
Abstract
Background
Gestational age is often used as a proxy for developmental maturity by clinicians and researchers alike. DNA methylation has previously been shown to be associated with age and has been used to accurately estimate chronological age in children and adults. In the current study, we examine whether DNA methylation in cord blood can be used to estimate gestational age at birth.
Results
We find that gestational age can be accurately estimated from DNA methylation of neonatal cord blood and blood spot samples. We calculate a DNA methylation gestational age using 148 CpG sites selected through elastic net regression in six training datasets. We evaluate predictive accuracy in nine testing datasets and find that the accuracy of the DNA methylation gestational age is consistent with that of gestational age estimates based on established methods, such as ultrasound. We also find that an increased DNA methylation gestational age relative to clinical gestational age is associated with birthweight independent of gestational age, sex, and ancestry.
Conclusions
DNA methylation can be used to accurately estimate gestational age at or near birth and may provide additional information relevant to developmental stage. Further studies of this predictor are warranted to determine its utility in clinical settings and for research purposes. When clinical estimates are available this measure may increase accuracy in the testing of hypotheses related to developmental age and other early life circumstances.
Similar content being viewed by others
Background
Differences in gestational age (GA) as small as one week have been shown to have significant impacts on neonatal morbidity and mortality, as well as long-term outcomes [1–6]. In light of this, the American College of Obstetricians and Gynecologists (ACOG) recently recommended revising the categorization of births from term (>37 weeks gestation) and preterm (≤37 weeks gestation) into several subcategories (early preterm, preterm, early term, full term, late term, and post term) that better reflect the developmental differences associated with GA at each of these time points [7, 8]. Accurate classification systems that reflect both developmental time and maturity may improve our ability to predict neonatal risk.
Traditionally, GA is estimated using one or more of the following methods: early obstetric ultrasound, last menstrual period (LMP), or neonatal estimation [9]. Ultrasound-based methods are considered to be the gold standard and have proven to be a better predictor of delivery date [10] as LMP estimates may be influenced by uncertainty regarding LMP dates, normal variations in ovulation timing, atypical bleeding, and contraceptive use [9]. Neonatal estimation, which is based on a combination of physical appearance, muscular tone, flexibility, and reflexes, is the only available method for determining GA after birth but is less precise than LMP and ultrasound [9, 11, 12]. In circumstances where LMP date is uncertain and ultrasounds are not available, a more accurate method for estimating GA may be beneficial.
Recently, DNA methylation (DNAm) has been used to accurately predict chronological age in children and adults [13–16]. Later work revealed that a methylation-based prediction of age may also associate with physiological consequences in adults when a study reported that an increased methylation age relative to chronological age was associated with an increase in mortality risk [17–22]. However, the predictors optimized in these studies were not designed to estimate GA and did not attempt to differentiate between different GA, as samples taken at birth were either assigned an age of zero or were excluded from the model [13, 14]. Because the accuracy and precision of a prediction model is, in general, weakest at the extremes of the distribution, a predictor developed from primarily adult samples would, by nature, be less accurate in neonates than one that is optimized for that purpose.
DNAm differences in specific CpG sites have been associated with GA at birth in multiple studies [23–26]. We hypothesize that a predictor designed specifically for use with umbilical cord blood or blood spots already routinely collected for newborn screening could allow for accurate neonatal estimation of GA that may also be informative of developmental stage. The objective of this study was to develop such a predictor to estimate GA from DNAm data using umbilical cord blood or blood spot samples and to assess its ability to predict other indicators of developmental maturity.
Results and Discussion
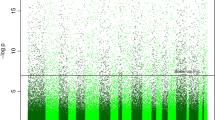
DNAm data from 1434 neonates, representing 15 independent cohorts, were used for this study. For each sample, HumanMethylation27 or HumanMethylation450 BeadChips (Table 1; Additional file 1: Table S1) were used to generate data from DNA extracted from umbilical cord blood or blood spots. Of the 16,676 CpG sites that passed quality control in the testing and training datasets referenced in Table 1, 3155 (19 %) were at least nominally associated with GA in an epigenome-wide association study (p < .05; Additional file 1: Figure S1), and adjustment for proportions of white blood cell subtypes and nucleated red blood cells had little effect on the results (Additional file 1: Figure S2). Associated CpGs were enriched for a range of biological processes, including cell proliferation and chordate embryonic development (Additional file 1: Table S2).
Predicting DNAm GA in neonates
To train the DNAm GA predictor, six independent cohorts were selected to sample a wide range of GAs and ancestries. Consistent with the approach described by Horvath [13], elastic net regression was used to select a set of 148 CpG sites (Additional file 2) predictive of GA from a set of 16,838 CpG sites that were available in all training datasets. Although some of the individual studies report associations between the perinatal environment and DNAm, no CpG site reported to associate with environmental exposures in these cohorts were among the sites selected for this predictor [27–30]. Overall, 90 out of 148 CpG sites selected for the predictor (61 %) showed some evidence for association with GA in the cell type-adjusted epigenome-wide association study (p < 0.05). In the training datasets, correlation between the resulting predictor (DNAm GA) and clinically estimated GA was 0.99 (Fig. 1a), indicating a strong fit of the model. The 148 CpG sites selected by the elastic net were uniformly distributed across the genome and were not located in genes more likely to be represented among specific biological pathways (data not shown). They were more likely to reside in CpG island shores than the remaining 16,690 CpG sites that were eligible for inclusion in the predictor (odds ratio (OR) = 1.73; p = 0.00096) and less likely to reside in CpG islands (OR = 0.53; p = 0.00019) or active promoters (OR = 0.59; p = 0.0028). The 148 sites showed no significant enrichment or depletion for CpG island shelves or enhancers (Additional file 1: Table S3). They were also not enriched or depleted for sites with genetic variants located in the probe sequence or sites previously reported to associate with African American or Caucasian race (Additional file 1: Table S3) [31–33].

Correlation between DNAm GA and GA. a DNAm GA and estimated clinical GA (EGA) are highly correlated in the training dataset: r = 0.99, median error (m.e.) = 0.35. b DNAm GA and estimated clinical GA were also highly correlated in the testing dataset: r = 0.91, median error = 1.24. Solid line = regression line; dotted line indicates equivalence
The predictive accuracy of the model was tested in 1135 samples from six independent datasets. The testing and training datasets had comparable GA distributions (Additional file 1: Figure S3). In the testing datasets, overall correlation between DNAm GA and GA was 0.91 (p < 2.20 × 10−16; Fig. 1b). Within individual test datasets, correlation between GA and DNAm GA remained high (0.52 < r < 0.65; Additional file 1: Figure S4) though appeared lower than in the combined dataset due to lower sample sizes and GA range. We were not able to obtain similar predictive power using the DNAm age predictor proposed by Horvath, which has a highly significant but much weaker correlation with GA (r = 0.14, p = 4.89 × 10−6; Additional file 1: Figure S5). This correlation coefficient is similar to that observed for prenatal brain samples (r = 0.15) [34].
We did not evaluate the Hannum predictor [14] since it is less accurate than the Horvath predictor in children [14, 35]. Of note, only six CpG sites included in the DNAm GA predictor overlap with CpG sites in the predictor designed by Horvath and no sites overlap with the predictor designed by Hannum. However, one would not necessarily expect overlap. Elastic net regression selects a parsimonious set of the full list of CpG sites and among highly correlated CpG sites only one may be chosen, introducing an element of chance into CpG selection. Moreover, the late gestational period is associated with unique developmental changes that cannot be discriminated by the adult predictor, which did not include measures of GA in its training dataset. Thus, this lack of overlap may indicate that the CpG sites predictive of GA in neonates are distinct from CpG sites predicting age in adults because of their association with changes specific to gestational development.
The average absolute difference between DNAm GA and GA in test samples was 1.49 weeks, with a standard deviation of 1.16 weeks. The median absolute difference (“median error”) between DNAm GA and GA was 1.24 weeks. This falls well within the range of error for clinical estimates of GA based on either LMP or ultrasound, as each of these clinical measures has an inherent variability due to recall bias and natural phenotypic variation associated with development [9, 10, 36]. However, it was interesting to note that DNAm GA correlated more strongly with clinical GA estimates based on ultrasound than those based exclusively on LMP (Additional file 1: Figure S6). Error rates for ultrasound range from 5–7 days if performed during the first trimester to 3.0–4.3 weeks when performed in the third trimester. This predictor is closer to clinical estimates of GA than post-birth measures using neonatal estimation, which can overestimate the GA of preterm neonates by up to 2.57 weeks [37–41].
The accuracy of this predictor is consistent with that of established clinical methods for estimating GA, though its accuracy can only be interpreted in the context of the available clinical measurements. Predictive accuracy was not influenced by neonatal sex as there was no difference between the median errors in males versus females (p = 0.76). The median error between DNAm GA and clinically estimated GA was 1.07 for the cord blood datasets and 1.57 for blood spot datasets. This discrepancy may be due to differences in the precision of GA, as sample collection for blood spots was performed up to 39 days after birth (Additional file 1: Figures S3 and S7). It is also possible that there may be differences in sample quality, as some blood spot samples were stored for more than 30 years, although there was no difference in the number of samples that failed quality control between the cord blood and blood spot datasets. The correlation between DNAm GA and clinically estimated GA was 0.94 (median error (m.e.) = 1.4) for samples processed on the HumanMethylation450 array and 0.55 (m.e. = 1.02) for samples processed on the HumanMethylation27 array (Additional file 1: Figure S8). This discrepancy is likely due to the differences in GA range between samples run on the two arrays (19.3 and 11.1 weeks, respectively). Finally, the partial correlation from regressions of DNAm GA on clinically estimated GA did not substantially change when cell composition covariates were included, suggesting that the accuracy of the predictor is not confounded by cellular heterogeneity (roriginal = 0.91, rcell type adjusted = 0.81).
To limit concerns regarding the potential for overfitting of the models, we next validated the predictor in a second testing dataset, comprised of 92 samples from three cohorts (FAP, GSE66459, and GSE69633) that were not included in the initial testing or training sets. Cohort demographics are provided in Additional file 1: Table S3. The correlation in these datasets is similar to that of the first testing dataset (r = 0.89, m.e. = 0.89; Additional file 1: Figure S9), further indicating that this model fits well when applied to novel datasets and should be generalizable to other studies.
Accuracy of DNAm GA in the same subjects
Serial blood sampling was conducted from two neonates admitted to the Neonatal Intensive Care Unit (NICU), independent of the testing and training samples. Seven timepoints were collected between birth at 25 weeks and discharge at 40 weeks. DNAm GA increased as expected over time from birth until term equivalency (Fig. 2). These pilot data demonstrate that the predictor has the sensitivity required to detect changes in DNAm GA in the magnitude of days or weeks and that methylation patterns change from birth to term equivalency in a predictable manner.

Reproducibility of DNAm GA. DNAm GA from birth until term equivalency for two subjects recruited through the EpiPrem study, gestational age at birth 25 weeks. DNAm GA increases appropriately with gestational age in weeks. Change in DNAm GA over equivalent weeks gestation
DNAm GA as a measure of developmental age
In adults, the difference between DNAm-based age estimates and chronological age associates with all cause mortality, HIV status, and Down syndrome [17, 42, 43]. This difference is usually described as age acceleration [13]. We calculated a similar measure, which we will subsequently refer to as “GA acceleration”, in our cohorts by using the residual of a linear model regressing DNAm GA on clinically estimated GA. Because accelerated GA may indicate increased developmental maturity, we sought to evaluate whether GA acceleration associated with perinatal measures of health and development in the cohorts with available data.
Birthweight is widely used as a proxy of developmental maturity in studies assessing the association between the prenatal environment and short-term or long-term neonatal risk, with those born at the lowest birthweight generally having the highest risk for mortality over the first year of life and for cardio-metabolic conditions as adults [44, 45]. Birthweight is positively correlated with GA so birthweight percentile, which is calculated based on birthweight averages for a given GA corrected for fetal sex, is commonly used as an indicator of perinatal health [46, 47]. Previous studies have shown that infants in the lowest birthweight percentiles have an increased risk of perinatal death and other adverse outcomes and are often defined as growth restricted [48, 49]. In cohorts with available data, GA acceleration significantly predicted birthweight percentile (p = 4.5 × 10−4; Fig. 3a) and birthweight (p = 0.033; Fig. 3b) after correcting for clinically estimated GA, race, estimated cell type proportions, and cohort. Consistent with the idea that DNAm GA may reflect maturity, the fitted regression model predicts approximately the 50th percentile to have GA acceleration of 0. Thus, neonates falling in the lowest birthweight percentiles show lower, while neonates falling in the highest percentiles show higher or accelerated GA. There was no association between GA acceleration calculated using the DNAm age predictor of Horvath [13] and either birthweight or birthweight percentile (Additional file 1: Figure S10).

GA acceleration associates with birthweight. The association between GA acceleration and a birthweight percentile (p = 4.5 × 10−4) or b birthweight (p = 0.033) adjusted for race, cellular composition, cohort, and gestational age in CANDLE, WMHP, and PROGRESS. Solid line = regression line
One study by Appleton and colleagues [50] suggests that socioeconomic adversity promotes adverse health outcomes through epigenetic programming of neonatal DNAm. We hypothesize that factors related to early life adversity might influence the developmental age of the neonate. One such factor is socioeconomic status, which is essential to examine as children born into socioeconomically disadvantaged families, often operationalized by insurance status (Medicaid versus private health insurance), have poorer health in childhood and early adulthood [51, 52]. In the most socioeconomically diverse cohort (CANDLE), GA acceleration associated with maternal Medicaid status (p = 0.023) after adjusting for race, clinically estimated GA, and estimated cell type proportions (Fig. 4). Specifically, methylation-based estimates of GA were lower than clinical estimates for the neonates of women on Medicaid compared with women with private health insurance. This association supports the hypothesis that prenatal adversity associates with changes in neonatal methylation consistent with a delayed developmental age, which may have consequences later in life.

Maternal insurance status and GA acceleration. Neonates born to mothers with private insurance have higher GA acceleration than neonates born to mothers on Medicaid (p = 0.023) after adjusting for race, gestational age, and cellular composition
Conclusions
GA can be accurately predicted between 24 and 44 weeks gestation using DNAm values obtained from both umbilical cord blood and blood spot samples. DNAm GA is more concordant with GA estimates performed with the gold standard of ultrasound than with estimates based on LMP. However, the question remains as to whether GA acceleration is truly a measure of maturity versus a reflection of the relative accuracy of DNAm GA compared with clinical estimates. We consider three possibilities for interpreting the difference between DNAm GA and clinically estimated GA. First, an accelerated GA may reflect differences in physiological development of the neonate such that neonates with a higher DNAm GA are more developmentally mature than their chronological age suggests. A second possibility is that the differences between DNAm GA and chronological GA reflect epigenetic programming by early life environmental exposures, such as maternal prenatal stress or pregnancy disorders, which may affect neonatal outcomes and development [53]. Finally, any difference may simply be reflective of the variable nature of clinical GA estimations; evaluation of DNAm GA in neonates conceived through in vitro fertilization would be helpful for delineating these different possibilities. These models may be interrelated, such that the true interpretation is likely a combination of these possibilities. A future study examining other prediction methods, including the use of non-linear models or transformations, may facilitate this interpretation by further delineating developmental differences between early and late GAs. Overall, our results suggest that DNAm GA and GA acceleration are promising tools for evaluating neonatal developmental maturity.
A targeted assay of the CpG sites necessary to compute DNAm GA could provide a rapid and robust estimator of GA at birth, and the framework described in this paper could be used to develop and validate a predictor based on other tissues that may be sampled prior to delivery, such as chorionic villi or amniotic fluid. Our results suggest that DNAm GA is highly reproducible and can predict measures of developmental maturity, such as birthweight, better than clinical estimates of GA alone. As such, it has the potential to serve as a biomarker for GA and the rate of neonatal development. Recent studies of GA and DNAm [23–26] support that shifts in methylation underlie the aging process, further supporting the development of methylation-based biomarkers. DNAm is a convenient molecular marker for GA in that umbilical cord blood and blood sampling are routinely performed to monitor neonatal health in humans, and it can be readily sampled repeatedly in the same person, as demonstrated by the time course data in the subjects from preterm birth through term equivalency.
As a biomarker, DNAm GA and GA acceleration would have numerous clinical, research, and forensic applications. It would serve as a molecular marker of GA that complements clinical estimates, when available, and provides additional information when clinical estimates are unavailable or unreliable. For example, it could be used to estimate GA in women who seek prenatal care late in pregnancy, are unsure of the date of their last menstrual period, or did not have ultrasounds performed early in pregnancy. DNAm GA is more precise than the estimation methods typically performed at birth, which rely on biometric measurements. Precise knowledge of GA would be most informative for neonates born extremely preterm, when parents and clinicians are confronted with decisions regarding active intensive care interventions versus providing comfort care. GA based on an epigenetic developmental profile may also complement clinical estimates of GA, providing a screening tool to identify children who may benefit from additional monitoring and care. Studies to explore the extent to which DNAm GA reflects developmental maturity, and thus may be a more reliable predictor of outcomes after preterm birth compared to time or growth-based methods, are needed.
DNAm GA may also serve as a surrogate marker for developmental maturity in research studies of neonatal development, interventions, and disease. Our results already demonstrate that it will be fruitful to study antenatal and perinatal factors that associate with DNAm GA and GA acceleration and to determine whether these metrics are better prognosticators of neonatal well-being than conventional measures. Future studies should evaluate the effects of maternal stress, nutrition, and interventions such as vitamin supplementation that are highly relevant to fetal development and pregnancy outcomes. Future research could also explore whether GA acceleration relates to risk of developing pediatric disorders, such as autism, and whether it can predict health outcomes later in life. Finally, establishing precise GA is important for forensic, anthropologic, or other medico-legal investigations. Indeed, DNAm-based predictors of adult age are already under investigation for forensic applications [54]. In summary, we have identified a potential biomarker for GA with an abundance of applications that warrant further investigation and development.
Methods
Description of cohorts
Training datasets were selected to include a wide range of GAs and ancestries. Publically available datasets were downloaded from the Gene Expression Omnibus (GEO): GSE36642, GSE62924 [27], GSE51180 [55], and GSE30870 [56]. Methylation data for all of these datasets were generated on either the Illumina Infinium HumanMethylation27 BeadChip or Infinium HumanMethylation450 BeadChip (Table 1). These methods have been shown to be highly reproducible and consistent with the results of other epigenetic methods [57, 58]. For umbilical cord blood samples, GA was defined as the GA at birth. For blood spot samples, GA was defined as the GA plus the number of days that occurred between birth and sampling. The individual cohorts are detailed in Additional file 1.
Quality control and normalization
All analyses were performed using R version 3.1.2. Datasets used in this study underwent several quality control measures. The DNAm age predictor developed by Horvath was initially run on all samples to establish predicted age and gender [13]. Samples with gender discordance or estimated age >1.5 years were excluded from further analysis. After this initial quality control step, datasets were subjected to standard quality control through the use of the R package CpGassoc [59]. A data frame consisting of β values (Methylated signal/(Methylated signal + Unmethylated signal)) was supplied as input to CpGassoc. Any data point with a detection p value above 0.001 was set to missing. CpG sites with >5 % missing data were excluded; subsequently, samples with >5 % missing data were excluded. These quality control measures were performed to ensure that the predictor is built based on high quality probes and samples. Any probe missing entirely from one of the datasets was excluded from the remaining datasets, so only probes passing quality control in all training datasets and probes present on both the HumanMethylation450 and HumanMethylation27 arrays were included, for a total of 16,838 probes. Finally, datasets were normalized according to Horvath’s modified beta-mixture quantile (BMIQ) normalization [13, 60]. While the original BMIQ is a within-sample normalization method to address probe type bias by modifying the type II distribution to match that of type I probes, Horvath modified this BMIQ procedure for a different purpose: the distribution of each given array is related to that of a “gold standard” array (defined here as the mean across all of the training datasets). Thus, Horvath’s modification of the BMIQ method could be interpreted as a form of between-sample normalization. All training datasets were normalized together, as a single group. After normalization, missing values for each sample were imputed by the k-nearest neighbors method where k = 10, using the R package impute so that no missing values remain in the dataset after pipeline completion [61]. Test datasets were normalized separately, following the same procedures as above. One test cohort, PROGRESS, which was processed with an out of band background correction, dye bias correction, and then the original BMIQ procedure, was excluded from the quality control pipeline as raw files were not available. Principal components analysis was used to assess the potential impact of BeadChip on the CpG sites selected for inclusion in the predictor. We did not observe clustering by chip (Additional file 1: Figure S11), suggesting that the chip was not a confounding factor.
Estimation of cellular composition
Proportions of white blood cells and nucleated red blood cells were estimated from genome-wide DNAm patterns using the method proposed by Houseman et al. [62], with reference samples from homogenous cell populations for white blood cells (CD4+ T cells, CD8+ T cells, natural killer cells, B cells, monocytes, and granulocytes), nucleated red blood cells [63, 64], and whole blood (GSE80310).
Epigenome-wide association study
The R package CpGassoc [59] was used to perform epigenome-wide association studies (EWAS) to assess associations between GA and DNAm. Two separate EWAS were performed, with and without the inclusion of cellular composition covariates. Each EWAS was performed as a meta-analysis across all cohorts by including indicators for each study as covariates. Test statistics from the two EWAS were plotted to assess the robustness of results to potential cell type heterogeneity.
Elastic net regression and age prediction
The six training datasets (GSE36642, WMHP1, GSE62924, NBC, GSE51180, and GSE30870) were combined to perform an elastic net regression of GA on the 16,838 CpG probes remaining after quality control and filtering. The regression was performed using the R package glmnet to select a parsimonious set of CpG sites predictive of GA. Following Horvath [13], the elastic net mixing parameter, alpha, was set to 0.5 allowing for equal contribution of the ridge and lasso methods [65]. The lambda parameter was chosen through a tenfold cross validation, which involves randomly partitioning the training dataset into ten equally sized subsamples. The cross-validation procedure is then performed ten times, retaining a different subsample as a validation dataset each time. In the procedure, data from the other nine subsamples are used to build a predictor based on a particular value of lambda, and the fit of the predictor is then tested in the omitted validation set. The mean squared error is calculated for the validation set in each iteration and then averaged over the ten subsamples. This procedure is performed for a sequence of lambda values to determine the lambda that yields the minimum mean squared error. No additional covariates were included in the analysis, consistent with the development of the DNAm age predictor by Horvath [13]. The training coefficient values and CpG probes selected from this regression were used to fit a linear model to generate predicted values of GA, based on a modified version of the R code in the DNAm age tutorial published by Horvath [13]. The accuracy of predicted values of GA was determined from correlation coefficients obtained through linear regression of DNAm GA and clinical GA.
Analysis of GA acceleration
GA acceleration was calculated as the residual from a linear regression of DNAm GA on clinical estimates of GA for the combined testing dataset. Analysis of DNAm GA with birthweight and birthweight percentile was then conducted using linear regression of birthweight and birthweight percentile on GA acceleration and covariates for race, estimated cell type proportions, and cohort. Clinically estimated GA was included as a covariate in the analysis for birthweight but not birthweight percentile as birthweight percentile is already adjusted for clinically estimated GA. Maternal insurance status (as a proxy for income) was analyzed in the CANDLE cohort through logistic regression of maternal insurance status on GA acceleration, adjusting for estimated clinical GA, race (African American versus Caucasian), and estimated cell type proportions.
Enrichment tests
To assess whether the CpG sites selected for the DNAm GA predictor were more likely than others to be located in functionally relevant regions, two approaches were used. First, CpG positions were intersected with the hg19 CpG island annotation track from the UCSC Genome Browser (http://genome.ucsc.edu) to define whether each site was located in a CpG island, CpG shore (±1.5 kb from island), or CpG shelf (±1.5 kb from shore). Second, the CpG positions were intersected with ENCODE’s ChromHMM annotation for lymphoblastoid cell line GM12878, which uses a hidden Markov model to assign genomic features based on the combinatorial pattern of various chromatin marks [66]. The ChromHMM annotation allowed identification of CpGs located in promoters and enhancers. Fisher’s exact test was used to assess whether there was significant enrichment of each feature in CpG sites selected for the predictor compared to the full set of 16,838 sites included in the elastic net model. A similar analysis was preformed to assess whether these CpG sites were enriched for sites containing a genetic variant in the 50-bp probe (using annotation derived from the 1000 Genomes Project) or sites previously reported to associate with race [31]. DAVID was used to evaluate whether CpG sites used to estimate DNAm GA were located in genes enriched for any biological pathways [32].
References
Young PC, Glasgow TS, Li X, Guest-Warnick G, Stoddard G. Mortality of late-preterm (near-term) newborns in Utah. Pediatrics. 2007;119:e659–665.
Engle WA. Morbidity and mortality in late preterm and early term newborns: a continuum. Clin Perinatol. 2011;38:493–516.
Yang S, Platt RW, Kramer MS. Variation in child cognitive ability by week of gestation among healthy term births. Am J Epidemiol. 2010;171:399–406.
Davis EP, Buss C, Muftuler LT, Head K, Hasso A, Wing DA, Hobel C, Sandman CA. Children’s brain development benefits from longer gestation. Front Psychol. 2011;2:1.
Hansen AK, Wisborg K, Uldbjerg N, Henriksen TB. Risk of respiratory morbidity in term infants delivered by elective caesarean section: cohort study. BMJ. 2008;336:85–7.
Parikh LI, Reddy UM, Mannisto T, Mendola P, Sjaarda L, Hinkle S, Chen Z, Lu Z, Laughon SK. Neonatal outcomes in early term birth. Am J Obstet Gynecol. 2014;211:265. e1–265.e11.
ACOG. Committee Opinion No 579: Definition of term pregnancy. Obstet Gynecol. 2013;122:1139–40.
Extremely Preterm Birth. http://www.acog.org/Patients/FAQs/Extremely-Preterm-Birth.
Lynch CD, Zhang J. The research implications of the selection of a gestational age estimation method. Paediatr Perinat Epidemiol. 2007;21 Suppl 2:86–96.
Mongelli M, Wilcox M, Gardosi J. Estimating the date of confinement: ultrasonographic biometry versus certain menstrual dates. Am J Obstet Gynecol. 1996;174:278–81.
Dubowitz LM, Dubowitz V, Goldberg C. Clinical assessment of gestational age in the newborn infant. J Pediatr. 1970;77:1–10.
Ballard JL, Khoury JC, Wedig K, Wang L, Eilers-Walsman BL, Lipp R. New Ballard Score, expanded to include extremely premature infants. J Pediatr. 1991;119:417–23.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011;6:e14821.
Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15:R24.
Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25.
Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016;15:149–54.
Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY). 2015;7:1159–70.
Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015;44:1388–96.
Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015;7:1198–211.
Breitling LP, Saum KU, Perna L, Schottker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016;8:21.
Parets SE, Conneely KN, Kilaru V, Fortunato SJ, Syed TA, Saade G, Smith AK, Menon R. Fetal DNA methylation associates with early spontaneous preterm birth and gestational age. PLoS One. 2013;8:e67489.
Schroeder JW, Conneely KN, Cubells JC, Kilaru V, Newport DJ, Knight BT, Stowe ZN, Brennan PA, Krushkal J, Tylavsky FA, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. 2011;6:1498–504.
Simpkin AJ, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Tilling K, Davey Smith G, Relton CL. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet. 2015;24(13):3752-63.
Lee H, Jaffe AE, Feinberg JI, Tryggvadottir R, Brown S, Montano C, Aryee MJ, Irizarry RA, Herbstman J, Witter FR, et al. DNA methylation shows genome-wide association of NFIX, RAPGEF2 and MSRB3 with gestational age at birth. Int J Epidemiol. 2012;41:188–99.
Rojas D, Rager JE, Smeester L, Bailey KA, Drobna Z, Rubio-Andrade M, Styblo M, Garcia-Vargas G, Fry RC. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci. 2015;143:97–106.
Smith AK, Conneely KN, Newport DJ, Kilaru V, Schroeder JW, Pennell PB, Knight BT, Cubells JC, Stowe ZN, Brennan PA. Prenatal antiepileptic exposure associates with neonatal DNA methylation differences. Epigenetics. 2012;7:458–63.
Schroeder JW, Smith AK, Brennan PA, Conneely KN, Kilaru V, Knight BT, Newport DJ, Cubells JF, Stowe ZN. DNA methylation in neonates born to women receiving psychiatric care. Epigenetics. 2012;7:409–14.
Nemoda Z, Massart R, Suderman M, Hallett M, Li T, Coote M, Cody N, Sun ZS, Soares CN, Turecki G, et al. Maternal depression is associated with DNA methylation changes in cord blood T lymphocytes and adult hippocampi. Transl Psychiatry. 2015;5:e545.
Adkins RM, Krushkal J, Tylavsky FA, Thomas F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res A Clin Mol Teratol. 2011;91:728–36.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol. 2014;38:231–41.
Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CC, O’Donovan MC, Bray NJ, Mill J. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25:338–52.
Simpkin AJ, Hemani G, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Sharp GC, Tilling K, Horvath S, et al. Prenatal and early life influences on epigenetic age in children: A study of mother-offspring pairs from two cohort studies. Hum Mol Genet. 2015;25(1):191-201.
Nguyen TH, Larsen T, Engholm G, Moller H. Evaluation of ultrasound-estimated date of delivery in 17,450 spontaneous singleton births: do we need to modify Naegele’s rule? Ultrasound Obstet Gynecol. 1999;14:23–8.
Moore KA, Simpson JA, Thomas KH, Rijken MJ, White LJ, Dwell SL, Paw MK, Wiladphaingern J, Pukrittayakamee S, Nosten F, et al. Estimating gestational age in late presenters to antenatal care in a resource-limited setting on the Thai-Myanmar border. PLoS One. 2015;10:e0131025.
Donovan EF, Tyson JE, Ehrenkranz RA, Verter J, Wright LL, Korones SB, Bauer CR, Shankaran S, Stoll BJ, Fanaroff AA, et al. Inaccuracy of Ballard scores before 28 weeks’ gestation. National Institute of Child Health and Human Development Neonatal Research Network. J Pediatr. 1999;135:147–52.
Sanders M, Allen M, Alexander GR, Yankowitz J, Graeber J, Johnson TR, Repka MX. Gestational age assessment in preterm neonates weighing less than 1500 grams. Pediatrics. 1991;88:542–6.
Karunasekera KA, Sirisena J, Jayasinghe JA, Perera GU. How accurate is the postnatal estimation of gestational age? J Trop Pediatr. 2002;48:270–2.
Dubowitz L, Ricciw D, Mercuri E. The Dubowitz neurological examination of the full-term newborn. Ment Retard Dev Disabil Res Rev. 2005;11:52–60.
Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;13(3):491-95.
Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015;212:1563–73.
Wilcox AJ, Russell IT. Birthweight and perinatal mortality: II. On weight-specific mortality. Int J Epidemiol. 1983;12:319–25.
Godfrey KM, Barker DJ. Fetal nutrition and adult disease. Am J Clin Nutr. 2000;71:1344S–52S.
Coory M. Does gestational age in combination with birthweight provide better statistical adjustment of neonatal mortality rates than birthweight alone? Paediatr Perinat Epidemiol. 1997;11:385–91.
Hertz-Picciotto I, Din-Dzietham R. Comparisons of infant mortality using a percentile-based method of standardization for birthweight or gestational age. Epidemiology. 1998;9:61–7.
Boulet SL, Alexander GR, Salihu HM, Kirby RS, Carlo WA. Fetal growth risk curves: defining levels of fetal growth restriction by neonatal death risk. Am J Obstet Gynecol. 2006;195:1571–7.
Patterson RM, Prihoda TJ, Gibbs CE, Wood RC. Analysis of birth weight percentile as a predictor of perinatal outcome. Obstet Gynecol. 1986;68:459–63.
Appleton AA, Armstrong DA, Lesseur C, Lee J, Padbury JF, Lester BM, Marsit CJ. Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS One. 2013;8:e74691.
Silva LM. Fetal origins of socioeconomic inequalities in early childhood health. The Netherlands: The Generation R Study, Erasmus University Rotterdam; 2009.
Kramer MS, Seguin L, Lydon J, Goulet L. Socio-economic disparities in pregnancy outcome: why do the poor fare so poorly? Paediatr Perinat Epidemiol. 2000;14:194–210.
Ching T, Ha J, Song MA, Tiirikainen M, Molnar J, Berry MJ, Towner D, Garmire LX. Genome-scale hypomethylation in the cord blood DNAs associated with early onset preeclampsia. Clin Epigenetics. 2015;7:21.
Yi SH, Xu LC, Mei K, Yang RZ, Huang DX. Isolation and identification of age-related DNA methylation markers for forensic age-prediction. Forensic Sci Int Genet. 2014;11:117–25.
Cruickshank MN, Oshlack A, Theda C, Davis PG, Martino D, Sheehan P, Dai Y, Saffery R, Doyle LW, Craig JM. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96.
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109:10522–7.
Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe H, Lehmann U. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes. 2012;5:210.
Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium((R)) assay. Epigenomics. 2009;1:177–200.
Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics. 2012;28:1280–1.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96.
Hastie T, Tibshirani R, Balasubramanian N, Chu G. impute: Imputation for microarray data. R package version 1.42.0. https://bioconductor.org/packages/release/bioc/html/impute.html.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361.
de Goede OM, Razzaghian HR, Price EM, Jones MJ, Kobor MS, Robinson WP, Lavoie PM. Nucleated red blood cells impact DNA methylation and expression analyses of cord blood hematopoietic cells. Clin Epigenetics. 2015;7:95.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9.
Acknowledgements
The authors gratefully acknowledge Dr. Isabel Iglesias Platas, Dr. Holger Heyn, and Dr. Manel Esteller for providing their unpublished data for the training dataset.
Funding
This research was supported by grants from the National Institutes of Minority and Health Disparities (R01MD009064 to AKS), the National Health and Medical Research Council of Australia (project grants 1083779, 491246; Centre of Research Excellence Grants 546519, 1060733, early career fellowship to JLYC 1053787), and the Urban Child Institute. Salary support for AKK, EMK, and SEP was provided, in part, by the National Institute of General Medical Sciences (T32GM008490) and the National Institute of Environmental Health Sciences (T32ES012870), respectively. Salary support for ACJ was provided by NIEHS grant K99 ES023450. Salary support for SH was provided by NIH/NIA 1U34AG051425-01. Support for PREDO was provided by the Academy of Finland (grants 127437, 129306, 130326, 134791, and 263924), Sigrid Juselius Foundation, Foundation for Pediatric Research, Novo Nordisk Foundation (to EK). Additional support provided by the Academy of Finland grants 121196 and 134791, Finnish Medical Society Duodecim, Government Special Subsidy for Health Sciences at Helsinki and Uusimaa Hospital District, Jane and Aatos Erkko Foundation, Päivikki and Sakari Sohlberg Foundation, and University of Helsinki Research Funds (to HL). The PROGRESS/ELEMENT study was supported by funding from the National Institute of Environmental Health Sciences (R01ES020268; R01ES021357) and by the National Institute of Public Health/Ministry of Health of Mexico. The American British Cowdray Hospital provided facilities used for PROGRESS/ELEMENT research. WMHP sample collection was supported by the Translational Research Center in Behavioral Sciences (TRCBS; P50 MH077928 to ZNS) and methylation assays were provided by the National Institute of Mental Health (RC1 MH088609 to AKS/PAB). The FAP study was supported by the Georgia Experimental Agriculture Station, HATCH #GEO00706 and the Interdisciplinary Proposal Developmental Program at the University of Georgia.
Availability of data and materials
R code and documentation for predicting DNAm GA is included as Additional files 3, 4, 5, 6, and 7 and requires Additional files 21, 22, and 24 from Horvath [13] or can be accessed at https://github.com/akknight/PredictGestationalAge under an open-source, MIT license (DOI 10.5281/zenodo.60519). The datasets supporting the conclusions of this article are available in NCBI Gene Expression Omnibus (GEO) under accession numbers: GSE64940 (CANDLE, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64940), GSE79056 (NBC, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79056), GSE80283 (VICS, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80283), GSE79969 (EpiPrem, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79969), GSE80310 (FAP, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80310), GSE36642 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36642), GSE62924 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62924), GSE51180 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51180), GSE30870 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30870), GSE66459 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse66459) GSE69633 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse69633), the European Genome Phenome Archive under accession number EGAS00001001898 (PREDO, https://www.ebi.ac.uk/ega/studies/EGAS00001001898). Relevant data from PROGRESS are provided as Additional file 8. Subject enrollment and ascertainment for the WMHP cohorts was obtained under NIH grant P50 MH-77928, which established a Data Enclave through which investigators can request clinical and molecular data from the center director (ZNS). This approach was approved by NIH due to 1) the sensitive privacy issues specific to clinical research data, 2) the vulnerable population to be studied, and 3) the inclusion of medical information such as obstetrical and labor and delivery records that makes complete de-identification difficult to achieve. Individual level data for DNSBtrios is not publically availably due to legal restrictions in the country where it was collected.
Authors’ contributions
AKK carried out the data analysis to develop and test the predictor of gestational age and drafted the manuscript. JMC and PD participated in methylation analysis of VICS. LWD and JLYC contributed to data collection (VICS). YJL optimized the DNA extraction of dried blood in VICS and was involved in manuscript revisions. CT participated in the planning and analysis (VICS) and provided methylation data as PI of the EpiPrem study. PMV, EK, KR, HL, EBB, SI, AKP, DC, EH, and JMTL participated in the design and execution of the PREDO study. PBM, SMW, and TMW initiated the DNSBtrios study. PBM identified the samples. SMW and TMW quality controlled the data. MBH, JBG, CSH, MVH, and DH prepared the DNSBtrios, extracted the DNA, processed the methylation arrays, quality controlled the cohort and prepared the data for inclusion in the study. ZNS and DJN initiated sample collection in WMHP. PAB, JFC, and AKS initiated studies of DNAm and contributed to generation and analysis of the methylation data from WHMP. SEP and RM participated in the execution of the NBC study. MMTR contributed to the design and acquisition of the PROGRESS cohort. KS cleaned the phenotypic data and oversaw the process of the PROGRESS database management. ACJ oversaw the quality control and preprocessing analysis of the PROGRESS methylation data. LT generated the plating scheme for PROGRESS samples and assisted in quality control. ROW designed the PROGRESS study as PI and oversaw collection of the cord blood samples. AAB oversaw the methylation analysis and methylation study design. NRB, KZL, and FAT participated in the design and execution of the CANDLE study. HJP and LBB initiated collection of the FAP cohort and participated in generation of the methylation data. VK assisted in quality control and analysis of the WMHP, NBC, and FAP cohorts. SH participated in the study design, validation of the predictor, and data interpretation. KNC and AKS conceived of the study, supervised the data analysis and interpretation, and made substantial revisions to the manuscript. All authors participated in interpretation of the data and manuscript revision. All authors read and approved the final manuscript.
Competing interests
The authors do not have any competing interests related to this work.
Ethics approval and consent to participate
This study was conducted in compliance with the Helsinki Declaration. All subjects have given written informed consent, except participants of DNSBtrios. Permission to include the Danish samples and phenotypes in the study without individual informed consent from participants was granted by the Danish Health Board according to the Law on Patient’s rights (law number 482 of July 1st, 1998) on the condition that individual level data not be made public and that no attempt to identify or contact participants be made. The CANDLE study was approved by the Institutional Review Board of the University of Tennessee Health Science Center (06-08495-FB). The EpiPrem and VICS studies were approved by the Human Research Ethics Committee at the Royal Women’s Hospital Melbourne (projects 13/43 and 08/06). The FAP study was approved by the University of Georgia Institutional Review Board on Human Subjects (STUDY0000050) and the Athens Regional Medical Center Institutional Review Board. The study was registered at ClinicalTrials.gov (NCT02124642). The NBC study was approved by the Western Institutional Review Board (PRC-1-2007). The PREDO study was approved by the Ethics Committee of Obstetrics and Gynecology at Hospital District of Helsinki and Uusimaa. The PROGESS study was approved by Mount Sinai Health System Program for the Protection of Human Subjects; HS# 12-00751. The WMHP studies were approved by the Emory University Institutional Review Board (IRB00004249).
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figures S1–S11
and Tables S1–S3. (PDF 3090 kb)
Additional file 2:
CpG sites and corresponding genes used to predict DNAm age. (XLSX 43 kb)
Additional file 3:
CpG sites and coefficients used to predict DNAm age. (CSV 3 kb)
Additional file 4:
Wrapper program for predicting gestational age. (R 6 kb)
Additional file 5:
R code to normalized and predict DNAm age. (R 6 kb)
Additional file 6:
Test dataset for predicting DNAm age. (CSV 1240 kb)
Additional file 7:
Instructions for predicting gestational age from cord blood and blood spot methylation. (DOCX 137 kb)
Additional file 8:
Relevant data from PROGRESS cohort. (CSV 282 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Knight, A.K., Craig, J.M., Theda, C. et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol 17, 206 (2016). https://doi.org/10.1186/s13059-016-1068-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-016-1068-z