
Abstract
Mass spectrometry- and nuclear magnetic resonance-based metabolomic studies comparing diseased versus healthy individuals have shown that microbial metabolites are often the compounds most markedly altered in the disease state. Recent studies suggest that several of these metabolites that derive from microbial transformation of dietary components have significant effects on physiological processes such as gut and immune homeostasis, energy metabolism, vascular function, and neurological behavior. Here, we review several of the most intriguing diet-dependent metabolites that may impact host physiology and may therefore be appropriate targets for therapeutic interventions, such as short-chain fatty acids, trimethylamine N-oxide, tryptophan and tyrosine derivatives, and oxidized fatty acids. Such interventions will require modulating either bacterial species or the bacterial biosynthetic enzymes required to produce these metabolites, so we briefly describe the current understanding of the bacterial and enzymatic pathways involved in their biosynthesis and summarize their molecular mechanisms of action. We then discuss in more detail the impact of these metabolites on health and disease, and review current strategies to modulate levels of these metabolites to promote human health. We also suggest future studies that are needed to realize the full therapeutic potential of targeting the gut microbiota.
Similar content being viewed by others
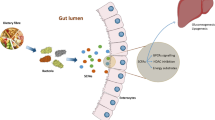


Alterations in microbial transformation of dietary components associate with disease
The symbiotic relationship between mammals and the trillions of microbial cells that reside in their gastrointestinal tracts relies on a complex molecular dialogue, with microbial metabolites acting as major mediators of this dialogue. Essential roles for several microbial metabolic pathways in host physiology have been long established, including in the production of vitamin K, the production of water-soluble B vitamins including biotin, folates, nicotinic acid, pyridoxine, riboflavin, cobalamin and panthotenic acid, the degradation of dietary oxalates, and modification of bile salts (reviewed in [1, 2]). However, intense interest in the gut microbiota over the past decade has led to the discovery of many new areas where bacterial transformation of dietary components may play critical roles in host health and disease. This increased understanding of diet–microbiota–host interactions suggests significant opportunities to create new therapeutic approaches, including selectively altering the microbial production of molecules to promote human health and prevent disease [3].
Elucidating target microbial metabolites that modulate host physiology requires identifying the major metabolites (and their downstream co-metabolites formed by the phase I/II xenobiotic metabolizing enzymes of their host) that differ between healthy and diseased individuals, and assessing the biological activities of these metabolites. A series of landmark metabolomics studies over the past decade have significantly advanced our understanding by using mass spectrometry (MS) or nuclear magnetic resonance (NMR) analysis to identify potentially important microbial metabolites that derive from the gut microbes [4–10], that are enriched or depleted in diseased individuals [11–30], or that can be used to predict physiological response to foods or other interventions [31, 32] (Table 1). These studies have identified a number of metabolites that may play important roles in human health and disease, including short-chain fatty acids (SCFAs) and long-chain fatty acid metabolites such as conjugated linoleic acid (CLA) and 10-hydroxy-cis-12-octadecenoate (HYA); trimethylamine (TMA) and trimethylamine N-oxide (TMAO); tryptophan metabolites such as indole, indole-3-propionate (IPA) and indoxyl-sulfate (IndsS); and tyrosine and phenylalanine metabolites such as hippuric acid, phenylacetylglycine, phenyl sulfate, para-cresyl sulfate (PCS), phenylpropionylglycine, cinnamoylglycine and equol sulfate. Many of the metabolites identified by these studies result from the transformation of specific dietary components by select species of microbes that express the necessary enzymes to act on these components. Thus, the variable presence of microbes utilizing these diet-dependent metabolic pathways may be key to understanding the variable host response to specific dietary components and susceptibility to disease [32].
This review will focus on several key metabolites formed by the gut microbiota from dietary components that have been revealed recently to produce remarkable effects on host physiology and that are currently being targeted or have high potential to be targeted as treatments for human disease. We will describe briefly the microbial origin of these metabolites and the biological actions of these metabolites on their host. We will then discuss in more detail current and potential therapeutic approaches to manipulate these metabolite levels and broader areas of research that are needed to understand the potential value of gut microbial metabolites.
Short-chain fatty acids
Biosynthesis and molecular mechanisms of action
SCFAs constitute the most abundant microbial metabolite, reaching concentrations of 50–130 mM in the proximal colon [33]. The biochemical pathways leading to the formation of these SCFAs by saccharolytic microbes are reviewed in [34]. Acetate, the most abundant SCFA, is produced by many microbial species as acetyl coenzyme A (acetyl-CoA) and is central to many metabolic pathways [35]. Propionate is synthesized predominantly through the succinate pathway [36], while butyrate is synthesized predominantly via butyryl-CoA:acetate CoA transferase [37]. Because the production of SCFAs depends on complex cross-feeding of substrates and disposal of waste products such as hydrogen and carbon dioxide gas among various species of the microbial community [34], there is not a simple linear relationship between gut SCFA levels and individual dietary components or bacterial strains. This is a key point when considering therapeutic attempts to increase SCFAs, particularly because these other waste products produce significant gastrointestinal distress for the host. Thus, simply administering a single dietary component or strain of bacteria may not have the intended effect on SCFAs, and careful confirmation of alterations in SCFA levels and other products are needed to interpret the results of such studies.
A number of molecular mechanisms of action have been ascribed to acetate, propionate and butyrate that may be relevant to their therapeutic potential to promote intestinal health, reduce inflammation, and inhibit cancer (Table 2). All three SCFAs are ligands for G-protein-coupled receptor 43 (GPR43; also known as FFA2) and GPR41 (also known as FFA3), although they range in potency [38, 39]. Butyrate is also a low-affinity ligand for GPR109A (also known as hydroxycarboxylic acid receptor) [40]. These three receptors are present throughout the gastrointestinal tract, as well as on immune cells and adipose tissues, and have been implicated in the regulation of inflammation and cancer. Additionally, both propionate and butyrate inhibit histone deacetylase (HDAC) activity and thereby alter gene expression, which appears to suppress tumor formation and inflammatory pathways in many tissues. In hepatocytes and adipocytes, all three SCFAs appear to modulate peroxisome proliferator-activated receptor-γ (PPAR-γ) expression (by an unknown, indirect mechanism), which leads to increased expression of uncoupling protein-2, reduced ATP levels, and activation of AMP kinase (AMPK) [41]. Similarly, propionate modulates PPAR-γ activity in intestinal cells, one effect of which is to increase expression of epithelial Kruppel-like factor 4 [42], a tumor suppressor transcription factor that may be important in preventing colorectal cancer. Butyrate also inhibits the NF-κB pathway (a prototypical proinflammatory signaling pathway that expresses genes for cytokines, chemokines and adhesion molecules) [43–47]. All three SCFAs are used as energy substrates, with propionate serving as a substrate for gluconeogenesis while acetate and butyrate serve as substrates for fatty acid synthesis. These various actions of SCFAs allow them to exert pluripotent effects that generally promote intestinal health, reduce inflammation and inhibit cancer, and, as will be discussed below, a number of studies have investigated the therapeutic potential of SCFAs or fermentable fibers. The results of these studies have often been equivocal, suggesting that a far better understanding of the appropriate doses and the precise mechanisms by which SCFAs act in various disease states is needed to design more appropriate interventions.
Effects on intestinal inflammation and colorectal cancer
A decrease in luminal SCFAs is associated with ulcerative colitis and intestinal inflammation, which can be ameliorated with dietary fiber or administration of SCFAs [48–50]. Reduced barrier function promotes intestinal inflammation, and butyrate promotes barrier function by inducing “physiological hypoxia” in intestinal cells via HDAC inhibition [51], which thereby stabilizes hypoxia inducible factor-1α to regulate a number of genes that improve epithelial barrier function [52]. Butyrate inhibition of HDAC also promotes intestinal immune tolerance through regulating the function of intestinal macrophages [53] and development of regulatory T cells through mechanisms that involve acetylation of forkhead box P3 (FOXP3) [54, 55] and activation of GPR43 [56]. Deletion of GPR43 exacerbates intestinal inflammation in mice [57], while GPR43 activation by acetate can also protect against colonic epithelial injury [58]. Butyrate can also modulate the expression of intestinal tight junction proteins, enhance epithelial cell proliferation, and inhibit apoptosis [59], possibly through its effects on glucagon-like peptide (GLP)-2 secretion, which is known to have a trophic effect on the epithelium [60].
Intestinal inflammation contributes to the development of colorectal cancer, and the contribution of SCFA-producing bacteria to the inhibition of colon carcinogenesis remains unresolved. Besides its anti-inflammatory effects, butyrate also exerts anti-proliferative and anti-cancer effects when tumor cell lines are exposed to it in vitro [61–63], primarily through HDAC inhibition [64, 65]. Epidemiological studies, although inconclusive, show an inverse relationship between the intake of dietary fiber and incidence of colon cancer [66–71], suggesting that increased colonic SCFAs as a result of fiber fermentation may be responsible for the protective effect. However, large randomized multicenter clinical trials, such as The Polyp Prevention Trial (n = 2079) [72] and the Wheat Bran Study (n = 1429), [73] showed no impact of a high-fiber diet on recurring polyp formation. However, other studies have shown inconsistent relationships between SCFAs and colon cancer development in humans [74–77] as well as in animals (for a critical evaluation of studies, see [78]). Butyrate has been shown to stimulate cell proliferation in a number of studies under conditions of energy deprivation [79, 80], which is likely due to butyrate being an energy source for colonic epithelial cells. Yet, under states of hyperproliferation, such as that induced by secondary bile acids [81] or in cancer cells maintained under high glucose [80], butyrate suppresses proliferation. These discrepancies can be partially explained by the fact that cancer cells predominantly use glucose rather than fatty acids such as butyrate as an energy source (the Warburg effect) [82], resulting in intracellular accumulation of butyrate that sufficiently inhibits HDAC and consequently cell growth [83]. Critical review of these conflicting and sometimes paradoxical results reached the conclusion that butyrate exerts anti-proliferative effects only at specific sensitive stages of the carcinogenesis, that these effects are dependent on delivery of butyrate to the colon, that the extent of butyrate production in the colon varies widely based on type of fiber, and that very high colonic concentrations of butyrate are required and may be difficult to reproducibly achieve in humans [78, 84]. More recent studies using mice to carefully control cancer phenotype have not resolved these issues. For instance, a mouse study used gnotobiotic mice treated with azoxymethane followed by dextran sodium sulfate (DSS) to induce colon cancer and then colonized these mice with butyrate-producing bacterium, and found that dietary fiber had a butyrate-dependent tumor-suppressive effect that required microbiota [83]. In this model, butyrate was metabolized less in tumors and functioned as a HDAC inhibitor. In contrast, another recent study showed that gut microbial production of butyrate stimulated polyp formation in a genetic mouse model of colorectal cancer (Apc Min/+ Msh2 −/−) [85]. Importantly, more than 10 % of colon cancers in humans carry lesions in adenomatous polyposis coli (APC) and DNA mismatch repair gene MutS homolog 2 (MSH2) [86]. Thus, whether consumption of dietary fiber to generate butyrate can be used therapeutically to prevent or treat colorectal cancer remains very much unresolved. Given the need for very high butyrate levels to be effective and the aversion most humans have for high amounts of fiber in their diets, alternative strategies such as fibers engineered to increase butyrate production with reduced hydrogen and carbon dioxide gas production may be required.
Peripheral inflammation
The anti-inflammatory effects of SCFAs extend beyond the gut, such as inhibiting vascular smooth muscle cell proliferation and migration [87], improving kidney function [88], conferring anti-inflammatory effects in the lung [89, 90], and protecting against inflammatory arthritis [91]. The mechanisms underlying these effects appear to center largely on HDAC inhibition and GPR43 activation, and thereby act via modulating immune cell activation. For example, mice fed a high-fiber diet have increased circulating SCFAs, which protected against allergic inflammation in the lungs by a mechanism that involved impairing the capacity of dendritic cells to instigate a TH2-cell-mediated allergic inflammation [89]. High fiber or acetate feeding was found to suppress allergic airway disease by HDAC inhibition and increased FOXP3 acetylation in adult mice, and this effect was conferrable to fetal mice, in which a high-fiber or acetate maternal diet was able to suppress the expression of certain genes related to asthma [90]. The Canadian Healthy Infant Longitudinal Development study found that infants at risk for asthma showed transient alterations in the composition of their gut microbiota compared to low-risk infants during the first 100 days of life [92]. These at-risk infants had reduced levels of microbial taxa involved in SCFA formation (specifically Lachnospira, Veillonella, Faecalibacterium and Rothia) and reduced fecal acetate. Inoculating germ-free mice with these four microbial taxa ameliorated airway inflammation in their offspring, demonstrating a causal role in suppressing inflammation [92]. Together, these results suggest the potential for introducing bacteria (or combinations of bacteria) that increase SCFA production as a measure to prevent the development of asthma and other related inflammatory diseases in both adults and children. Realization of this potential will require optimization of appropriate microbiota for supplementation and subsequent clinical trials.
Effects on regulation of appetite and energy homeostasis
The landmark study by Turnbaugh and colleagues showing that transfer of microbiota from obese mice into germ-free mice increased adiposity and cecal levels of SCFAs relative to the transfer of microbiota from lean mice [12] has led to intense interest in the role of the microbial production of SCFAs in the regulation of appetite and energy homeostasis. Turnbaugh and colleagues attributed the obesegenic potential of transferred microbiota to its increased capacity to ferment dietary components to SCFAs, resulting in increased energy harvest. Subsequent studies showed obese humans have higher fecal SCFA levels than lean individuals [93] and that that roux-en-Y gastric bypass surgery, a highly effective treatment for obesity and type 2 diabetes, causes a significant change in fecal microbial profiles in humans and rodents and results in reduced levels of SCFAs [94–97]. While these results suggest that elevated microbial production of SCFAs promote obesity, a number of studies support an opposite conclusion. For instance, microbiota transfer experiments into germ-free mice from human twin donors where one was obese and one lean showed that, as with obese mice, phenotypes transferred with the microbiota; however, in this case transfer from lean donors resulted in higher cecal propionate and butyrate levels. Furthermore, diets enriched in inulin or other non-digestible fibers that increase formation of SCFAs consistently inhibit obesity in humans [98, 99]. Finally, direct administration of SCFAs, particularly butyrate, inhibits weight gain, adiposity, and insulin resistance in mice fed a high-fat diet (HFD) [41, 100–103].
A recent meta-analysis of various studies in this field by Byrne and colleagues led them to conclude that increasing SCFA levels had an overall net benefit on obesity due to their effects on satiety and reduced food intake, increased energy expenditure and thermogenesis, and inhibition of lipogenesis and cholesterol synthesis [104]. One molecular mechanism underlying the anti-obesity effect of SCFAs is improved barrier function, which prevents the passage of bacterial toxins into the circulation, inducing metabolic endotoxemia, obesity, and insulin resistance [105–107]. Additionally, SCFA activation of AMPK and GPR43 induces multiple responses that can reduce adiposity, including increasing fatty oxidation, decreasing glucose levels, and increasing secretion of satiety-inducing peptides such as GLP-1, peptide YY (PYY), and gastric inhibitory polypeptide (GIP) (reviewed in [34]).
Since, on the whole, increasing the microbial production of SCFAs appears to be a reasonable therapeutic intervention for the treatment of obesity, future studies are needed to determine how to effectively carry out such long-term interventions in humans. For a significant fraction of the human population, consumption of large amounts of non-digestible dietary fibers such as inulin is significantly hampered by undesirable gastrointestinal effects such as bloating, abdominal cramping, flatulence and diarrhea. These adverse effects result in part from the simultaneous formation of hydrogen gas and carbon dioxide during fermentation. Therefore, optimization of the microbiota or substrates to enhance SCFA production and to minimize released hydrogen and carbon dioxide gas will be critical for the wide-spread application of this treatment in the general population.
Trimethylamine and trimethylamine N-oxide
TMA and its co-metabolite TMAO were identified by screening metabolites associated with cardiovascular disease (CVD), and TMA was shown to require gut bacteria for its formation [15]. Cleavage of choline to TMA and acetaldehyde by two enzymes originally identified in Desulfovibrio desulfuricans, CutC and CutD, allow choline to be used as an energy source [108]. Recent studies found homologous genes in a variety of Proteobacteria and Firmicutes, and to a much lesser extent Actinobacteria, suggesting spread via horizontal gene transfer [109]. TMA was also recently shown to form from l-carnitine and choline via an analogous reaction catalyzed by the YeaW and YeaX enzymes originally characterized in Escherichia coli [110], and by CntA and CntB, originally characterized in Acinetobacter baumannii [111]. After formation and absorption in the colon, TMA passes into the portal circulation, which directs blood into the liver, where it is oxidized to TMAO by flavin-containing mono-oxygenase 3 (FMO3) [112]. Analysis of genetic variation among inbred strains of mice indicates that plasma TMAO levels significantly correlate with FMO3 activity [112]. Oral antibiotics block the increase in TMAO that normally occurs after dietary challenge with either choline or carnitine, demonstrating that the generation of TMAO requires microbial bacteria [15, 113, 114].
TMAO levels predict risk for atherosclerosis [15, 112, 115], and are elevated in patients with chronic kidney disease (CKD) [116] and obesity [17, 98], and decreased in ulcerative colitis [117]. TMAO directly induces CVD, as administration of TMAO itself or of sufficient choline or l-carnitine to raise TMAO levels can all increase atherosclerosis in Apoe −/− mice [15, 114]. The specific molecular mechanisms by which TMAO exerts its pathological effects are currently unknown. Accumulation of TMAO in the kidney may alter osmotic balance and elevated TMAO levels associate in animal models with markers of renal damage such fibrosis and dysfunction [116]. Thus far, no receptor for TMAO has been identified. TMA, but not TMAO, acts as a ligand for trace amine-associated receptor 5 (TAAR5) [118], but TAAR5 appears to be exclusively expressed in the olfactory sensory neurons. Administration of TMAO to Apoe −/− mice inhibits reverse cholesterol transport from macrophages in vivo [114], but treating macrophages directly with TMAO in cell culture does not increase their ability to take up cholesterol or inhibit their ability to efflux cholesterol to ApoA1 or HDL [119]. Reduction of FMO3 activity (which increases TMA levels and decreases TMAO levels) decreases intestinal cholesterol absorption, reduces hepatic biliary secretion and LXR signaling, and increases cholesterol disposal via transintestinal cholesterol efflux (active secretion of cholesterol from the small intestine) [120]. Administering antibiotics blocks these effects, while TMAO supplementation does not, suggesting that the effects of reducing FMO3 activity resulted from increased TMA or another microbial substrate of FMO3 [120]. Thus, studies elucidating the molecular targets of TMAO and the potential roles of TMA are greatly needed.
Without identified TMAO molecular targets, interventions to reduce CVD must focus on reducing TMAO levels. Reducing dietary choline or l-carnitine would lower TMAO levels, but may have undesirable effects. In particular, supplementation with lower levels of l-carnitine than needed for TMAO formation may improve cardiovascular function [121]. A meta-analysis of 13 controlled trials (n = 3629) showed that l-carnitine supplementation reduces all-cause mortality by 27 % [122]. While potentially beneficial for cardiovascular health, choline deficiency markedly increases risk for non-alcoholic liver disease. Inhibiting FMO3 to reduce TMAO levels is also undesirable, as accumulation of TMA results in fish malodor disorder. Because of these limitations, current pharmaceutical development is focusing on a revolutionary approach: non-lethal targeting of microbes by selectively inhibiting pathways detrimental to their host, such as microbial CutC/D, CntA/B and YeaW/X. A structural analog of choline, 3,3-dimethyl-1-butanol (DMB), non-lethally inhibits microbial CutC/D and reduces TMAO levels in mice fed a high-choline or l-carnitine diet [123]. Importantly, DMB inhibits macrophage foam cell formation and atherosclerotic lesion development in Apoe −/− mice [123]. Future clinical trials are needed to determine the safety and efficacy of CutC/D inhibitors in reducing TMAO levels and disease in humans, as well as whether resistance to their effects will occur with long-term treatment strategies. Nevertheless, this revolutionary strategy of selective, non-lethal inhibition of microbial function likely represents an important new front in the pharmacological treatment of human diseases.
Tryptophan metabolites: indole and indole derivatives
Tryptophan is an essential amino acid found in a variety of foods such as red meat, fish and eggs. Commensal bacteria expressing tryptophanase catabolize tryptophan to indole, a quorum-sensing compound for bacteria [124] (Fig. 1). Lactobacillus spp. convert tryptophan to indole-3-aldehyde (I3A) through unidentified enzymes [125]. Clostridium sporogenes convert tryptophan to IPA [6], likely via a tryptophan deaminase. After absorption from the intestinal tract into portal circulation, the liver converts indole to IndS.

Molecular mechanisms of action of indole and its metabolites on host physiology and disease. Tryptophan in the colonic lumen is catabolized by bacteria to yield indole and indole derivatives. Indole-3-propionate (IPA) acts on intestinal cells via pregnane X receptors (PXR) to maintain mucosal homeostasis and barrier function. IPA can also act on other organs such as the brain, where it confers neuroprotective effects against ischemia-induced neuronal damage or against Alzheimer’s disease. Indole-3-aldehyde (I3A) acts on the aryl hydrocarbon receptor (AhR) found on intestinal immune cells and increases interleukin-22 (IL-22) production. Activation of AhR plays a crucial role in gut immunity, such as in maintaining the epithelial barrier function and promoting immune tolerance to promote microbial commensalism while protecting against pathogenic infections. Indole has a number of roles, such as a signaling molecule to intestinal L cells to produce glucagon-like protein 1 (GLP-1) or as a ligand for AhR. Indole is also metabolized by the liver to indoxyl sulfate, where an excess is detrimental to human health. Accumulation of indoxyl sulfate in physiologic fluid is toxic and associated with vascular disease and renal dysfunction. AST-120, an orally administered intestinal sorbent, adsorbs indole and decreases serum concentrations of indoxyl sulfate, and is a potential treatment for managing chronic kidney disease
Indole and its metabolites affect host physiology via a number of molecular mechanisms (Fig. 1). Indole and I3A are agonists for the aryl hydrocarbon receptor (AhR), a transcription factor that regulates interleukin (IL)-22 expression, increases TH17-cell activity, and helps maintain intraepithelial lymphocytes [125]. Indole upregulates the expression of tight junction proteins and modulates the expressions of pro- and anti-inflammatory genes in intestinal epithelial cells [126, 127]. These activities of AhR help ensure that commensal bacteria outcompete pathogenic bacteria in the gut microbiota [128], and the absence of AhR increases the severity of DSS-induced colitis [129] and response to Citrobacter rodentium infection [130] (a model of human enteropathogenic E. coli infections). In addition to these effects, recent studies show that indole also modulates GLP-1 release from L cells [131], so that indole formation may contribute to satiety and inhibition of obesity. Other recent studies demonstrate that IPA is a pregnane X receptor (PXR) agonist, particularly in the presence of indole [132]. A wide-range of PXR agonists inhibit NF-κB [133], and downregulation of intestinal tumor necrosis factor (TNF)-α and upregulation of junction proteins by IPA requires PXR [132]. IPA also potently scavenges hydroxyl radicals [134], thereby protecting against oxidative injury in various animal models [134–137]. Thus, future studies are needed to determine if enhancing IPA formation by bacteria or directly administering IPA is beneficial in inflammatory conditions such as inflammatory bowel disease and colorectal cancer.
While indole appears to be primarily beneficial, its metabolite IndS is a uremic toxin that accumulates in patients with CKD [138]. IndS is also associated with accelerated glomerular sclerosis [139], enhanced endothelial dysfunction [140], enhanced monocyte adhesion to the vascular endothelium [141], and increased oxidative stress [141, 142]. The oral charcoal adsorbent AST-120 binds indoles in the gut lumen and reduces plasma IndS levels, thereby reducing kidney damage and atherosclerosis associated with kidney injury [143]. Future studies are needed to determine if diverting tryptophan metabolism away from IndS towards IPA will be beneficial in renal disease or other conditions.
Tyrosine metabolites: para-cresyl sulfate and 4-ethylphenylsulfate
PCS and 4-ethylphenyl sulfate (EPS) are structurally similar uremic toxins formed by hepatic sulfation of the microbial metabolites para-cresol and 4-ethylphenol, respectively. The lack of PCS or EPS in the plasma and urine of germ-free mice demonstrates their microbial origins. Inactivating mutants of the hydroxyphenylacetate decarboxylase operon genes (hpdB/C/A) from Clostridium difficile prevent fermentation of tyrosine or its metabolite hydroxyphenylacetate to para-cresol [144]. Few other gut bacteria encode HpdB/C/A [144]. Bacterial pathways for formation of 4-ethylphenol have not yet been characterized, but the wine spoilage yeast Brettanomyces generates 4-ethylphenol from the tyrosine metabolite para-coumaric acid that is present in many foods via cinnamate decarboxylase and vinyl phenol reductase. 4-Ethylphenol also forms from orally administered genistein, a phytoestrogen found in soy, by uncharacterized but presumably microbial pathways [145].
Both PCS and EPS accumulate in patients with severe CKD undergoing hemodialysis [146]. PCS levels predict clinical outcomes in patients with CKD [147] and correlate with cardiovascular mortality in CKD patients [148, 149]. While conventional dialysis fails to remove PCS, treatment with the oral adsorbent AST-120 [150] or with the prebiotic arabino-xylo-oligosaccharide [151] lowers plasma PCS levels. Vegetarians have lower levels of PCS than omnivores [152]. There are very few studies of EPS. EPS levels are elevated in a rat model of chronic renal failure and AST-120 treatment lowers these levels [153]. EPS levels increase 46-fold in a mouse model of autism and treatment with Bacteroides fragilis blocks this increase [28]. Administration of EPS to mice results in anxiety-like behaviors [28].
Molecular mechanisms of action ascribed to PCS include direct damage of cell membranes [154], induction of apoptotic pathways [155], activation of NADPH oxidase 4 (NOX4) resulting in reactive oxygen species (ROS) formation [156], activation of JNK and p38-MAPK [157], activation of Rho-kinase (ROCK) leading to endothelial damage [158], activation of epidermal growth factor (EGF) receptor leading to expression of matrix metalloproteinases 2 and 9 [159], and inhibition of a variety of drug-metabolizing enzymes including CYP2E1, CYP3A4, UGT1A1, UGT1A9, and UGT2B7 [160]. Given its chemical similarity to PCS, EPS is expected to exert similar effects, but no specific molecular targets have been demonstrated to date. Future studies are needed to identify pharmaceutical inhibitors of the PCS and EPS biosynthetic pathways and whether such inhibitors have beneficial effects in disease.
Essential fatty acid-derived metabolites
The microbiota of ruminants have long been known to transform the essential fatty acids linoleic acid (LA) and linolenic acid to CLAs such as cis-9 and trans-11 CLA, and conjugate linolenic acids (CLnAs) such as cis-9, trans-11 and cis-15 CLnA, respectively [161–163], via the action of isomerases. However, recent studies found that the microbiota of mice and humans, particularly Lachnospiraceae, Lactobacillus spp. and Bifidobacteria, possess the capacity to generate both CLAs and CLnAs [164–166]. In Lactobacillus, intermediates for the formation of conjugated fatty acids include the oxygenated metabolites HYA and 10-hydroxyoctadecanoate (HYB) [167, 168]. The enzymes involved in the transformation of LA to CLAs by Lactobacillus were recently characterized and include myosin-cross-reactive antigen, short-chain dehydrogenase/oxidoreductase, and acetoacetate decarboxylase [169].
Conjugated fatty acids exert many highly beneficial effects, including reduction of adiposity, improved insulin sensitivity, reduced carcinogenesis, and reduced atherosclerosis (reviewed in [170]). CLAs and CLnAs act via PPAR-γ (reviewed in [171]), PPAR-α [172], and inhibition of cyclooxygenases and lipoxygenases [173, 174]. Whether typical intestinal microbiota generate sufficient CLA/CLnA to exert the extraintestinal effects seen with CLA/CLnA supplementation is unclear, as feeding essential fatty acids increases gut but not circulating levels of CLAs and CLnAs [164]. Like CLAs and CLnAs, HYA also exerts anti-inflammatory activities, including downregulating lipopolysaccharide (LPS)-induced maturation of dendritic cells, blocking TNF-induced barrier impairment, and protecting against DSS-induced intestinal injury [175, 176]. HYA acts via the GPR40–MEK–ERK pathway [176]. Future studies are needed to determine if increasing microbial HYA production can be used therapeutically.
Translation to future diagnostics and therapeutics
In previous sections, we have touched briefly on potential future studies for individual metabolites, but there are additional developments needed in broad areas of research and understanding to fully realize the potential of gut microbial metabolites for disease treatment. We will conclude by highlighting four of these needed developments.
First, the development of minimal sets of biomarker microbial metabolites that identify particular disease states or that distinguish between closely related disease conditions. The analysis carried out by de Preter and colleagues for inflammatory bowel disease is proof of principal for this strategy [22], and similar approaches for highly heterogeneous conditions such as autism spectrum disorder, in which the microbiota has also been implicated [177], might be even more valuable. This also applies to the identification of individuals who might be at risk for disease, such as was found for individuals who carried high levels of bacterial strains that converted cholesterol to coprostanol that made them more vulnerable to C. difficile infections. For translation to actual treatment, measurements will need to be carried out in clinical laboratories in which immunoassay arrays, rather than the more sophisticated MS or NMR methods available in research settings, will likely continue to be the primary methods available. Thus, identifying the minimal number of biomarker metabolites needed to selectively assess a condition is critical. Similar strategies can be used to determine the efficacy and safety of interventions.
Second, the development of algorithms to predict personalized responses to dietary and pharmaceutical interventions based on microbial metabolites. An exciting example of this approach was recently reported by Zeevi and colleagues, who demonstrated that the highly variable glycemic response of different individuals to the same foods could be predicted using their gut microbiota and other data [32]. Similarly, being able to predict the responses of specific metabolites such as SCFAs to individual foods using tools such as CASINO [31] may be critical for allowing individuals with intolerance for particular dietary components to successfully use functional foods to increase colonic levels of SCFAs. Algorithm-based personalization seems essential for any nutrition-based approaches, given the variability of microbial composition among individuals.
Third, the development of readily generalizable methods to increase gut microbial production of beneficial metabolites, either by selectively increasing the abundance of native species that produce that metabolite or by engineering endogenous gut microbiota to produce it in high levels. An example of this latter approach is our study using heterologous expression of the satiety factor N-acylphosphatidylethanolamine in commensal E. coli (strain Nissle 1917), leading to inhibition of obesity in mice fed a HFD [178]. Such strategies might be helpful to produce sufficient IPA, CLA or HYA to block inflammatory diseases, but could also be utilized to test novel metabolites as they are identified. One advantage of engineered bacteria may be the ability to produce beneficial metabolites in bacterial strains that colonize well in the gut of a diseased individual in the place of native bacteria that produce these same beneficial metabolites but poorly colonize in the diseased gut.
Fourth, the development of non-lethal specific inhibitors for various microbial pathways that produce harmful metabolites, similar to work done with CutC/D. In particular, inhibition of the formation of para-creysl and 4-ethylphenol appear amendable to this strategy. This revolutionary approach to controlling harmful bacterial metabolites seems unlikely to result in the rapid evolution of resistance that occurs with standard antibiotics, since there is a much more limited fitness advantage of carrying resistance. If this is the case, then long-term use of such metabolic pathway inhibitors will have great potential benefit in chronic diseases.
Conclusions and future perspectives
The past decade has seen remarkable progress in our understanding of the significant role that gut microbial metabolites play in modulating the health of their hosts. MS and NMR studies have identified a significant number of microbial metabolites that differ in disease conditions, and these same methods are now being exploited to better identify subtle differences in closely related diseases. Some of these identified metabolites, such as TMAO, IndS and PCS, appear to directly increase susceptibility to disease, while others, such as SCFA, IPA, CLA and HYA, appear to exert protective effects. Much work remains to fully characterize the physiological effects of these and the many other microbial metabolites that may be important in human health. It seems highly likely that future studies will identify many other disease states in which gut microbial metabolites are significantly enriched or depleted. It is important to keep in mind that by themselves such studies do not demonstrate causality. Thus, it seems there is a considerable need for carefully controlled studies to determine the physiological effects of each identified microbial metabolite and its specific mechanisms of action. Furthermore, in order to fully exploit the potential of the gut microbiota for disease prevention, we need a much greater understanding of how dietary components and host genetics affect the production of various metabolites. Finally, translation of these findings to clinical practice will require the development of widely available clinical chemistry methods to detect changes in an individual’s key metabolites. Despite these tremendous challenges to fully exploiting the gut microbiota for human health, the remarkable progress of the last decade suggests that such approaches have significant potential to revolutionize therapeutic approaches to human disease.
Abbreviations
- AhR:
-
aryl hydrocarbon receptor
- AMPK:
-
AMP kinase
- CKD:
-
chronic kidney disease
- CLA:
-
conjugated linoleic acid
- CLnA:
-
conjugated linolenic acid
- CoA:
-
coenzyme A
- CVD:
-
cardiovascular disease
- DMB:
-
3,3-dimethyl-1-butanol
- DSS:
-
dextran sodium sulfate
- EPS:
-
4-ethylphenylsulfate
- GIP:
-
gastric inhibitory polypeptide
- GLP:
-
glucagon-like peptide
- GPR:
-
G-protein-coupled receptor
- HDAC:
-
histone deacetylase
- HFD:
-
high-fat diet
- HYA:
-
10-hydroxy-cis-12-octadecenoate
- HYB:
-
10-hydroxyoctadecanoate
- I3A:
-
indole-3-aldehyde
- IL:
-
interleukin
- IndS:
-
indoxyl sulfate
- IPA:
-
indole-3-propionate
- LA:
-
linoleic acid
- LPS:
-
lipopolysaccharide
- MS:
-
mass spectrometry
- NMR:
-
nuclear magnetic resonance
- PCS:
-
para-cresyl sulfate
- PPAR-γ:
-
peroxisome proliferator-activated receptor-γ
- PYY:
-
Peptide YY
- PXR:
-
pregnane X receptor
- ROCK:
-
Rho-kinase
- ROS:
-
reactive oxygen species
- SCFA:
-
short-chain fatty acid
- TMA:
-
trimethylamine
- TMAO:
-
trimethylamine N-oxide
- TNF:
-
tumor necrosis factor
References
LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160–8. doi:10.1016/j.copbio.2012.08.005.
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59. doi:10.1194/jlr.R500013-JLR200.
Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, et al. Therapeutic modulation of microbiota-host metabolic interactions. Sci Transl Med. 2012;4(137):137rv6. doi:10.1126/scitranslmed.3004244.
Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. doi:10.1038/msb.2008.56.
Yap IK, Li JV, Saric J, Martin FP, Davies H, Wang Y, et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J Proteome Res. 2008;7(9):3718–28. doi:10.1021/pr700864x.
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106(10):3698–703. doi:10.1073/pnas.0812874106.
Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51(5):1101–12. doi:10.1194/jlr.M002774.
Swann JR, Tuohy KM, Lindfors P, Brown DT, Gibson GR, Wilson ID, et al. Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. J Proteome Res. 2011;10(8):3590–603. doi:10.1021/pr200243t.
Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, et al. The footprints of gut microbial-mammalian co-metabolism. J Proteome Res. 2011;10(12):5512–22. doi:10.1021/pr2007945.
Marcobal A, Kashyap P, Nelson T, Aronov P, Donia M, Spormann A, et al. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013;7:10. doi:10.1038/ismej.2013.89.
Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103(33):12511–6. doi:10.1073/pnas.0601056103.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi:10.1038/nature05414.
Waldram A, Holmes E, Wang Y, Rantalainen M, Wilson ID, Tuohy KM, et al. Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. J Proteome Res. 2009;8(5):2361–75. doi:10.1021/pr8009885.
Calvani R, Miccheli A, Capuani G, Tomassini Miccheli A, Puccetti C, Delfini M, et al. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int J Obes. 2010;34(6):1095–8. doi:10.1038/ijo.2010.44.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi:10.1038/nature09922.
Walker A, Pfitzner B, Neschen S, Kahle M, Harir M, Lucio M, et al. Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J. 2014;8(12):2380–96. doi:10.1038/ismej.2014.79.
Elliott P, Posma JM, Chan Q, Garcia-Perez I, Wijeyesekera A, Bictash M, et al. Urinary metabolic signatures of human adiposity. Sci Transl Med. 2015;7(285):285ra62. doi:10.1126/scitranslmed.aaa5680.
Gralka E, Luchinat C, Tenori L, Barbara E, Thurnheer M, Schultes B. Metabolomic fingerprint of severe obesity is dynamically affected by bariatric surgery in a procedure-dependent manner. Am J Clin Nutr. 2015;102(6):9. doi:10.3945/ajcn.115.110536.
Paul HA, Bomhof MR, Vogel HJ, Reimer RA. Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci Rep. 2016;6:20683. doi:10.1038/srep20683.
Jansson J, Willing B, Lucio M, Fekete A, Dicksved J, Halfvarson J, et al. Metabolomics reveals metabolic biomarkers of Crohn's disease. PLoS One. 2009;4(7), e6386. doi:10.1371/journal.pone.0006386.
Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT, et al. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res. 2011;10(9):4208–18. doi:10.1021/pr2003598.
De Preter V, Machiels K, Joossens M, Arijs I, Matthys C, Vermeire S, et al. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut. 2015;64(3):447–58. doi:10.1136/gutjnl-2013-306423.
Kieffer DA, Piccolo BD, Vaziri ND, Liu S, Lau WL, Khazaeli M, et al. Resistant starch alters gut microbiome and metabolomics profiles concurrent with amelioration of chronic kidney disease in rats. Am J Physiol Renal Physiol. 2016. doi:10.1152/ajprenal.00513.2015.
Theriot CM, Koenigsknecht MJ, Carlson Jr PE, Hatton GE, Nelson AM, Li B, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. doi:10.1038/ncomms4114.
Theriot C, Bowman A, Young V. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 2016. doi:10.1128/mSphere.00045-15.
Antharam VC, McEwen DC, Garrett TJ, Dossey AT, Li EC, Kozlov AN, et al. An integrated metabolomic and microbiome analysis identified specific gut microbiota associated with fecal cholesterol and coprostanol in Clostridium difficile infection. PLoS One. 2016;11(2), e0148824. doi:10.1371/journal.pone.0148824.
Ming X, Stein TP, Barnes V, Rhodes N, Guo L. Metabolic perturbance in autism spectrum disorders: a metabolomics study. J Proteome Res. 2012;11(12):5856–62. doi:10.1021/pr300910n.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–63. doi:10.1016/j.cell.2013.11.024.
Frohlich EE, Farzi A, Mayerhofer R, Reichmann F, Jacan A, Wagner B, et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun. 2016. doi:10.1016/j.bbi.2016.02.020.
Vanden Bussche J, Marzorati M, Laukens D, Vanhaecke L. Validated high resolution mass spectrometry-based approach for metabolomic fingerprinting of the human gut phenotype. Anal Chem. 2015;87(21):10927–34. doi:10.1021/acs.analchem.5b02688.
Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22(2):320–31. doi:10.1016/j.cmet.2015.07.001.
Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163(5):1079–94. doi:10.1016/j.cell.2015.11.001.
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28(10):1221–7.
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lip Res. 2013;54(9):2325–40. doi:10.1194/jlr.R036012.
Thauer RK, Moller-Zinkhan D, Spormann AM. Biochemistry of acetate catabolism in anaerobic chemotrophic bacteria. Annu Rev Microbiol. 1989;43:43–67. doi:10.1146/annurev.mi.43.100189.000355.
Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014;8(6):1323–35. doi:10.1038/ismej.2014.14.
Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol. 2002;68(10):5186–90.
Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278(13):11312–9. doi:10.1074/jbc.M211609200.
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278(28):25481–9. doi:10.1074/jbc.M301403200.
Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69(7):2826–32. doi:10.1158/0008-5472.CAN-08-4466.
den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes. 2015;64(7):2398–408. doi:10.2337/db14-1213.
Priyamvada S, Anbazhagan A, Chatterjee I, Alrefai W, Dudeja P, Borthakur A. Gut bacterial metabolite propionate upregulates intestinal epithelial kruppel-like factor 4 expression via a PPAR-γ-dependent mechanism. FASEB J. 2015;29(1):854.4.
Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-κB activity in a human colonic epithelial cell line. Gastroenterology. 2000;118(4):724–34.
Segain JP, Raingeard de la Bletiere D, Bourreille A, Leray V, Gervois N, Rosales C, et al. Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn's disease. Gut. 2000;47(3):397–403.
Menzel T, Luhrs H, Zirlik S, Schauber J, Kudlich T, Gerke T, et al. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm Bowel Dis. 2004;10(2):122–8.
Luhrs H, Gerke T, Muller JG, Melcher R, Schauber J, Boxberge F, et al. Butyrate inhibits NF-κB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol. 2002;37(4):458–66.
Luhrs H, Gerke T, Boxberger F, Backhaus K, Melcher R, Scheppach W, et al. Butyrate inhibits interleukin-1-mediated nuclear factor-κB activation in human epithelial cells. Dig Dis Sci. 2001;46(9):1968–73.
Breuer RI, Soergel KH, Lashner BA, Christ ML, Hanauer SB, Vanagunas A, et al. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: a randomised, placebo controlled trial. Gut. 1997;40(4):485–91.
Vernia P, Marcheggiano A, Caprilli R, Frieri G, Corrao G, Valpiani D, et al. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment Pharmacol Ther. 1995;9(3):309–13.
Vernia P. Butyrate in the treatment of ulcerative colitis. Digest Liver Dis Suppl. 2007;1(1):27–30. doi:10.1016/S1594-5804(08)60008-X.
Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, et al. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer. 2011;128(11):2591–601. doi:10.1002/ijc.25599.
Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17(5):662–71. doi:10.1016/j.chom.2015.03.005.
Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–52. doi:10.1073/pnas.1322269111.
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–5. doi:10.1038/nature12726.
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. doi:10.1038/nature12721.
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–73. doi:10.1126/science.1241165.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. doi:10.1038/nature08530.
Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734. doi:10.1038/ncomms7734.
Kinoshita M, Suzuki Y, Saito Y. Butyrate reduces colonic paracellular permeability by enhancing PPARγ activation. Biochem Biophys Res Commun. 2002;293(2):827–31. doi:10.1016/S0006-291X(02)00294-2.
Watanabe N, Matsuyama T, Namba M, Miyagawa J, Itoh H, Komatsu R, et al. Trophic effect of glucagon-(1-21)-peptide on the isolated rat ileal mucosal cells. Biochem Biophys Res Commun. 1988;152(3):1038–44.
Lupton JR. Microbial degradation products influence colon cancer risk: the butyrate controversy. J Nutrition. 2004;134(2):479–82.
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–39. doi:10.1016/j.immuni.2013.12.007.
Fung KY, Cosgrove L, Lockett T, Head R, Topping DL. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br J Nutrition. 2012;108(5):820–31. doi:10.1017/S0007114512001948.
Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutrition. 2003;133(7 Suppl):2485S–93S.
Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92(15):1210–6.
Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496–501.
Cassidy A, Bingham SA, Cummings JH. Starch intake and colorectal cancer risk: an international comparison. Br J Cancer. 1994;69(5):937–42.
Howe GR, Benito E, Castelleto R, Cornee J, Esteve J, Gallagher RP, et al. Dietary intake of fiber and decreased risk of cancers of the colon and rectum: evidence from the combined analysis of 13 case-control studies. J Natl Cancer Inst. 1992;84(24):1887–96.
Park Y, Hunter DJ, Spiegelman D, Bergkvist L, Berrino F, van den Brandt PA, et al. Dietary fiber intake and risk of colorectal cancer: a pooled analysis of prospective cohort studies. JAMA. 2005;294(22):2849–57. doi:10.1001/jama.294.22.2849.
Aune D, Chan DS, Lau R, Vieira R, Greenwood DC, Kampman E, et al. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ. 2011;343:d6617. doi:10.1136/bmj.d6617.
Chen HM, Yu YN, Wang JL, Lin YW, Kong X, Yang CQ, et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am J Clin Nutr. 2013;97(5):1044–52. doi:10.3945/ajcn.112.046607.
Lanza E, Yu B, Murphy G, Albert PS, Caan B, Marshall JR, et al. The polyp prevention trial continued follow-up study: no effect of a low-fat, high-fiber, high-fruit, and -vegetable diet on adenoma recurrence eight years after randomization. Cancer Epidemiol Biomarkers Prev. 2007;16(9):1745–52. doi:10.1158/1055-9965.EPI-07-0127.
Alberts DS, Martinez ME, Roe DJ, Guillen-Rodriguez JM, Marshall JR, van Leeuwen JB, et al. Lack of effect of a high-fiber cereal supplement on the recurrence of colorectal adenomas. N Engl J Med. 2000;342(16):1156–62. doi:10.1056/NEJM200004203421602.
Weaver GA, Krause JA, Miller TL, Wolin MJ. Short chain fatty acid distributions of enema samples from a sigmoidoscopy population: an association of high acetate and low butyrate ratios with adenomatous polyps and colon cancer. Gut. 1988;29(11):1539–43.
Clausen MR, Bonnen H, Mortensen PB. Colonic fermentation of dietary fibre to short chain fatty acids in patients with adenomatous polyps and colonic cancer. Gut. 1991;32(8):923–8.
Kashtan H, Stern HS, Jenkins DJ, Jenkins AL, Thompson LU, Hay K, et al. Colonic fermentation and markers of colorectal-cancer risk. Am J Clin Nutr. 1992;55(3):723–8.
Vernia P, Cittadini M. Short-chain fatty acids and colorectal cancer. Eur J Clin Nutr. 1995;49 Suppl 3:S18–21.
Sengupta S, Muir JG, Gibson PR. Does butyrate protect from colorectal cancer? J Gastroenterol Hepatol. 2006;21(1 Pt 2):209–18. doi:10.1111/j.1440-1746.2006.04213.x.
Kripke SA, Fox AD, Berman JM, Settle RG, Rombeau JL. Stimulation of intestinal mucosal growth with intracolonic infusion of short-chain fatty acids. JPEN J Parenter Enteral Nutr. 1989;13(2):109–16.
Singh B, Halestrap AP, Paraskeva C. Butyrate can act as a stimulator of growth or inducer of apoptosis in human colonic epithelial cell lines depending on the presence of alternative energy sources. Carcinogenesis. 1997;18(6):1265–70.
Velazquez OC, Seto RW, Bain AM, Fisher J, Rombeau JL. Deoxycholate inhibits in vivo butyrate-mediated BrDU labeling of the colonic crypt. J Surg Res. 1997;69(2):344–8. doi:10.1006/jsre.1997.5075.
Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–26. doi:10.1016/j.molcel.2012.08.033.
Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014;4(12):1387–97. doi:10.1158/2159-8290.cd-14-0501.
Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. The role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–19. doi:10.1111/j.1365-2036.2007.03562.x.
Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158(2):288–99. doi:10.1016/j.cell.2014.04.051.
Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology. 2010;56(2):167–79. doi:10.1111/j.1365-2559.2009.03392.x.
Ho KJ, Xiong L, Hubert NJ, Nadimpalli A, Wun K, Chang EB, et al. Vancomycin treatment and butyrate supplementation modulate gut microbe composition and severity of neointimal hyperplasia after arterial injury. Physiol Rep. 2015;3(12). doi:10.14814/phy2.12627.
Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. 2015;26(8):1877–88. doi:10.1681/ASN.2014030288.
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–66. doi:10.1038/nm.3444.
Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. 2015;6:7320. doi:10.1038/ncomms8320.
Vieira AT, Macia L, Galvão I, Martins FS, Canesso MCC, Amaral FA, et al. A role for gut microbiota and the metabolite-sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol. 2015;67(6):1646–56. doi:10.1002/art.39107.
Arrieta M-C, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7(307):307ra152. doi:10.1126/scitranslmed.aab2271.
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–5. doi:10.1038/oby.2009.167.
Li JV, Ashrafian H, Bueter M, Kinross J, Sands C, le Roux CW, et al. Metabolic surgery profoundly influences gut microbial-host metabolic cross-talk. Gut. 2011;60(9):1214–23. doi:10.1136/gut.2010.234708.
Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59(12):3049–57. doi:10.2337/db10-0253.
Liou AP, Paziuk M, Luevano Jr JM, Machineni S, Turnbaugh PJ, Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Trans Med. 2013;5(178):178ra41. doi:10.1126/scitranslmed.3005687.
Tremaroli V, Karlsson F, Werling M, Stahlman M, Kovatcheva-Datchary P, Olbers T, et al. Roux-en-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metab. 2015;22(2):228–38. doi:10.1016/j.cmet.2015.07.009.
Zhang C, Yin A, Li H, Wang R, Wu G, Shen J, et al. Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine. 2015;2(8):966–82. doi:10.1016/j.ebiom.2015.07.007.
Chassaing B, Miles-Brown J, Pellizzon M, Ulman E, Ricci M, Zhang L, et al. Lack of soluble fiber drives diet-induced adiposity in mice. Am J Physiol Gastrointest Liver Physiol. 2015;309(7):G528–41. doi:10.1152/ajpgi.00172.2015.
Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. 2012;3(4):279–88. doi:10.4161/gmic.19625.
Arora T, Loo RL, Anastasovska J, Gibson GR, Tuohy KM, Sharma RK, et al. Differential effects of two fermentable carbohydrates on central appetite regulation and body composition. PLoS One. 2012;7(8), e43263. doi:10.1371/journal.pone.0043263.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17. doi:10.2337/db08-1637.
Vinolo MA, Rodrigues HG, Festuccia WT, Crisma AR, Alves VS, Martins AR, et al. Tributyrin attenuates obesity-associated inflammation and insulin resistance in high-fat-fed mice. Am J Physiol Endocrinol Metab. 2012;303(2):E272–82. doi:10.1152/ajpendo.00053.2012.
Byrne CS, Chambers ES, Morrison DJ, Frost G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int J Obesity. 2015;39(9):1331–8. doi:10.1038/ijo.2015.84.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. doi:10.2337/db06-1491.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81. doi:10.2337/db07-1403.
Cani PD. Crosstalk between the gut microbiota and the endocannabinoid system: impact on the gut barrier function and the adipose tissue. Clin Microbiol Infect. 2012;18 Suppl 4:50–3. doi:10.1111/j.1469-0691.2012.03866.x.
Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9(7):1408–13. doi:10.1021/cb500113p.
Martinez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, et al. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio. 2015;6(2). doi:10.1128/mBio.00042-15.
Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, et al. γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of l-carnitine to TMAO. Cell Metab. 2014;20(5):799–812. doi:10.1016/j.cmet.2014.10.006.
Zhu Y, Jameson E, Crosatti M, Schafer H, Rajakumar K, Bugg TD, et al. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc Natl Acad Sci U S A. 2014;111(11):4268–73. doi:10.1073/pnas.1316569111.
Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49–60. doi:10.1016/j.cmet.2012.12.011.
Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–84. doi:10.1056/NEJMoa1109400.
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–85. doi:10.1038/nm.3145.
Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35(14):904–10. doi:10.1093/eurheartj/ehu002.
Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–55. doi:10.1161/CIRCRESAHA.116.305360.
Wilson A, Teft WA, Morse BL, Choi YH, Woolsey S, DeGorter MK, et al. Trimethylamine-N-oxide: a novel biomarker for the identification of inflammatory bowel disease. Dig Dis Sci. 2015;60(12):3620–30. doi:10.1007/s10620-015-3797-3.
Wallrabenstein I, Kuklan J, Weber L, Zborala S, Werner M, Altmuller J, et al. Human trace amine-associated receptor TAAR5 can be activated by trimethylamine. PLoS One. 2013;8(2), e54950. doi:10.1371/journal.pone.0054950.
Collins HL, Drazul-Schrader D, Sulpizio AC, Koster PD, Williamson Y, Adelman SJ, et al. l-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE−/− transgenic mice expressing CETP. Atherosclerosis. 2016;244:29–37. doi:10.1016/j.atherosclerosis.2015.10.108.
Warrier M, Shih Diana M, Burrows Amy C, Ferguson D, Gromovsky Anthony D, Brown Amanda L, et al. The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10(3):326–38. doi:10.1016/j.celrep.2014.12.036.
Ussher JR, Lopaschuk GD, Arduini A. Gut microbiota metabolism of l-carnitine and cardiovascular risk. Atherosclerosis. 2013;231(2):456–61. doi:10.1016/j.atherosclerosis.2013.10.013.
DiNicolantonio JJ, Lavie CJ, Fares H, Menezes AR, O'Keefe JH. l-carnitine in the secondary prevention of cardiovascular disease: systematic review and meta-analysis. Mayo Clin Proc. 2013;88(6):544–51. doi:10.1016/j.mayocp.2013.02.007.
Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163(7):1585–95. doi:10.1016/j.cell.2015.11.055.
Kim J, Park W. Indole inhibits bacterial quorum sensing signal transmission by interfering with quorum sensing regulator folding. Microbiology. 2013;159(Pt 12):2616–25. doi:10.1099/mic.0.070615-0.
Zelante T, Iannitti Rossana G, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39(2):372–85. doi:10.1016/j.immuni.2013.08.003.
Bansal T, Alaniz RC, Wood TK, Jayaraman A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc Natl Acad Sci U S A. 2010;107(1):228–33. doi:10.1073/pnas.0906112107.
Shimada Y, Kinoshita M, Harada K, Mizutani M, Masahata K, Kayama H, et al. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS One. 2013;8(11), e80604. doi:10.1371/journal.pone.0080604.
Romani L, Zelante T, De Luca A, Iannitti RG, Moretti S, Bartoli A, et al. Microbiota control of a tryptophan-AhR pathway in disease tolerance to fungi. Eur J Immunol. 2014;44(11):3192–200. doi:10.1002/eji.201344406.
Li Y, Innocentin S, Withers David R, Roberts Natalie A, Gallagher Alec R, Grigorieva Elena F, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147(3):629–40. http:10.1016/j.cell.2011.09.025.
Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334(6062):1561–5. doi:10.1126/science.1214914.
Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014;9(4):1202–8. doi:10.1016/j.celrep.2014.10.032.
Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet Alexandre P, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity;41(2):296–310. doi:10.1016/j.immuni.2014.06.014.
Zhou C, Tabb MM, Nelson EL, Grün F, Verma S, Sadatrafiei A, et al. Mutual repression between steroid and xenobiotic receptor and NF-κB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116(8):2280–9. doi:10.1172/JCI26283.
Chyan Y-J, Poeggeler B, Omar RA, Chain DG, Frangione B, Ghiso J, et al. Potent neuroprotective properties against the Alzheimer β-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J Biol Chem. 1999;274(31):21937–42. doi:10.1074/jbc.274.31.21937.
Hwang IK, Yoo KY, Li H, Park OK, Lee CH, Choi JH, et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J Neurosci Res. 2009;87(9):2126–37. doi:10.1002/jnr.22030.
Karbownik M, Stasiak M, Zasada K, Zygmunt A, Lewinski A. Comparison of potential protective effects of melatonin, indole-3-propionic acid, and propylthiouracil against lipid peroxidation caused by potassium bromate in the thyroid gland. J Cell Biochem. 2005;95(1):131–8. doi:10.1002/jcb.20404.
Karbownik M, Stasiak M, Zygmunt A, Zasada K, Lewinski A. Protective effects of melatonin and indole-3-propionic acid against lipid peroxidation, caused by potassium bromate in the rat kidney. Cell Biochem Funct. 2006;24(6):483–9. doi:10.1002/cbf.1321.
Lin C-J, Chen H-H, Pan C-F, Chuang C-K, Wang T-J, Sun F-J, et al. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J Clin Lab Anal. 2011;25(3):191–7. doi:10.1002/jcla.20456.
Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med. 1994;124(1):96–104.
Faure V, Dou L, Sabatier F, Cerini C, Sampol J, Berland Y, et al. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J Thromb Haemost. 2006;4(3):566–73. doi:10.1111/j.1538-7836.2005.01780.x.
Ito S, Osaka M, Higuchi Y, Nishijima F, Ishii H, Yoshida M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J Biol Chem. 2010;285(50):38869–75. doi:10.1074/jbc.M110.166686.
Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5(6):1302–8. doi:10.1111/j.1538-7836.2007.02540.x.
Yamamoto S, Zuo Y, Ma J, Yancey PG, Hunley TE, Motojima M, et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol Dial Transplant. 2011;26(8):2491–7. doi:10.1093/ndt/gfq759.
Dawson LF, Donahue EH, Cartman ST, Barton RH, Bundy J, McNerney R, et al. The analysis of para-cresol production and tolerance in Clostridium difficile 027 and 012 strains. BMC Microbiol. 2011;11:86. doi:10.1186/1471-2180-11-86.
Yasuda T, Ueda J, Ohsawa K. Urinary metabolites of genistein administered orally to rats. Chem Pharm Bull. 2001;49(11):1495–7.
Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, Niwa T. Correlation between serum levels of protein-bound uremic toxins in hemodialysis patients measured by LC/MS/MS. Mass Spectrom (Tokyo). 2013;2(Spec Iss):S0017. doi:10.5702/massspectrometry.S0017.
Lin CJ, Pan CF, Chuang CK, Sun FJ, Wang DJ, Chen HH, et al. P-cresyl sulfate is a valuable predictor of clinical outcomes in pre-ESRD patients. Biomed Res Int. 2014;2014:526932. doi:10.1155/2014/526932.
Lin CJ, Chuang CK, Jayakumar T, Liu HL, Pan CF, Wang TJ, et al. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch Med Sci. 2013;9(4):662–8. doi:10.5114/aoms.2013.36901.
Lin CJ, Wu V, Wu PC, Wu CJ. Meta-analysis of the associations of p-cresyl sulfate (PCS) and indoxyl sulfate (IS) with cardiovascular events and all-cause mortality in patients with chronic renal failure. PLoS One. 2015;10(7), e0132589. doi:10.1371/journal.pone.0132589.
Lee CT, Hsu CY, Tain YL, Ng HY, Cheng BC, Yang CC, et al. Effects of AST-120 on blood concentrations of protein-bound uremic toxins and biomarkers of cardiovascular risk in chronic dialysis patients. Blood Purif. 2014;37(1):76–83. doi:10.1159/000357641.
Koppe L, Pillon NJ, Vella RE, Croze ML, Pelletier CC, Chambert S, et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J Am Soc Nephrol. 2013;24(1):88–99. doi:10.1681/ASN.2012050503.
Patel KP, Luo FJ, Plummer NS, Hostetter TH, Meyer TW. The production of p-cresol sulfate and indoxyl sulfate in vegetarians versus omnivores. Clin J Am Soc Nephrol. 2012;7(6):982–8. doi:10.2215/CJN.12491211.
Kikuchi K, Itoh Y, Tateoka R, Ezawa A, Murakami K, Niwa T. Metabolomic search for uremic toxins as indicators of the effect of an oral sorbent AST-120 by liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(29):2997–3002. doi:10.1016/j.jchromb.2010.09.006.
Idziak M, Pedzisz P, Burdzinska A, Gala K, Paczek L. Uremic toxins impair human bone marrow-derived mesenchymal stem cells functionality in vitro. Exp Toxicol Pathol. 2014;66(4):187–94. doi:10.1016/j.etp.2014.01.003.
Poveda J, Sanchez-Nino MD, Glorieux G, Sanz AB, Egido J, Vanholder R, et al. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol Dial Transplant. 2014;29(1):56–64. doi:10.1093/ndt/gft367.
Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013;83(4):582–92. doi:10.1038/ki.2012.448.
Tanaka H, Iwasaki Y, Yamato H, Mori Y, Komaba H, Watanabe H, et al. p-Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone. 2013;56(2):347–54. doi:10.1016/j.bone.2013.07.002.
Meijers BK, Van Kerckhoven S, Verbeke K, Dehaen W, Vanrenterghem Y, Hoylaerts MF, et al. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54(5):891–901. doi:10.1053/j.ajkd.2009.04.022.
Sun CY, Young GH, Hsieh YT, Chen YH, Wu MS, Wu VC, et al. Protein-bound uremic toxins induce tissue remodeling by targeting the EGF receptor. J Am Soc Nephrol. 2015;26(2):281–90. doi:10.1681/ASN.2014010021.
Barnes KJ, Rowland A, Polasek TM, Miners JO. Inhibition of human drug-metabolising cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in vitro by uremic toxins. Eur J Clin Pharmacol. 2014;70(9):1097–106. doi:10.1007/s00228-014-1709-7.
Lawson RE, Moss AR, Givens DI. The role of dairy products in supplying conjugated linoleic acid to man's diet: a review. Nutr Res Rev. 2001;14(1):153–72. doi:10.1079/NRR200121.
Parodi PW. Conjugated linoleic acid and other anticarcinogenic agents of bovine milk fat. J Dairy Sci. 1999;82(6):1339–49. doi:10.3168/jds.S0022-0302(99)75358-0.
Gholami Z, Khosravi-Darani K. An overview of conjugated linoleic acid: microbial production and application. Mini Rev Med Chem. 2014;14(9):734–46.
Druart C, Neyrinck AM, Vlaeminck B, Fievez V, Cani PD, Delzenne NM. Role of the lower and upper intestine in the production and absorption of gut microbiota-derived PUFA metabolites. PLoS One. 2014;9(1), e87560. doi:10.1371/journal.pone.0087560.
O'Shea EF, Cotter PD, Stanton C, Ross RP, Hill C. Production of bioactive substances by intestinal bacteria as a basis for explaining probiotic mechanisms: bacteriocins and conjugated linoleic acid. Int J Food Microbiol. 2012;152(3):189–205. doi:10.1016/j.ijfoodmicro.2011.05.025.
Gorissen L, Leroy F, De Vuyst L, De Smet S, Raes K. Bacterial production of conjugated linoleic and linolenic acid in foods: a technological challenge. Crit Rev Food Sci Nutr. 2015;55(11):1561–74. doi:10.1080/10408398.2012.706243.
Kishino S, Takeuchi M, Park S-B, Hirata A, Kitamura N, Kunisawa J, et al. Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc Natl Acad Sci U S A. 2013;110(44):17808–13. doi:10.1073/pnas.1312937110.
Ogawa J, Kishino S, Ando A, Sugimoto S, Mihara K, Shimizu S. Production of conjugated fatty acids by lactic acid bacteria. J Biosci Bioeng. 2005;100(4):355–64. doi:10.1263/jbb.100.355.
Yang B, Chen H, Gu Z, Tian F, Ross RP, Stanton C, et al. Synthesis of conjugated linoleic acid by the linoleate isomerase complex in food-derived lactobacilli. J Appl Microbiol. 2014;117(2):430–9. doi:10.1111/jam.12524.
Belury MA. Dietary conjugated linoleic acid in health: physiological effects and mechanisms of action. Ann Rev Nutr. 2002;22(1):505–31. doi:10.1146/annurev.nutr.22.021302.121842.
Yuan G, Chen X, Li D. Modulation of peroxisome proliferator-activated receptor γ (PPARγ) by conjugated fatty acid in obesity and inflammatory bowel disease. J Agric Food Chem. 2015;63(7):1883–95. doi:10.1021/jf505050c.
Moya-Camarena SY, Heuvel JPV, Blanchard SG, Leesnitzer LA, Belury MA. Conjugated linoleic acid is a potent naturally occurring ligand and activator of PPARα. J Lipid Res. 1999;40(8):1426–33.
Mashhadi Z, Boeglin WE, Brash AR. Robust inhibitory effects of conjugated linolenic acids on a cyclooxygenase-related linoleate 10S-dioxygenase: comparison with COX-1 and COX-2. Biochim Biophys Acta. 2015;1851(10):1346–52. doi:10.1016/j.bbalip.2015.07.004.
Bialek A, Jelinska M, Tokarz A. Influence of maternal diet enrichment with conjugated linoleic acids on lipoxygenase metabolites of polyunsaturated fatty acids in serum of their offspring with 7,12-dimethylbenz[a]anthracene induced mammary tumors. Prostaglandins Other Lipid Mediat. 2015;116–117:10–8. doi:10.1016/j.prostaglandins.2014.10.001.
Bergamo P, Luongo D, Miyamoto J, Cocca E, Kishino S, Ogawa J, et al. Immunomodulatory activity of a gut microbial metabolite of dietary linoleic acid, 10-hydroxy-cis-12-octadecenoic acid, associated with improved antioxidant/detoxifying defences. J Funct Foods. 2014;11:192–202. doi:10.1016/j.jff.2014.10.007.
Miyamoto J, Mizukure T, Park SB, Kishino S, Kimura I, Hirano K, et al. A gut microbial metabolite of linoleic acid, 10-hydroxy-cis-12-octadecenoic acid, ameliorates intestinal epithelial barrier impairment partially via GPR40-MEK-ERK pathway. J Biol Chem. 2015;290(5):2902–18. doi:10.1074/jbc.M114.610733.
Buie T. Potential etiologic factors of microbiome disruption in autism. Clin Ther. 2015;37(5):976–83. doi:10.1016/j.clinthera.2015.04.001.
Chen Z, Guo L, Zhang Y, Walzem RL, Pendergast JS, Printz RL, et al. Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. J Clin Invest. 2014;124(8):3391–406. doi:10.1172/JCI72517.
MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: relevance to autism spectrum disorder. Behav Brain Res. 2011;217(1):47–54.
Viladomiu M, Hontecillas R, Bassaganya-Riera J. Modulation of inflammation and immunity by dietary conjugated linoleic acid. Eur J Pharmacol. 2015. doi:10.1016/j.ejphar.2015.03.095.
Acknowledgements
This work was supported in part by funds from the National Institutes of Health grant AT007830.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LSZ and SSD wrote the manuscript. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, L.S., Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med 8, 46 (2016). https://doi.org/10.1186/s13073-016-0296-x
Published:
DOI: https://doi.org/10.1186/s13073-016-0296-x