Purification Kit (Promega; Madison, WI, USA) according to the manufacturer´s instructions. HPV detection and genotyping were performed with an INNO‑LiPA genotyping kit (Fujirebio Europe, Gent, Belgium) without amendment and according to the manufacturer's protocol.
Methylation-specific PCR
Methylation analysis of the AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17 genes was performed by MSP and densitometric analysis was performed with ImageJ software (NIH; Bethesda, Maryland, USA). Primer sequences are shown in Table 4. Briefly, 2 μg of genomic DNA was modified using the EZ DNA Methylation-Gold™ Kit (Zymo Research; Irvine, CA, USA). PCR was performed using Amplitaq Gold Master Mix (Applied Biosystems; Foster City, CA, USA) according to the manufacturer’s protocol. Amplification conditions were as follows: denaturation, 95 °C for 10 min; 30 to 35 cycles of amplification: 30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C; and a final extension of 72 °C for 10 min. DNA from human leukocytes methylated in vitro with M.SssI (New England Biolabs, Inc., Ipswich, USA) was used as a positive control, and reactions without DNA were used as a negative control. Additional controls were included, selection was made from information collected from cell lines of different tissues and pathological states in the genome browser. RRBS and Methyl 450 K Array methylation dates were charge for the CpG island of GAPDH (unmethylated) and OXT (Methylated) genes. Furthermore, OXT has been used as a control methylated DNA by MeDIP assay [54] (Additional file 2: Fig. S1).
Semiquantification of methylation levels
We performed semiquantification of methylation levels by densitometry analysis with ImageJ. The quantitative values of each sample and of each gene were obtained, and the minimum and maximum values of each sample group (non-SIL, LSIL, HSIL and CC) were calculated tertiles. With these data, we established three categories: unmethylated (first tertile), methylated (second tertile) and hypermethylated (third tertile). In addition, for CpG island methylation phenotype (CIMP) analysis, we assigned an arbitrary value to each category: 0 for unmethylated, 1 for methylated and 2 for hypermethylated. Thus, with the sum of arbitrary values of the eight genes by each patient, CIMP categories were established: CIMP negative (0 to 1), CIMP low (2 to 5) and CIMP high (> 6).
Statistical analysis
All results are expressed as the mean ± standard deviation. The chi-squared and Kruskal–Wallis tests were used to determine the differences between non-SIL, LSIL, HSIL and CC. The associations between CIMP status and the risk of developing CC, HSIL, and LSIL were estimated by odds ratios (ORs) using the STATA 10.0 software package (StataCorp, College Station, TX, USA). Ninety-five percent confidence intervals (95% CIs) and p values are reported for the OR.
Availability of data and materials
Not applicable.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50.
Castellsague X. Natural history and epidemiology of HPV infection and cervical cancer. Gynecol Oncol. 2008;110(3 Suppl 2):S4-7.
Illades-Aguiar B, Cortes-Malagon EM, Antonio-Vejar V, Zamudio-Lopez N, Alarcon-Romero Ldel C, Fernandez-Tilapa G, et al. Cervical carcinoma in Southern Mexico: Human papillomavirus and cofactors. Cancer Detect Prev. 2009;32(4):300–7.
Fang J, Zhang H, Jin S. Epigenetics and cervical cancer: from pathogenesis to therapy. Tumour Biol. 2014;35(6):5083–93.
Zhu H, Zhu H, Tian M, Wang D, He J, Xu T. DNA methylation and hydroxymethylation in cervical cancer: diagnosis. Prognosis and Treatment Front Genet. 2020;11:347.
Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis. 2000;21(3):461–7.
Ehrlich M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics. 2019;14(12):1141–63.
Feng C, Dong J, Chang W, Cui M, Xu T. The progress of methylation regulation in gene expression of cervical cancer. Int J Genomics. 2018;2018:8260652.
Peralta-Arrieta I, Hernandez-Sotelo D, Castro-Coronel Y, Leyva-Vazquez MA, Illades-Aguiar B. DNMT3B modulates the expression of cancer-related genes and downregulates the expression of the gene VAV3 via methylation. Am J Cancer Res. 2017;7(1):77–87.
Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416(6880):552–6.
Verlaat W, Van Leeuwen RW, Novianti PW, Schuuring E, Meijer C, Van Der Zee AGJ, et al. Host-cell DNA methylation patterns during high-risk HPV-induced carcinogenesis reveal a heterogeneous nature of cervical pre-cancer. Epigenetics. 2018;13(7):769–78.
Cardoso MFS, Castelletti CHM, Lima-Filho JL, Martins DBG, Teixeira JAC. Putative biomarkers for cervical cancer: SNVs, methylation and expression profiles. Mutat Res. 2017;773:161–73.
Del Pino M, Sierra A, Marimon L, Marti Delgado C, Rodriguez-Trujillo A, Barnadas E, et al. CADM1, MAL, and miR124 promoter methylation as biomarkers of transforming cervical intrapithelial lesions. Int J Mol Sci. 2019;20(9):10.
Li C, Ke J, Liu J, Su J. DNA methylation data-based molecular subtype classification related to the prognosis of patients with cervical cancer. J Cell Biochem. 2020;121(3):2713–24.
Verlaat W, Snoek BC, Heideman DAM, Wilting SM, Snijders PJF, Novianti PW, et al. Identification and validation of a 3-gene methylation classifier for HPV-based cervical screening on self-samples. Clin Cancer Res. 2018;24(14):3456–64.
Zhang J, Yao T, Lin Z, Gao Y. Aberrant methylation of MEG3 functions as a potential plasma-based biomarker for cervical cancer. Sci Rep. 2017;7(1):6271.
Louvanto K, Aro K, Nedjai B, Butzow R, Jakobsson M, Kalliala I, et al. Methylation in predicting progression of untreated high-grade cervical intraepithelial neoplasia. Clin Infect Dis. 2020;70(12):2582–90.
Jia M, Gao X, Zhang Y, Hoffmeister M, Brenner H. Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: a systematic review. Clin Epigenetics. 2016;8:25.
Suzuki H, Yamamoto E, Maruyama R, Niinuma T, Kai M. Biological significance of the CpG island methylator phenotype. Biochem Biophys Res Commun. 2014;455(1–2):35–42.
Chen YC, Huang RL, Huang YK, Liao YP, Su PH, Wang HC, et al. Methylomics analysis identifies epigenetically silenced genes and implies an activation of beta-catenin signaling in cervical cancer. Int J Cancer. 2014;135(1):117–27.
Tan DS, Holzner M, Weng M, Srivastava Y, Jauch R. SOX17 in cellular reprogramming and cancer. Semin Cancer Biol. 2020;67(Pt 1):65–73.
Fu DY, Wang ZM, Li C, Wang BL, Shen ZZ, Huang W, et al. Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Res Treat. 2010;119(3):601–12.
Li L, Yang WT, Zheng PS, Liu XF. SOX17 restrains proliferation and tumor formation by down-regulating activity of the Wnt/beta-catenin signaling pathway via trans-suppressing beta-catenin in cervical cancer. Cell Death Dis. 2018;9(7):741.
Moshi JM, Hoogduin KJ, Ummelen M, Henfling MER, van Engeland M, Wouters KAD, et al. Switches of SOX17 and SOX2 expression in the development of squamous metaplasia and squamous intraepithelial lesions of the uterine cervix. Cancer Med. 2020;9(17):6330–43.
Wardle FC. Master control: transcriptional regulation of mammalian Myod. J Muscle Res Cell Motil. 2019;40(2):211–26.
Clarke MA, Luhn P, Gage JC, Bodelon C, Dunn ST, Walker J, et al. Discovery and validation of candidate host DNA methylation markers for detection of cervical precancer and cancer. Int J Cancer. 2017;141(4):701–10.
Sood S, Patel FD, Srinivasan R, Dhaliwal LK. Chemoradiation therapy induces in vivo changes in gene promoter methylation & gene transcript expression in patients with invasive cervical cancer. Indian J Med Res. 2018;147(2):151–7.
Wu F, Qin Y, Jiang Q, Zhang J, Li F, Li Q, et al. MyoD1 suppresses cell migration and invasion by inhibiting FUT4 transcription in human gastric cancer cells. Cancer Gene Ther. 2020;27(10–11):773–84.
Mahdian R, Nodouzi V, Asgari M, Rezaie M, Alizadeh J, Yousefi B, et al. Expression profile of MAGI2 gene as a novel biomarker in combination with major deregulated genes in prostate cancer. Mol Biol Rep. 2014;41(9):6125–31.
Chang CC, Wang HC, Liao YP, Chen YC, Weng YC, Yu MH, et al. The feasibility of detecting endometrial and ovarian cancer using DNA methylation biomarkers in cervical scrapings. J Gynecol Oncol. 2018;29(1):e17.
Valle BL, Rodriguez-Torres S, Kuhn E, Diaz-Montes T, Parrilla-Castellar E, Lawson FP, et al. HIST1H2BB and MAGI2 methylation and somatic mutations as precision medicine biomarkers for diagnosis and prognosis of high-grade serous ovarian cancer. Cancer Prev Res (Phila). 2020;13(9):783–94.
Holubekova V, Mendelova A, Grendar M, Mersakova S, Kapustova I, Jasek K, et al. Methylation pattern of CDH1 promoter and its association with CDH1 gene expression in cytological cervical specimens. Oncol Lett. 2016;12(4):2613–21.
Siegel EM, Riggs BM, Delmas AL, Koch A, Hakam A, Brown KD. Quantitative DNA methylation analysis of candidate genes in cervical cancer. PLoS ONE. 2015;10(3):e0122495.
Tanaka H, Kanda M, Koike M, Iwata N, Shimizu D, Ezaka K, et al. Adherens junctions associated protein 1 serves as a predictor of recurrence of squamous cell carcinoma of the esophagus. Int J Oncol. 2015;47(5):1811–8.
Lai HC, Wang YC, Yu MH, Huang RL, Yuan CC, Chen KJ, et al. DNA methylation as a biomarker for the detection of hidden carcinoma in endometrial atypical hyperplasia. Gynecol Oncol. 2014;135(3):552–9.
Lin N, Di C, Bortoff K, Fu J, Truszkowski P, Killela P, et al. Deletion or epigenetic silencing of AJAP1 on 1p36 in glioblastoma. Mol Cancer Res. 2012;10(2):208–17.
Ooki A, Maleki Z, Tsay JJ, Goparaju C, Brait M, Turaga N, et al. A panel of novel detection and prognostic methylated DNA markers in primary non-small cell lung cancer and serum DNA. Clin Cancer Res. 2017;23(22):7141–52.
Oldrini B, Vaquero-Siguero N, Mu Q, Kroon P, Zhang Y, Galan-Ganga M, et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat Commun. 2020;11(1):3883.
Huang J, Luo JY, Tan HZ. Associations of MGMT promoter hypermethylation with squamous intraepithelial lesion and cervical carcinoma: A meta-analysis. PLoS ONE. 2019;14(10):e0222772.
Sun LL, Liu Y, Sun X, Pan L, Wu D, Wang YD. Limited Role of Promoter Methylation of MGMT and C13ORF18 in Triage of Low-Grade Squamous Intraepithelial Lesion. Chin Med J (Engl). 2018;131(8):939–44.
Malpeli G, Innamorati G, Decimo I, Bencivenga M, Nwabo Kamdje AH, Perris R, et al. Methylation dynamics of RASSF1A and its impact on cancer. Cancers (Basel). 2019;11(7):10.
Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007;23(1–2):73–87.
Raos D, Ulamec M, Katusic Bojanac A, Bulic-Jakus F, Jezek D, Sincic N. Epigenetically inactivated RASSF1A as a tumor biomarker. Bosn J Basic Med Sci. 2020;21:10.
Andreeva AV, Kutuzov MA. Cadherin 13 in cancer. Genes Chromosomes Cancer. 2010;49(9):775–90.
Wang Y, Zhang L, Yang J, Li B, Wang J. CDH13 promoter methylation regulates cisplatin resistance of non-small cell lung cancer cells. Oncol Lett. 2018;16(5):5715–22.
Xu D, Yuan H, Meng Z, Yang C, Li Z, Li M, et al. Cadherin 13 inhibits pancreatic cancer progression and epithelial-mesenchymal transition by Wnt/beta-Catenin signaling. J Cancer. 2020;11(8):2101–12.
Ye M, Huang T, Li J, Zhou C, Yang P, Ni C, et al. Role of CDH13 promoter methylation in the carcinogenesis, progression, and prognosis of colorectal cancer: A systematic meta-analysis under PRISMA guidelines. Medicine (Baltimore). 2017;96(4):e5956.
Sen P, Ganguly P, Ganguly N. Modulation of DNA methylation by human papillomavirus E6 and E7 oncoproteins in cervical cancer. Oncol Lett. 2018;15(1):11–22.
Salmeron-Barcenas EG, Illades-Aguiar B, Del Moral-Hernandez O, Ortega-Soto A, Hernandez-Sotelo D. HOTAIR knockdown decreased the activity Wnt/beta-catenin signaling pathway and increased the mRNA levels of its negative regulators in hela cells. Cell Physiol Biochem. 2019;53(6):948–60.
Yang M, Wang M, Li X, Xie Y, Xia X, Tian J, et al. Wnt signaling in cervical cancer? J Cancer. 2018;9(7):1277–86.
McMellen A, Woodruff ER, Corr BR, Bitler BG, Moroney MR. Wnt signaling in gynecologic malignancies. Int J Mol Sci. 2020;21(12):10.
Bahrami A, Hasanzadeh M, ShahidSales S, Yousefi Z, Kadkhodayan S, Farazestanian M, et al. Clinical significance and prognosis value of Wnt signaling pathway in cervical cancer. J Cell Biochem. 2017;118(10):3028–33.
Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66.
Acknowledgements
During the development of the investigation, Loaeza-Loaeza J was a grant recipient of CONACYT.
Funding
The present study was supported by the National Council of Science and Technology (grant no. 201579).
Author information
Authors and Affiliations
Contributions
JLL and BIA designed and performed the experiments. MLV, BIA and DHS conceived and designed the study. OdM, YCC, RDM, JOO and DHS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Bioethics and Research Committee of the Universidad Autónoma de Guerrero approved the study, which was performed in accordance with the recommendations of the Declaration of Helsinki (2013) for biomedical research involving human subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1. Prevalence of genotypes HPV in single, multiple and mix HPV infection in normal cervix, precancerous lesions and cervical cancer; Table S2. Methylation status of CDH1 and risk of cervical lesion; Table S3. Methylation status of AJAP1 and risk of cervical lesion; Table S4. Methylation status of MAGI2 and risk of cervical lesion; Table S5. Methylation status of MYOD1 and risk of cervical lesion; Table S6. Methylation status of SOX17 and risk of cervical lesion; Table S7. Methylation status of MGMT and risk of cervical lesion; Table S8. Methylation status of RASSF1A and risk of cervical lesion; Table S9. Methylation status of CDH13 and risk of cervical lesion.
Additional file 2: Fig. S1
. Analysis of the methylation levels of control genes in cervical tissue. Methylation was analyzed in 5 samples negative for squamous intraepithelial lesions (non-SILs), 5 low-grade squamous intraepithelial lesions (LSILs), 5 high-grade squamous intraepithelial lesions (HSILs) and 5 cervical cancers (CCs). a) MSP products in agarose gels for methylated GAPDH (168 bp) and methylated OXT (131 bp). b) Densitometry analysis from GAPDH and OXT methylation. The data are presented as mean ± standard deviation at 5 samples in each group.
Additional file 3: Fig. S2
. Methylation and expression heatmaps of AJAP1, SOX17, CDH13, MAGI2, MGMT, CDH1, RASSF1, and MYOD1 genes in public database (Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma, TCGA, Firehose Legacy). a) Methylation data of 309 patients. b) Expression level of analyzed genes. The yellow box shows a representative patient’s group for AJAP1 gene with high methylation levels and low expression levels.
Additional file 4: Fig. S3
. Analysis of the expression levels of the AJAP1, CDH13, MAGI2, SOX17, MGMT, and MYOD1 genes in cervical cancer cell lines. The dates are presented as the fold change in cancer cell line relative to HaCaT cell line. *p<0.05
Additional file 5: Fig. S4
. Analysis of the methylation levels of the AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17 genes in each of the non-SIL and LSIL samples. The sum of arbitrary values of methylation and CIMP status is shown.
Additional file 6: Fig. S5
. Analysis of the methylation levels of the AJAP1, CDH1, CDH13, MAGI2, MGMT, MYOD1, RASSF1A and SOX17 genes in each of the HSIL and CC samples. The sum of arbitrary values of methylation and CIMP status is shown.
Additional file 7: Fig. S6
. Expression of analyzed genes in TCGA dataset. The mRNA levels of analyzed genes were investigated in normal and cancer samples. Cancer included cervical squamous cell carcinoma and endocervical adenocarcinoma tumor samples. *p<0.05
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Loaeza-Loaeza, J., Illades-Aguiar, B., del Moral-Hernández, O. et al. The CpG island methylator phenotype increases the risk of high-grade squamous intraepithelial lesions and cervical cancer. Clin Epigenet 14, 4 (2022). https://doi.org/10.1186/s13148-021-01224-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01224-0