
Abstract
Background
Preterm birth and shorter duration of pregnancy are associated with increased morbidity in neonatal and later life. As the epigenome is known to have an important role during fetal development, we investigated associations between gestational age and blood DNA methylation in children.
Methods
We performed meta-analysis of Illumina’s HumanMethylation450-array associations between gestational age and cord blood DNA methylation in 3648 newborns from 17 cohorts without common pregnancy complications, induced delivery or caesarean section. We also explored associations of gestational age with DNA methylation measured at 4–18 years in additional pediatric cohorts. Follow-up analyses of DNA methylation and gene expression correlations were performed in cord blood. DNA methylation profiles were also explored in tissues relevant for gestational age health effects: fetal brain and lung.
Results
We identified 8899 CpGs in cord blood that were associated with gestational age (range 27–42 weeks), at Bonferroni significance, P < 1.06 × 10− 7, of which 3343 were novel. These were annotated to 4966 genes. After restricting findings to at least three significant adjacent CpGs, we identified 1276 CpGs annotated to 325 genes. Results were generally consistent when analyses were restricted to term births. Cord blood findings tended not to persist into childhood and adolescence. Pathway analyses identified enrichment for biological processes critical to embryonic development. Follow-up of identified genes showed correlations between gestational age and DNA methylation levels in fetal brain and lung tissue, as well as correlation with expression levels.
Conclusions
We identified numerous CpGs differentially methylated in relation to gestational age at birth that appear to reflect fetal developmental processes across tissues. These findings may contribute to understanding mechanisms linking gestational age to health effects.
Similar content being viewed by others

Background
Preterm birth (birth before 37 weeks’ gestation) is associated with increased neonatal morbidity and mortality [1, 2], as well as later health [3,4,5,6]. In children born at very young gestational ages, bronchopulmonary dysplasia, retinopathy and neurodevelopmental impairment are major health challenges [7,8,9,10,11,12]. Lower lung function is observed in children born moderately preterm, i.e. between 32 and 36 completed weeks, compared to those born at term [13]. Even variation in gestational age within the normal range (37–41 weeks) is related to various health outcomes, including neurological and cognitive development [14,15,16,17] and respiratory disease [4]. Mechanisms for many of these findings are not well understood.
The epigenome is known to have an important role during fetal development. The best studied epigenetic modification is methylation. DNA methylation patterns have been associated with environmental factors relevant to preterm birth, including smoking, air pollution exposure, microbial and maternal nutritional factors [18,19,20,21,22]. Such exposure-related epigenetic patterns potentially influence gene expression profiles and/or susceptibility to chronic disease during the lifecourse [23, 24]. Further, DNA methylation in whole blood at birth may also reflect development across fetal life. It is possible that DNA methylation changes at birth may contribute to the myriad immediate and late health outcomes that have been associated with gestational age.
Knowledge about DNA methylation and gene expression profiles associated with length of gestation may help to better understand both the molecular basis of abnormal processes related to prematurity as well as normal human development. Several studies have reported associations of gestational age among both term and preterm births with cord blood DNA methylation [25,26,27,28,29]. In the largest EWAS to date (n = 1753 newborns), 5474 CpGs in cord blood were associated with gestational age [30]. While these individual studies have identified widespread associations of DNA methylation patterns at birth with gestational age, meta-analysis of results from multiple individual cohorts increases sample size and, thus, greatly increases power to detect robust differential methylation signals.
We examined DNA methylation levels in newborns in relation to gestational age in a large-scale meta-analysis and also examined functional effects on expression of nearby genes of potential relevance for later health. We meta-analysed harmonized cohort specific EWAS results of the association of gestational age with cord blood DNA methylation levels from the Pregnancy And Childhood Epigenetics (PACE) Consortium of pregnancy and childhood cohorts [31]. We also examined associations with continuous gestational age limited to term newborns. CpGs that were differentially methylated in cord blood in relation to gestational age were then analysed in two fetal tissues (lung and brain), with relevance for health impacts of low gestational age [7,8,9,10,11,12]. We conducted analyses to explore whether associations of CpG methylation with gestational age persisted in older children aged 4–18 years. DNA methylation status at the identified CpGs was analysed for association with gene expression patterns of nearby genes in cord blood during different developmental stages. Finally, we performed pathway and functional network analysis of identified genes to gain insight into the biological implications of our findings.
Methods
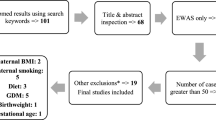
Figure 1 gives an outline of the design of this study.

An overview of the study design
Study population
A total of 11,000 participants in 26 independent cohorts were included in our study. In the “all births model” meta-analysis, we included n = 6885 newborns from 20 cohorts. In our main “no complications model”, we excluded participants with maternal complications (maternal pre-eclampsia or diabetes or hypertension) and caesarean section delivery or delivery start with induction, leaving 3648 newborns from 17 cohorts for this analysis (Additional file 1: Table S1). For the additional look-up of persistent differential methylation at later ages, we used participants from 4 cohorts with whole blood DNA methylation in early childhood (4–5 years; n = 453), 5 cohorts with whole blood DNA methylation at school age (7–9 years; n = 899) and 5 cohorts with whole blood DNA methylation in adolescence (16–18 years; n = 1129). Detailed methods for each cohort are provided in Additional file 2: Supplementary information. All cohorts acquired ethics approval and informed consent from participants prior to data collection through local ethics committees (Additional file 2: Supplementary information).
Gestational age
In each cohort, information on gestational age at birth was obtained from birth certificates (n = 725), medical records using ultrasound estimation (n = 1931), or last menstrual period date (n = 468), or combined estimate from ultrasound and last menstrual period date (n = 6630), or otherwise from self-administrated questionnaires (n = 1246). Gestational age was analysed in days. Women with a gestational age of more than 42 weeks (294 days) were excluded from all models. Additionally, multiple births were also excluded from the analysis.
Methylation measurements and quality control
DNA methylation from newborns and older children was measured using the Illumina450K platform. Each cohort conducted their own quality control and normalization of DNA methylation data, as detailed in Additional file 1: Table S2. Cohorts corrected for batch effects in their data using surrogate variables, ComBat [32], or by including a batch covariate in their models. To reduce the impact of severe outliers in the DNA methylation data on the meta-analysis, cohorts trimmed the methylation beta values by removing, for each CpG, observations more than three times the interquartile range below the 25th percentile or above the 75th percentile [33]. Cohorts retained all CpGs that passed quality control and removed CpGs that were mapped to the X (n = 11,232) or Y (n = 416) chromosomes and control probes (n = 65), leaving a maximum total of 473,864 CpGs included in the meta-analysis.
Cohort-specific statistical analyses
Each cohort performed independent EWAS according to a common, pre-specified analysis plan. Robust linear regression (rlm in the MASS R package [34]) was used to model gestational age as the exposure and DNA methylation beta values as the outcome. In the primary analysis, gestational age was used as a continuous variable excluding cohorts that had term-only infants. In secondary models, we modeled term-only children defined as a gestational age ≥ 37 weeks (≥ 259 days), but less or equal with 42 weeks. All models were adjusted for sex, maternal age (years), maternal social class (variable defined by each individual cohort; Additional file 1: Table S2), maternal smoking status (the preferred categorization was into three groups: no smoking in pregnancy, stopped smoking in early pregnancy, smoking throughout pregnancy, but a binary categorization of any versus no smoking was also acceptable), parity (the preferred categorization was into two groups: no previous children, one or more previous children), birth weight in grams, age of the child (years) included for older children, batch or surrogate variables. Optionally, cohorts could include ancestry, and/or selection covariates, if relevant to their study. We also adjusted for potential confounding by cell type using estimated cell type proportions calculated from a cord blood cell type reference panel [35] for newborn cohorts or the adult blood cell type reference panel [36] for cohorts with older children using the estimateCellCounts function in the minfi R package [37].
Meta-analysis
We performed fixed-effects meta-analysis weighted by the inverse of the variance with METAL [38]. A shadow meta-analysis was also conducted independently by a second study group (see author contribution) and the results were compared [39] (and confirmed). All downstream analyses were conducted using R version 2.5.1 or later [40]. Multiple testing was accounted for by applying the Bonferroni correction level for 473,864 tests (P < 1.06 × 10− 7). A random effects model was performed using the METASOFT tool [41]. We explored heterogeneity between studies using the I2 statistic [42]. A priori, we defined I2 > 50% as reflecting a high level of between-study variation. In case of I2 > 50%, we replaced values with random effects estimates as these are attenuated in the face of heterogeneity and thus more conservative. To focus functional analyses and bioinformatics efforts on genes and loci that were found to be robustly associated with gestational age, we selected regions that had at least three adjacent Bonferroni significant CpGs (P < 1.06 × 10− 7) [43]. Genome-wide DNA methylation meta-analysis summary statistics corresponding to the main analysis presented in this manuscript are available at figshare (https://doi.org/10.6084/m9.figshare.11688762.v1) [44].
Analyses of differentially methylated regions
Differentially methylated regions (DMRs) were identified using two methods available for meta-analysis results comb-p [45] and DMRcate [46]. Input parameters used for the DMR calling in both algorithms are provided in Additional file 2: Supplementary information. Comb-p uses a one-step Šidák correction [45] and DMRcate uses an FDR correction [46] per default. The selected regions were defined based on the following criteria: the minimum number of CpGs in a region had to be 2, regional information can be combined from probes within 1000 bp and the multiple-testing corrected P < 0.01 (Šidák-corrected P < 0 .01 from comb-p and FDR < 0.01 from DMRcate).
Analyses of embryonic DNA methylation
DNA methylation from lung tissue of 74 foetuses (estimated ages 59 to 122 days post conception [47]) were used for analyses of differentially methylated CpGs (three or more adjacent Bonferroni significant CpGs, P < 1.06 × 10− 7; n = 1276) from the newborn meta-analysis. A linear regression model adjusted for sex and in utero smoke exposure (IUS) was applied. A Bonferroni look-up level correction (0.05/1030; P < 4.85 × 10− 5) considered as significance threshold, followed by a comparison of the direction of effect with that in the cord blood meta-analysis. We also performed look-up analyses of selected 1276 CpGs in another organ, fetal brain tissue, from 179 foetuses collected between 23 and 184 days post-conception [48]. For these analyses, we kept the available Bonferroni correction P < 1.06 × 10− 7 as significance threshold, followed by a comparison of the direction of effect with that in the cord blood meta-analysis.
Look-up analyses in older ages
Differentially methylated CpGs (three or more adjacent CpGs below the Bonferroni correction P < 1.06 × 10− 7; n = 1276) from the newborn meta-analyses were analysed with a look-up approach using data from four early childhood, five school age, and five adolescence cohorts. Cohorts included the same covariates in these analyses as in the cord blood analyses and child age. We performed fixed effects inverse variance weighted meta-analyses using METAL [38] for these three age groups. For this hypothesis-driven analysis, CpG methylation association with gestational age was considered statistically significant at nominal P < 0.05, followed by a comparison of the direction of effect with that in the cord blood meta-analysis.
Longitudinal analysis
Longitudinal DNA methylation data from birth to early childhood and from birth to adolescence were analysed for the three or more adjacent Bonferroni significant 1276 CpGs found to be associated with gestational age. DNA methylation from two time points (birth and 4 years) in INMA and three time points (birth, 7 and 17 years) in ALSPAC were analysed separately. To estimate changes in DNA methylation, we applied linear mixed models with repeated measurement taking into account the within-person time effect. The models were adjusted for covariates and estimated cell count similar to cross-sectional analysis. Interaction terms between age and gestational age were included in the model to capture differences in methylation change between birth and 4 years, birth and 7 years and 7 and 17 years per day increase in gestational age at delivery, respectively. The stable CpGs that did not change significantly from birth to adolescence had no association with age (at nominal P < 0.05), and no interaction between gestational age and childhood age (at nominal P < 0.05).
Enrichment and functional analysis
CpGs were annotated using FDb.InfiniumMethylation.hg19 R package, with enhanced annotation for nearest genes within 10 Mb of each site, as previously described [20]. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the overrepresentation analysis (ORA) tool ConsensusPathDB (http://consensuspathdb.org/ [49, 50]). P values for enrichment were adjusted for multiple testing using the FDR method.
DNA methylation in relation to gene expression
Correlations between DNA methylation and gene expression levels were tested using paired DNA methylation and gene expression data in publicly available datasets. We tested transcript levels of genes within a 500-kb region of the 1276 three adjacent CpGs (250 kb upstream and 250 kb downstream). The mRNA gene expression (Affymetrix Human Transcriptome Array 2.0) and methylation (Illumina Infinium® HumanMethylation450 BeadChip assay) were measured in cord-blood samples from 38 newborns [51,52,53]. First, we created residuals for mRNA expression and residuals for DNA methylation and used linear regression models to evaluate correlations between expression residuals and DNA methylation residuals. These residual models were adjusted for covariates, estimated white blood cell proportions, and technical variation. We corrected these analyses for multiple testing using Bonferroni correction.
Results
Study characteristics
We meta-analysed Illumina’s HumanMethylation450-array results from 17 independent cohorts with data on newborn DNA methylation status, and 10 cohorts with data on DNA methylation in older children (age 4 to 18 years), including 4 cohorts with DNA methylation data both at birth and at an older age (Fig. 1). Table 1 summarizes the characteristics of participating cohorts. A summary of methods used by each cohort is provided in Additional file 1: Tables S1 and S2. In our main “no complications” model, we excluded participants exposed to maternal pregnancy complications (maternal diabetes, hypertension or pre-eclampsia) and whose labour was induced or who were delivered by caesarean section. With continuous gestational age in the number of days as the exposure (gestational age range 186–294 days corresponding to 27–42 weeks), we analysed results from 3648 newborns and from 2481 older children. This model was selected as the main model because associations of DNA methylation with gestational age related to pregnancy complications or potentially influenced by obstetric interventions may be less reflective of normal developmental processes than newborns with spontaneous uncomplicated delivery. However, we also analysed a larger dataset of 6885 newborns from 20 independent cohorts, including pregnancies with pregnancy complications and obstetric interventions, referred to as the “all births model” (see below).
Associations between gestational age and newborn DNA methylation
We identified 8899 CpGs in cord blood that were associated with gestational age (range 27–42 weeks), at Bonferroni significance, P < 1.06 × 10–7, of which 3343 were novel. These were annotated to 4966 genes. CpGs associated with gestational age had a modest predominance of negative (60%) versus positive (40%) direction of effect, with an overall absolute median difference in mean methylation of 0.36% per gestational week, IQR = [0.26%–0.49%] (Fig. 2a). In general, results were highly homogeneous; evidence of high between-study heterogeneity, using a criterion of I2 > 50%, was seen for only 319 of the 8899 CpGs (Additional file 1: Table S3). Leave one out analyses did not indicate an influential effect on meta-analysis results of any single study. However, we replaced fixed effects values with random effects estimates for those CpGs with between study I2 > 50%, as these are more conservative in the case of heterogeneity.

A, B Volcano (A) and Manhattan (B) plots for the meta-analysis of gestational age and offspring DNA methylation association at birth, after adjustment for covariates and estimated cell proportions. The effect size represents methylation change per gestational week
Differentially methylated CpGs spanned all chromosomes (Fig. 2b). The CpG with the lowest P value (P = 2.7 × 10− 129 for cg16103712; Table 2) was annotated to MATN2 on chr 8, and the difference in mean methylation at this CpG was 2.13% lower per additional gestational week (equal to 0.30% per day). The CpG with the largest negative association was cg04347477, annotated to NCOR2 on chr 12 (Table 3), with a lower mean methylation of 2.53% per additional gestational week. B3GALT4 (chr 6) had the largest number of significant CpGs negatively associated with gestational age (21 out of 52 (40%) tested CpGs annotated to B3GALT4). The largest positive association was observed for cg13036381 annotated to LOC401097 (chr 3) (Table 3) with a difference in mean methylation of 1.95% per additional gestational week. DDR1 (chr 6) had the largest number of significant CpGs positively associated with gestational age (26/95 (27%) CpGs). A complete list of associated CpGs is presented in Additional file 1: Table S3 and the CpG variation across cohorts in Additional file 3: Figure S1 (top CpGs).
We performed a sensitivity analysis by excluding cohorts that were included in previous EWAS of gestational age [29, 30] (three cohorts: MoBa1, MoBa2 and ALSPAC) in order to evaluate associations not driven by previous results, and found a high correlation (r = 0.89) of effect estimates (Additional file 3: Figure S2) compared with results from all cohorts included in the no complication model.
Next, we performed a meta-analysis of the larger dataset of 6885 participants from 20 studies without excluding maternal complications and caesarean section delivery or induced delivery. In this “all births model”, 17,095 CpGs located in or near 7931 genes were associated with gestational age after Bonferroni correction (P < 1.06 × 10− 7). Not surprisingly given the higher levels of statistical significance in this much larger data set, we found somewhat more between-study heterogeneity than in the no complications model, but high levels (I2 > 50%) were observed for only 1784 out of these 17,095 CpGs (Additional file 1: Table S4). We also observed a considerable overlap of CpGs between the two models with 93% of the 8899 CpGs in the no complication model also reaching Bonferroni significance in the all birth model and showing the same direction of effect.
CpG localization and regulatory region analyses
The 8899 differentially methylated CpGs in relation to continuous gestational age in the no complications model were enriched for localization to CpG island shores (33% of the 8899 CpGs are in shores, whereas 23% of all CpGs on the 450 K array are in shores, Penrichment = 4.1× 10− 100, Fig. 3), open sea (45% versus 37%, Penrichment = 1.4 × 10− 63), enhancers (37% versus 22%, Penrichment = 1.05 × 10− 236), DNase hypersensitivity sites (18% versus 12%, Penrichment = 1.3× 10− 56) and CpG island shelves (12% versus 10%, Penrichment = 1.2 × 10− 11) (Fig. 3). In contrast, we found relative depletion in CpG islands (10% versus 31%, Penrichment = 2.2 × 10− 308), FANTOM 4 promoters (2.3% versus 6.7%, Penrichment = 6.7 × 10− 79) and promoter-associated regions (11% versus 19%, Penrichment = 2.2 × 10− 104).

Position enrichment analyses for CpGs. Salmon: all CpGs in the Illumina450k annotation file, green: CpGs significantly associated with GA after Bonferroni correction (P < 1.06 × 10− 7) and blue: three or more adjacent CpGs associated with GA after Bonferroni correction (P < 1.06 × 10− 7). “**” represent significant two-sided doubling mid P value of the hypergeometric test
Analysis restricted to term-births
To evaluate whether observed DNA methylation differences in relation to continuous gestational age were driven by preterm birth, we repeated the no complication model including only infants born at term (gestational age 37 to 42 weeks). In this analysis, we meta-analysed results from 18 cohorts (one additional cohort with term-birth data only was included; GEN3G) (n = 3593). We identified 5930 sites significantly associated with gestational age at Bonferroni correction (P < 1.06 × 10− 7, median difference in mean methylation per additional gestational week = 0.43%, IQR = [0.32%–0.58%]). The vast majority (5399; 91%) of these differentially methylated CpGs overlapped with those found in the main analyses (no complications model) without exclusion of those born preterm (Fig. 4).

Overlap between Bonferroni-significant CpG sites from two different analyses after exclusion of maternal and delivery start with induction or caesarean section (“no complication” model). The blue colour represents the continuous gestational age main model, and the green represents the continuous model restricted to term only. Overlap of findings alters the colour
Selection of CpGs for downstream analyses
Given the large number of significant associations in our main model (8899 CpGs), we focused subsequent analyses on loci including at least three adjacent CpGs that survived Bonferroni correction [43]. There were 1276 differentially methylated CpGs in 325 unique genes that fulfilled this criterion (Additional file 1: Table S5). As in the overall data, we observed a slight predominance of negative (n = 702; 55%) versus positive (n = 574; 45%) directions of effect (Fig. 2a). The lowest P value, P = 1.2 × 10− 93, was observed for cg04276536 (CCDC102A, chromosome 16). As for the full EWAS results, the largest negative and positive association effect sizes were observed for cg04347477 (NCOR2) and cg13036381 (LOC401097), respectively. These 1276 CpGs had the same CpG localization enrichment pattern as the full set of Bonferroni-significant CpGs (n = 8899), except that there was a relative depletion in CpG island shelves (7.6% versus 10% overall, Penrichment = 2.3 × 10− 12) and open sea (32% versus 37%, Penrichment = 2.4 × 10− 12) (Fig. 3).
Differentially methylated region (DMR) analyses
Using two different methods for DMR analysis of gestational age in relation to newborn DNA methylation, we identified 4479 significant (Šidák-corrected P < 0.01) DMRs from the comb-p method and 14,671 significant (FDR P < 0.01) DMRs from DMRcate, respectively, including 2375 DMRs (representing 11,861 CpGs) that were significant based on both approaches (Additional file 1: Table S6). Out of the 8899 Bonferroni significant single CpGs, 2289 CpGs overlapped with CpGs in identified in the combined DMR analyses (11,861 CpGs). Moreover, from loci included by the three or more adjacent CpG selection (n = 1276), 521 CpGs overlapped with those identified in the combined DMR analyses. Of note, out of the 1276 CpGs, 1223 and 1231 CpGs were captured by DMRs identified using the comb-p and DMRcate independent approaches, respectively.
Assessment of CpG methylation in earlier embryonic stages
We examined whether the CpGs detected in cord blood (that originate from embryonic germ layer mesoderm) were differentially methylated in relation to gestational age in other fetal tissues, lung and brain that originate from the two other embryonic germ layers, ectoderm and endoderm, respectively, collected prenatally [47, 48]. To this end, we performed look-up analyses in DNA methylation data for 74 fetal lung samples representing gestational age 59 to 122 days (~ 8 to 17 completed gestational weeks) [47]. Out of the 1276 CpGs, selected based on three or more adjacent CpGs from our no complications model, 1030 CpGs were available in the fetal lung dataset. We observed associations at Bonferroni look-up level correction significance (0.05/1030; P < 4.85 × 10− 5) between DNA methylation levels in fetal lung tissue and gestational age at tissue collection for 151 (15%) CpGs (Additional file 1: Table S7). Of these 151 (58 negatively and 93 positively associated), 78 showed the same direction of association with gestational age in cord blood and fetal lung tissue. The look-up analyses of fetal brain tissue were undertaken in 179 samples representing 23 to 184 days (~ 3 to 26 completed weeks) [48]. Out of the 1276 CpGs, we found significant associations (using Bonferroni correction P < 1.06 × 10− 7 cut-off since only this data was available for analyses; Additional file 1: Table S8) for 268 CpGs (21%) in relation to gestational age at tissue collection. Of these 268 sites, 227 had same direction of effect in the cord blood and fetal brain data. We found enrichment more than expected by chance for our cord blood gestational age associated CpGs (n = 1276) in fetal lung (P = 2.1 × 10− 4) and brain (P = 3.9 × 10− 57) tissue. Thirty CpGs showed significant associations with gestational age in all three tissues (cord blood, fetal lung and fetal brain).
Assessment of CpG methylation in older children
We examined whether the differentially methylated CpGs detected in cord blood samples were associated with gestational age at birth in whole blood from older children. We conducted three separate meta-analyses (no complications model) reflecting different age periods in a total of 2481 children: (i) Early childhood (4–5 years; n = 453 from 4 cohorts); (ii) school age (7–9 years; n = 899 from 5 cohorts) and (iii) adolescence (16–18 years; n = 1129 from 5 cohorts), Additional file 1: Table S1. Of the 1276 three or more adjacent genome-wide significant CpGs from our analyses in cord blood, 1258 CpGs were available for analyses in all older age groups. Out of these CpGs, we observed 40 sites in early childhood, 60 sites in school age, and 60 sites in adolescence to be associated with gestational age at the nominal significance level, P < 0.05 with the same direction of effect (Additional file 1: Table S9). However, no CpG survived Bonferroni look-up level correction (0.05/1258; P < 3.97 × 10− 5). One CpG (cg26385222 annotated to TMEM176B) previously associated with gestational age at birth [27] was nominally significant in all age groups with same direction of effect.
Longitudinal analysis
The results of the longitudinal analyses of blood DNA methylation in the INMA Study (n = 177 with paired samples from birth and 4 years) and the ALSPAC Study (n = 281 with samples collected at birth, 7 and 17 years) are provided in Additional file 1: Table S10. The vast majority of gestational age associated CpGs (n = 1054/1276; 83%) underwent changes in methylation levels with age. Both increasing and decreasing patterns of change during early childhood (4 years) were observed, followed by stabilization during school age (7 years). For example, for cg08943494 in PRR5L on chr 11, an initial level of 61.5% and 51.4% in cord blood DNA methylation in INMA and ALSPAC respectively, decreased by 8.2% per year on average during early childhood in INMA and by 3.3% per year on average up to school age in ALSPAC, but then negligible further changes were seen from 7 to 17 years (Fig. 5A). In contrast, increasing levels were seen for cg18183624 (chr 17; IGF2BP1), from an initial 48.8% and 38.7% in cord blood DNA methylation in INMA and ALSPAC, respectively, with a 5.1% per year on average between birth to 4 years in INMA and 1.9% per year on average between birth to 7 years, but after that no changes from 7 to 17 years. (Fig. 5B).

Change in DNA methylation during childhood and adolescence for selected CpG sites associated with gestational age. A Decreasing methylation levels from birth to childhood (A.1) and stabilization during adolescence (A.2). B Increasing methylation levels from birth to childhood and stabilization during adolescence. C Stable CpGs that did not change during childhood or adolescence; (1) INMA from birth to early childhood and (2) ALSPAC from birth to adolescence. The figures show representative single CpGs for each category (A–C)
Of the 1054 CpGs displaying changes in DNA methylation levels with age, there were 589 CpGs where gestational age was associated with changes in DNA methylation levels (i.e. where an interaction between gestational age and age was found) from birth to 4 years (INMA) and 460 CpGs with changes from birth to 7 years (ALSPAC). However, only 30 of the 1054 CpGs changed significantly in DNA methylation between 7 and 17 years (ALSPAC), suggesting that gestational age-related changes in DNA methylation levels had largely stabilized by age 7.
We identified 222 stable CpGs out of 1276 (17%) that did not change appreciably from birth to adolescence. As an example, the stable DNA methylation at cg27058497 (RUNX3, chromosome 1) is shown in Fig. 5C. A much lower proportion of the gestational age associated CpGs were stable from birth to adolescence compared to all CpGs on the array (17% versus 71%, Penrichment = 2.23× 10− 308).
Enrichment for biological processes and pathways
Using the complete list of 8899 CpGs annotated to 4966 genes, these were enriched for 1784 GO terms including regulation of cellular and biological processes, system development, different signaling pathways and organ development (Additional file 1: Table S11). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses revealed 124 significant terms at FDR < 0.05 representing a variety of human diseases, most notably various cancers, viral infections, metabolic processes and immune-related disorders (Additional file 1: Table S12). The 325 genes annotated to the 1276 CpGs, selected by virtue of three or more CpGs being localized to the same gene, were enriched for 198 Gene Ontology (GO) terms very similar to those identified using Bonferroni significant CpGs (Additional file 1: Table S13). When restricting analyses to the 222 longitudinally stable CpGs, corresponding to 139 genes, 13 significant KEGG terms were revealed, primarily representing infection- and immune-related disorders (Additional file 1: Table S14). For 186 genes annotated to the 1054 CpGs changing with postnatal age, only one KEGG terms were identified as statistically significant (P = 1.2 × 10− 3 for the term MAPK signaling pathways; Additional file 1: Table S14).
Correlation of DNA methylation and gene expression
For the 1276 CpGs differentially methylated in relation to gestational age with at least 3 adjacent CpGs, we assessed correlations between DNA methylation and gene expression (cis-eQTMs). From a publicly available dataset of expression and DNA methylation measured in 38 cord blood samples [51,52,53], 1174 out of the 1276 CpGs were located within a 500-kb (+/− 250 kb) window of a transcript cluster. Of these 1174, 246 unique CpGs (367 total CpG-transcript associations) correlated significantly with gene expression (Bonferroni P < 0.05, Additional file 1: Table S15). Forty-six percent of these DNA methylation-expression correlations were negative, with the lowest P = 3.55 × 10− 6 coeff = − 6.03 for cg01332054 and SEMA7A expression and the largest negative effect estimate (− 12.69) for cg26179948 and JAZF1 expression (Additional file 3: Figure S3 A, B). Fifty-four percent were positive, with the lowest P = 1.04 × 10− 5 coeff = 2.88 for cg20139800 and MOG expression and the largest positive effect estimate (19.35) for cg03665259 and CDSN expression (Additional file 3: Figure S3 C, D).
Discussion
In this large consortium-based meta-analysis, we identified 8899 sites across the genome where gestational age at birth was associated with cord blood DNA methylation. We also identified numerous unique differentially methylated regions (DMRs) associated with gestational age by applying two independent methods. The results were consistent when restricted to births at term, demonstrating that the majority of our results were not driven by preterm births. We confirmed many of the findings from previously published EWAS of gestational age [23, 26, 27, 29, 30, 67] and found a very high correlation between the significant CpG point estimates in previously published datasets compared to our study (e.g. corr = 0.92 between Hannon et al. CpGs and our data; Additional file 1: Table S16), but importantly, we also found 3343 CpGs corresponding to 2577 genes that had not been described previously. There was a general lack of stability of the cord blood findings into childhood and adolescence. However, there was a significant overlap of differentially methylated CpGs in cord blood, fetal brain and lung tissues.
We found that various functional elements were enriched among gestational age-associated CpGs. CpG island shores, enhancers and DNase I hypersensitive sites were particularly susceptible to DNA methylation changes in relation to gestational age, suggesting that these differentially methylated sites are of functional importance [68].
We found clear overlap of differentially methylated CpGs in cord blood, fetal brain and fetal lung tissues in relation to gestational age. Thus, our cord blood findings seem to partly capture the epigenomic plasticity of prenatal development across tissues. The gene with the largest negative magnitude of association with cord blood DNA methylation in relation to gestational age, NCOR2, was also differentially methylated in brain and lung fetal tissues. NCOR2 is involved in vitamin A metabolism and has previously been associated in GWAS with lung function [69]. Vitamin A supplementation is suggested to reduce the risk of bronchopulmonary dysplasia in extremely preterm-born children [70]. Differential methylation of NCOR2 in neurons associated with ageing has been reported [71]. The gene with the second largest magnitude of negative association with methylation at birth, PRR5L, has been linked in GWAS to allergic diseases, found downregulated (expression) in osteoarthritis, and differentially methylated in type II diabetes [72,73,74]. The gene with the lowest P value in our EWAS, MATN2 plays a critical role in the differentiation and maintenance of skeletal muscles, peripheral nerves, liver and skin during development and regeneration [75] and is suggested as a potential biomarker in the early stage of osteoarthritis [76].
Differentially methylated CpGs associated with gestational age in cord blood were also present in our childhood and adolescence analyses. The only CpG (cg26385222, TMEM176B) that was associated with gestational age at all three time points (birth, childhood and adolescence) has been associated with gestational age in cord blood in previous studies [27]. The protein encoded by TMEM176B has also been suggested as a potential biomarker for various cancers [77]. The low number of significant associations with gestational age at older ages with no CpG surviving multiple test correction may be partially explained by smaller sample sizes in childhood and adolescence than at birth and by the fact that many later exposures may obscure the association. However, in agreement with the cross-sectional analyses, our longitudinal analyses showed that DNA methylation at gestational age-associated CpGs typically undergoes dynamic changes during early childhood to a much higher degree than overall for CpGs on the 450K array. For the majority of these dynamics CpGs, change was most prominent during the first years of life, with many sites tending stabilize in methylation levels by school age. We also identified a subset of the CpGs differential methylated at birth (17%) which seem stable over time. For these CpGs, the early alteration of methylation levels by length of gestation was found stable postnatally across childhood and into adolescence.
In recent analyses by Xu et al, 14,150 CpGs related to childhood age were identified [78] and we found 280 overlapping with these CpGs among our 1276 CpG list. Moreover, a study by Acevedo et al. showed 794 age-modified CpGs within 3 to 60 months after birth and 57 CpGs were overlapping with our 1276 CpG list [79]. Thus, a proportion of gestational age-related CpGs are also associated with postnatal ageing. But similar to results from Simpkin et al. [80], we observed very little overlap (only 3 CpGs) with the CpGs used to derive epigenetic age by the Hannum and Horvath approach [81, 82] or the epigenetic clock for gestational age at birth (10 CpGs overlapping) [28]. It should be noted that these studies primarily used the Illumina 27K array for analyses, which makes comparison difficult.
In the functional analyses, we observed significant enrichment for several GO terms related to embryonic development, regulation of process and immune system development. The pathway analyses identified a subset of these genes linked to diseases also associated with low gestational age, for example asthma [83], inflammatory bowel disease [84], type I/II diabetes [85] and cancer (leukaemia) [86]. Importantly, genes annotated to CpGs found stable across childhood also showed enrichment for infection- and immune-related conditions. Whether cord blood DNA methylation at these CpGs affects later disease risk remains to be studied. Interestingly, differentially methylated loci in relation to asthma development have been recently identified in newborns [87]. The stable CpG cg27058497 (RUNX3) has been associated with in utero tobacco smoking exposure [88], childhood asthma [89], oesophagus squamous cell carcinoma [90] and chronic fatigue syndrome [91]. Despite adjustment for maternal smoking in our gestational age EWAS model, we observed overlap between all FDR hits from our gestational age EWAS with those FDR hits presented in the maternal smoking related DNA methylation [20] with an overlap of 2302/47,324 CpGs (4.9%, Penrichment < 2.2 × 10− 308). This overlap likely reflects some pregnant women under reporting their smoking behaviour and the fact that smoking-related CpGs capture quantitative smoking history better than self-report [92, 93]. However, we cannot rule out the possibility that some overlapping CpGs could be involved in biologic pathways linking smoking to the well-established consequence of shorter gestational length [94]. Other potential confounders not accounted for in this study such as maternal obesity and alcohol intake may influence offspring DNA methylation although we have found in the PACE consortium that their impact on methylation [95, 96] is very modest compared with maternal smoking in pregnancy which was included in our models.
This paper aimed at identifying CpGs associated with gestational age while adjusting for birth weight. In a recent PACE paper, we found 1071 CpGs at Bonferroni significant levels association with birth weight [97]. Even after adjustment of birth weight in our gestational age EWAS, we observed overlap between the birth weight EWAS and the current gestational age EWAS for 373/1071 CpGs (34.9% Penrichment < 2.2 × 10− 308). These two perinatal factors, birth weight and gestational age, may have a shared impact on DNA methylation in newborns. However, it is difficult to disentangle the effects of these correlated factors.
To further investigate a potential functional impact of our differentially methylated CpGs, we examined correlations with gene expression in cord blood. We found multiple cis-eQTMs among the gestational age-related CpGs where methylation was strongly correlated with gene expression in cord blood, implying that the identified CpGs may have a direct functional effect in newborns. IGF2BP1, known to be involved in adiposity and cardiometabolic disease risk [98], and to play an essential role in embryogenesis and carcinogenesis [99, 100], was the most significant positively differentially methylated CpG in cord blood. Low gestational age is a well-established risk factor for later cardiometabolic disease [101]. Our expression findings likely reflect relevant for health outcomes associated with low gestational age.
There are potential study limitations in our study including heterogeneity in normalization and quality control (QC) protocols since individual cohorts performed their own QC and normalization. However, one of our previous EWAS meta-analysis reported robust results comparing the non-normalized methylation and different data processing methods used across the cohorts for normalization [20]. Furthermore, between-study heterogeneity at our pre-specified threshold was observed for only a minority of differentially methylated CpGs. Cohorts collected gestational age data from medical records, birth certificates or questionnaires in two ways, either ultrasound estimates and/or according to last menstrual period (or combined estimates), which may introduce bias. However, gestational age determined by ultrasound correlates well with last menstrual period data [102]. Despite a large sample size, we had few extreme premature births included in our dataset. Interpretation of effects of DNA methylation on gene expression was done for cis-effects only, not trans-effects. Since our analyses were primarily cross-sectional, we cannot infer the temporality in the associations and we cannot assume associations are causal [103]. We recognize the possibility that the observed methylation patterns represent fetal maturity, accompanying a “normal” developmental process or determining time in utero; it was however not possible to include foetuses who did not survive pregnancy most of whom will have been delivered very early. The majority of study participants were of European ancestry, and very few cohorts were Hispanic. We were unable to explore ethnic differences in detail since that would require large sample sizes for each ethnic group. However, when analyses were restricted to European-ancestry cohorts, the results were essentially identical with correlation coefficient 0.97 (Additional file 3: Figure S4) to those with all cohorts included. Finally, we acknowledge a potential limitation by applying a filter (regions with at least three or more adjacent CpGs with a Bonferroni-corrected P value < 0.05) in order to capture a set of genes robustly affected by gestational age, which may have led to potentially important single CpGs not being included in the functional analyses. In addition, genes with few CpGs represented on the 450K array are likely under-represented in the downstream analyses. The strengths of our study are large sample size, the comprehensive analyses using robust statistical methods, as well as the availability of samples at multiple ages and our ability to compare our findings with those in fetal tissue datasets. To account for potential cell type effects, we adjusted our models for estimated cell counts using cord blood and adult whole blood references [35, 36]. However, we acknowledge the limitations of available blood cell type reference data sets and recognize that some of the signals we identified as effects of gestational age might reflect differences in cell type composition that we did not completely control. Larger panels that better capture cell type composition across the range of gestational age would be a useful advance. Although we present data on all available participants in our all births model, we based our study conclusions on the main no complication model results, after excluding samples related to delivery induced by medical interventions (induction and/or caesarean section) and maternal complications.
Conclusions
We show that DNA methylation at numerous CpG sites and DMRs across the genome is associated with gestational age at birth. Our results provide a comprehensive catalogue of differential methylation in relation to this important factor, which may serve as utility to the growing community of researchers studying the developmental origins of adult disease. Identified CpGs were linked to multiple functional pathways related to human diseases and enriched for several categories of biological processes critical to fetal development. As such, many sites might capture epigenomic plasticity of fetal development across tissues. We also found that blood DNA methylation levels in identified CpGs change over time for a majority of CpGs and that levels stabilize after school age. Taken together, our findings provide new insight into epigenetics related to preterm birth and gestational age.
References
Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet (London, England). 2008;371:75–84.
Engle WA. Morbidity and mortality in late preterm and early term newborns: a continuum. Clin Perinatol. 2011;38:493–516.
Leung JY, Lam HS, Leung GM, Schooling CM. Gestational age, birthweight for gestational age, and childhood hospitalisations for asthma and other wheezing disorders. Paediatr Perinat Epidemiol. 2016;30:149–59.
Raby BA, et al. Low-normal gestational age as a predictor of asthma at 6 years of age. Pediatrics. 2004;114:e327–32.
Been JV, et al. Preterm birth and childhood wheezing disorders: a systematic review and meta-analysis. PLoS Med. 2014;11:e1001596.
den Dekker HT, et al. Early growth characteristics and the risk of reduced lung function and asthma: a meta-analysis of 25,000 children. J Allergy Clin Immunol. 2016;137:1026–35.
Parets SE, Bedient CE, Menon R, Smith AK. Preterm birth and its long-term effects: methylation to mechanisms. Biology. 2014;3:498–513.
Kwinta P, Pietrzyk JJ. Preterm birth and respiratory disease in later life. Expert Rev Respir Med. 2010;4:593–604.
Hille ET, et al. Functional outcomes and participation in young adulthood for very preterm and very low birth weight infants: the Dutch project on preterm and small for gestational age infants at 19 years of age. Pediatrics. 2007;120:e587–95.
Geldof CJ, van Wassenaer AG, de Kieviet JF, Kok JH, Oosterlaan J. Visual perception and visual-motor integration in very preterm and/or very low birth weight children: a meta-analysis. Res Dev Disabil. 2012;33:726–36.
Kerkhof GF, Breukhoven PE, Leunissen RW, Willemsen RH, Hokken-Koelega AC. Does preterm birth influence cardiovascular risk in early adulthood? J Pediatr. 2012;161:390–6.e391.
Aarnoudse-Moens CS, Weisglas-Kuperus N, van Goudoever JB, Oosterlaan J. Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics. 2009;124:717–28.
Thunqvist P, et al. Lung function at 8 and 16 years after moderate-to-late preterm birth: a prospective cohort study. Pediatrics. 2016;137(4).
Ghartey K, et al. Neonatal respiratory morbidity in the early term delivery. Am J Obstet Gynecol. 2012;207:292.e291–294.
Noble KG, Fifer WP, Rauh VA, Nomura Y, Andrews HF. Academic achievement varies with gestational age among children born at term. Pediatrics. 2012;130:e257–64.
Talge NM, Allswede DM, Holzman C. Gestational age at term, delivery circumstance, and their association with childhood attention deficit hyperactivity disorder symptoms. Paediatr Perinat Epidemiol. 2016;30:171–80.
Yang S, Bergvall N, Cnattingius S, Kramer MS. Gestational age differences in health and development among young Swedish men born at term. Int J Epidemiol. 2010;39:1240–9.
Gruzieva O, et al. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspect. 2017;125:104–10.
Joubert BR, et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 2016;7:10577.
Joubert BR, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98:680–96.
Gruzieva O, et al. Prenatal particulate air pollution and DNA methylation in newborns: an epigenome-wide meta-analysis. Environ Health Perspect. 2019;127:57012.
Pan WH, et al. Exposure to the gut microbiota drives distinct methylome and transcriptome changes in intestinal epithelial cells during postnatal development. Genome Med. 2018;10:27.
Cruickshank MN, et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96.
Cutfield WS, Hofman PL, Mitchell M, Morison IM. Could epigenetics play a role in the developmental origins of health and disease? Pediatr Res. 2007;61:68r–75r.
Lee H, et al. DNA methylation shows genome-wide association of NFIX, RAPGEF2 and MSRB3 with gestational age at birth. Int J Epidemiol. 2012;41:188–99.
Schroeder JW, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. 2011;6:1498–504.
Parets SE, et al. Fetal DNA methylation associates with early spontaneous preterm birth and gestational age. PLoS One. 2013;8:e67489.
Knight AK, et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016;17:206.
Simpkin AJ, et al. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet. 2015;24:3752–63.
Bohlin J, et al. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016;17:207.
Felix JF, et al. Cohort Profile: Pregnancy And Childhood Epigenetics (PACE) Consortium. Int J Epidemiol. 2018;47:22–23u.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Hoaglin DC, Iglewicz B, Tukey JW. Performance of some resistant rules for outlier labeling. J Am Stat Assoc. 1986;81:991–9.
Venables WR, Ripley BD. Modern Applied Statistics with S. New York: Springer-Verlag; 2002.
Bakulski KM, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11:354–62.
Reinius LE, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361.
Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford, England). 2014;30:1363–9.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics (Oxford, England). 2010;26:2190–1.
Rice K, Higgins JP, Lumley T. A re-evaluation of fixed effect(s) meta-analysis. J R Statist Soc A. 2018;181:205–27.
R Core Team. R Foundation for Statistical Computing; Vienna: R: A language and environment for statistical computing; 2013. http://www.R-project.org/.
Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–98.
Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58.
Hannula-Jouppi K, et al. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics. 2014;9:351–65.
Merid SK et al. Summary statistics Data sets. figshare. 2020. https://doi.org/10.6084/m9.figshare.11688762.v1.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics (Oxford, England). 2012;28:2986–8.
Peters TJ, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Chhabra D, et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics. 2014;9:1473–84.
Spiers H, et al. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25:338–52.
Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB—a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–8.
Kamburov A, et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–7.
Rojas D, et al. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci. 2015;143:97–106.
Rager JE, et al. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environ Mol Mutagen. 2014;55:196–208.
Barrett T, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–5.
Ma X, et al. Ethnic difference in daycare attendance, early infections, and risk of childhood acute lymphoblastic leukemia. Cancer Epidemiol Biomarkers Prev. 2005;14:1928–34.
McConnell R, et al. Traffic, susceptibility, and childhood asthma. Environ Health Perspect. 2006;114:766–72.
Eskenazi B, et al. CHAMACOS, a longitudinal birth cohort study: lessons from the fields. J Childrens Health. 2003;1:3–27.
Heude B, et al. Cohort profile: the EDEN mother-child cohort on the prenatal and early postnatal determinants of child health and development. Int J Epidemiol. 2016;45:353–63.
Vineis P, et al. The exposome in practice: design of the EXPOsOMICS project. Int J Hyg Environ Health. 2017;220:142–51.
Kruithof CJ, et al. The generation R study: biobank update 2015. Eur J Epidemiol. 2014;29:911–27.
Guxens M, et al. Cohort profile: the INMA--INfancia y Medio Ambiente--(environment and childhood) project. Int J Epidemiol. 2012;41:930–40.
Everson TM, et al. DNA methylation loci associated with atopy and high serum IgE: a genome-wide application of recursive random Forest feature selection. Genome Med. 2015;7:89.
Girchenko P, et al. Cohort profile: prediction and prevention of preeclampsia and intrauterine growth restriction (PREDO) study. Int J Epidemiol. 2017;46:1380–1381g.
Oken E, et al. Cohort profile: project viva. Int J Epidemiol. 2015;44:37–48.
Xu CJ, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. 2018;6:379–88.
Jarvelin MR, Hartikainen-Sorri AL, Rantakallio P. Labour induction policy in hospitals of different levels of specialisation. Br J Obstet Gynaecol. 1993;100:310–5.
Straker L, et al. Cohort Profile: The Western Australian Pregnancy Cohort (Raine) Study-Generation 2. Int J Epidemiol. 2017;46:1384–1385j.
Hannon E, et al. Variable DNA methylation in neonates mediates the association between prenatal smoking and birth weight. Philos Trans Ro Soc Lond. 2019;374:20180120.
Ziller MJ, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–81.
Minelli C, et al. Association of Forced Vital Capacity with the developmental gene NCOR2. PLoS One. 2016;11:e0147388.
Garg BD, Bansal A, Kabra NS. Role of vitamin A supplementation in prevention of bronchopulmonary dysplasia in extremely low birth weight neonates: a systematic review of randomized trials. J Matern Fetal Neonatal Med. 2019;32:2608-15.
Gasparoni G, et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenetics Chromatin. 2018;11:41.
Ferreira MAR, et al. Eleven loci with new reproducible genetic associations with allergic disease risk. J Allergy Clin Immunol. 2019;143:691-9.
Wang X, Ning Y, Guo X. Integrative meta-analysis of differentially expressed genes in osteoarthritis using microarray technology. Mol Med Rep. 2015;12:3439–45.
Al Muftah WA, et al. Epigenetic associations of type 2 diabetes and BMI in an Arab population. Clin Epigenetics. 2016;8:13.
Korpos E, Deak F, Kiss I. Matrilin-2, an extracellular adaptor protein, is needed for the regeneration of muscle, nerve and other tissues. Neural Regen Res. 2015;10:866–9.
Zhang S, et al. Matrilin-2 is a widely distributed extracellular matrix protein and a potential biomarker in the early stage of osteoarthritis in articular cartilage. Biomed Res Int. 2014;2014:986127.
Cuajungco MP, et al. Abnormal accumulation of human transmembrane (TMEM)-176A and 176B proteins is associated with cancer pathology. Acta Histochem. 2012;114:705–12.
Xu CJ, et al. The emerging landscape of dynamic DNA methylation in early childhood. BMC Genomics. 2017;18:25.
Acevedo N, et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin Epigenetics. 2015;7:34.
Simpkin AJ, et al. Prenatal and early life influences on epigenetic age in children: a study of mother-offspring pairs from two cohort studies. Hum Mol Genet. 2016;25:191–201.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Goyal NK, Fiks AG, Lorch SA. Association of late-preterm birth with asthma in young children: practice-based study. Pediatrics. 2011;128:e830–8.
Sonntag B, et al. Preterm birth but not mode of delivery is associated with an increased risk of developing inflammatory bowel disease later in life. Inflamm Bowel Dis. 2007;13:1385–90.
Li S, et al. Preterm birth and risk of type 1 and type 2 diabetes: systematic review and meta-analysis. Obes Rev. 2014;15:804–11.
Wang YF, Wu LQ, Liu YN, Bi YY, Wang H. Gestational age and childhood leukemia: A meta-analysis of epidemiologic studies. Hematology (Amsterdam, Netherlands). 2018;23:253–62.
Reese SE, et al. Epigenome-wide meta-analysis of DNA methylation and childhood asthma. J Allergy Clin Immunol. 2019;143:2062-74.
Maccani JZ, Koestler DC, Houseman EA, Marsit CJ, Kelsey KT. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics. 2013;5:619–30.
Yang IV, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015;136:69–80.
Zheng Y, Zhang Y, Huang X, Chen L. Analysis of the RUNX3 gene methylation in serum DNA from esophagus squamous cell carcinoma, gastric and colorectal adenocarcinoma patients. Hepato-gastroenterology. 2011;58:2007–11.
de Vega WC, Herrera S, Vernon SD, McGowan PO. Epigenetic modifications and glucocorticoid sensitivity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). BMC Med Genet. 2017;10:11.
Reese SE, et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ Health Perspect. 2017;125:760–6.
Valeri L, et al. Misclassified exposure in epigenetic mediation analyses. Does DNA methylation mediate effects of smoking on birthweight? Epigenomics. 2017;9:253–65.
Warren GW, Alberg AJ, Kraft AS, Cummings KM. The 2014 surgeon General's report: "the health consequences of smoking--50 years of progress": a paradigm shift in cancer care. Cancer. 2014;120:1914–6.
Sharp GC, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26:4067–85.
Sharp GC, et al. Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics. 2018;10:27–42.
Kupers LK, et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat Commun. 2019;10:1893.
Lu Y, et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun. 2016;7:10495.
Mahaira LG, et al. IGF2BP1 expression in human mesenchymal stem cells significantly affects their proliferation and is under the epigenetic control of TET1/2 demethylases. Stem Cells Dev. 2014;23:2501–12.
Huang X, et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol Oncol. 2018;11:88.
Cooper R, Atherton K, Power C. Gestational age and risk factors for cardiovascular disease: evidence from the 1958 British birth cohort followed to mid-life. Int J Epidemiol. 2009;38:235–44.
Hoffman CS, et al. Comparison of gestational age at birth based on last menstrual period and ultrasound during the first trimester. Paediatr Perinat Epidemiol. 2008;22:587–96.
Dyke SOM, et al. Points-to-consider on the return of results in epigenetic research. Genome Med. 2019;11:31.
Acknowledgements
For all studies, detailed information can be found in Additional file 2: Supplementary information.
Funding
This study was specifically funded by a grant from the European Research Council (TRIBAL, grant agreement 757919). For all studies, detailed information can be found in Additional file 2: Supplementary information. Open access funding provided by Uppsala University.
Availability of data and materials
Genome-wide DNA methylation meta-analysis summary statistics corresponding to the main analysis presented in this manuscript are available at figshare (https://doi.org/10.6084/m9.figshare.11688762.v1) [44]. Individual cohort level data may be available by application to the relevant institutions after obtaining required approvals. All datasets used are previously published as described in Felix et al. [31]. Additional details and references to the study cohorts are available in Additional file 2.
Author information
Authors and Affiliations
Contributions
EM and SJL conceived and designed the study with input from the project group (SKM, GHK, JF, M-FH, AG, NH, MW, OS, PB, JK, SER, C-JX, AC, OG, CAM, CS, AK and LKK). GCS (ALSPAC and GOYA), SKM (BAMSE, EDEN and PIAMA), RR (CBC), OS (CHAMACOS), LG (CHS), PJ (EXPOSOMICS: Environage, PiccoliPlus and RHEA), LKK (GECKO), CA (Gen3G), FOV (Generation R), LAS (INMA), FIR (IOW F1), HZ (IOW F2), SER (MoBa1 and MoBa2), AN (MoBa3), MW (NFBC86), DC (PREDO), AC (Project Viva) and PEM (Raine) conducted the cohort-specific analyses. Longitudinal analyses were performed by SKM (INMA, with support from MB) and GSC (ALSPAC). ATK performed analyses on fetal lung data sets. SKM meta-analyses all results with AN as shadow analyst. SKM performed expression and DNA methylation follow-up analyses and bioinformatics analysis. SKM, EM and SJL wrote the first draft of the manuscript. All authors (SKM, AN, GCS, LKK, ATK, RR, LG, IAM, PJ, MP, MK, CA, FOV, NK, LAS, FIR, HZ, SS, DC, SLR-S, PEM, DAL, GP, CVB, KH, NB, LG, TSN, EC, PP, LD, EAN, MB, SLE, WK, SZ, CMP, ZH, M-RJ, JL, AAB, DA, PK, CLR, AB, BE, MHS, PV, HS, LB, VWJ, TIAS, MV, SHA, JWH, SEH, PM, TD, EBB, DLD, JMV, JN, KGT, IK, JLW, BH, JS, WN, MCM-K, KR, EO, R-CH, STW, JMA, JB, AK, CS, CA, AC, OG, C-JX, SER, JK, PB, OS, MW,NH, AG, M-FH, JFF, GHK, SJL, EM) read and critically revised subsequent drafts, and approved the final version. Correspondence and material requests should be addressed to EM (erik.melen@ki.se).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All cohorts acquired ethics approval and informed consent from participants prior to data collection through local ethics committees; detailed information for each cohort can be found in Additional file 2: Supplementary information. Our research conformed to the principles of the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
DA Lawlor declares grants from Medtronic Ltd. and Roche Diagnostics and EBB; A Ghantous is identified as personnel of the IARC, the author alone is responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the IARC. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Cohort-specific results from epigenome-wide association analyses of gestational age. Table S2. Normalization technique and phenotype definitions used by each cohort. Table S3. Bonferroni-significant CpGs from the meta-analysis on the association between continuous gestational age (no complications model) and offspring DNA methylation at birth adjusted for estimated cell counts. Table S4. Bonferroni-significant CpGs from the meta-analysis on the association between continuous gestational age (all births model) and offspring DNA methylation at birth adjusted for estimated cell counts. Table S5. Gene regions that had at least three consecutive Bonferroni significant CpG sites from the continuous gestational age analyses (no complications model). Table S6. DMRs (n = 2375) for gestational age in relation to newborn methylation (no complication model) identified by using both comb-p (P < 0.01) and DMRcate (FDR < 0.01) methods. Table S7. DNA methylation analyses in fetal lung tissue using the no complication gestational age three or more consecutive CpG list. Table S8. DNA methylation analyses in fetal brain tissue using the no complication gestational age three or more consecutive CpG list. Table S9. Methylation look-up analyses in older children using the no complication gestational age three or more consecutive CpG list. Table S10. Longitudinal analysis of methylation levels in the INMA and ALSPAC studies using the no complication gestational age three or more consecutive CpG list. Table S11. Gene Ontology (GO) term enrichment analyses for bonferroni-significant CpGs from the meta-analysis (no complications model). Table S12. KEGG pathway analyses for bonferroni-significant CpGs from the meta-analysis (no complications model). Table S13. Gene Ontology (GO) term enrichment analyses for three or more CpGs being localized to the same gene. Table S14. KEGG pathway analyses for stable and dynamic CpGs. Table S15. Correlation between methylation and gene expression levels in cord blood (cis-effects). Table S16. The replication of bonferroni-significant CpGs from the meta-analysis (no complications model) in previous publication.
Additional file 2.
Supplementary information.
Additional file 3:
Figure S1. Forest plot for the top 10 Bonferroni-significant CpGs from the meta-analysis on the association between continuous GA and offspring DNA methylation at birth adjusted for estimated cell proportions. Figure S2. Sensitivity analysis: Correlation of the point estimates for the no complications model main association of DNA methylation with gestational age (y-axis representing 3648 participants from 17 cohorts) with point estimates for a meta-analysis after excluding three cohorts (MoBa1, MoBa2 and ALSPAC) that were included in a previous publication1,2 (x-axis representing 2190 participants from 14 cohorts). Figure S3. Correlations between methylation and gene expression levels for selected four pairs. First, we created residuals for mRNA expression and residuals for DNA methylation and used linear regression models to evaluate correlations between expression residuals and methylation residuals. These residual models were adjusted for covariates, estimated white blood cell proportions, and technical variation. Figure S4. Sensitivity analysis: Correlation of the point estimates for the no complications model main association of DNA methylation with gestational age (y-axis representing 3648 participants from 17 cohorts) with point estimates for a meta-analysis after excluding Non-European three cohorts (CBC, CHS and CHAMACOS) (x-axis representing 3290 participants from 14 cohorts).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Merid, S.K., Novoloaca, A., Sharp, G.C. et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med 12, 25 (2020). https://doi.org/10.1186/s13073-020-0716-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-020-0716-9