
Abstract
We report unbiased metagenomic detection of chikungunya virus (CHIKV), Ebola virus (EBOV), and hepatitis C virus (HCV) from four human blood samples by MinION nanopore sequencing coupled to a newly developed, web-based pipeline for real-time bioinformatics analysis on a computational server or laptop (MetaPORE). At titers ranging from 107–108 copies per milliliter, reads to EBOV from two patients with acute hemorrhagic fever and CHIKV from an asymptomatic blood donor were detected within 4 to 10 min of data acquisition, while lower titer HCV virus (1 × 105 copies per milliliter) was detected within 40 min. Analysis of mapped nanopore reads alone, despite an average individual error rate of 24 % (range 8–49 %), permitted identification of the correct viral strain in all four isolates, and 90 % of the genome of CHIKV was recovered with 97–99 % accuracy. Using nanopore sequencing, metagenomic detection of viral pathogens directly from clinical samples was performed within an unprecedented <6 hr sample-to-answer turnaround time, and in a timeframe amenable to actionable clinical and public health diagnostics.
Similar content being viewed by others

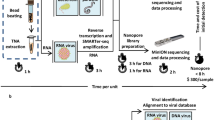

Background
Acute febrile illness has a broad differential diagnosis and can be caused by a variety of pathogens. Metagenomic next-generation sequencing (NGS) is particularly attractive for diagnosis and public health surveillance of febrile illness because the approach can broadly detect viruses, bacteria, and parasites in clinical samples by uniquely identifying sequence data [1, 2]. Although currently limited by sample-to-answer turnaround times typically exceeding 20 hr (Fig. 1a), we and others have reported that unbiased pathogen detection using metagenomic NGS can generate actionable results in timeframes relevant to clinical diagnostics [3–6] and public health [7, 8]. However, timely analysis using second-generation platforms such as Illumina and Ion Torrent has been hampered by the need to wait until a sufficient read length has been achieved for diagnostic pathogen identification, as sequence reads for these platforms are generated in parallel and not in series.

Metagenomic sequencing workflow for MinION nanopore sequencing compared to Illumina MiSeq sequencing. a Overall workflow. b Steps in the MetaPORE real-time analysis pipeline. The turnaround time for sample-to-detection nanopore sequencing, defined here as the cumulative time taken for nucleic acid extraction, reverse transcription, library preparation, sequencing, MetaPORE bioinformatics analysis, and pathogen detection, was under 6 hr, while Illumina sequencing took over 20 hr. The time differential is accounted for by increased times for library quantitation, sequencing, and bioinformatics analysis with the Illumina protocol. *Assumes a 12-hr 50-bp single-end MiSeq run of ~12–15 million reads, with 50 bp the minimum estimated read length needed for accurate pathogen identification. **Denotes estimated average SURPI bioinformatics analysis run length for MiSeq data [19]. The stopwatch is depicted as a 12-hr clock
Nanopore sequencing is a third-generation sequencing technology that has two key advantages over second-generation technologies – longer reads and the ability to perform real-time sequence analysis. To date, the longer nanopore reads have enabled scaffolding of prokaryotic and eukaryotic genomes and sequencing of bacterial and viral cultured isolates [9–13], but the platform’s capacity for real-time metagenomic analysis of primary clinical samples has not yet been leveraged. As of mid-2015, the MinION nanopore sequencer is capable of producing at least 100,000 sequences with an average read length of 5 kb, in total producing up to 1 Gb of sequence in 24 hr on one flow cell [14]. Here we present nanopore sequencing for metagenomic detection of viral pathogens from clinical samples with a sample-to-answer turnaround time of under 6 hr (Fig. 1a). We also present MetaPORE, a real-time web-based sequence analysis and visualization tool for pathogen identification from nanopore data (Fig. 1b).
Methods
Ethics statement
The chikungunya virus (CHIKV) plasma sample was collected from a donor from Puerto Rico, who provided written consent for use of samples and de-identified clinical metadata in medical research [15]. For the Ebola virus (EBOV) samples, patients provided oral consent for collection and analysis of their blood, as was the case for previous outbreaks [16, 17]. Consent was obtained either at the homes of patients or in hospital isolation wards by a team that included staff members of the Ministry of Health in the Democratic Republic of the Congo (DRC). The hepatitis C virus (HCV) sample was a banked aliquot from a patient with known hepatitis C infection at the University of California, San Francisco (UCSF), and sequence analysis was performed under a waiver of consent granted by the UCSF Institutional Review Board.
MAP program
Since July 2014, our lab has participated in the MinION Access Program (MAP), an early access program for beta users of the Oxford Nanopore MinION. Program participants receive free flow cells and library preparation kits for testing and validation of new protocols and applications on the MinION platform. During our time in the MAP program, we have seen significant progress in sequencing yield, although the quality of flow cells has varied considerably and individual read error rates remain high (Table 1).
Nucleic acid extraction
Frozen surplus plasma samples were collected during the peak weeks of the 2014 CHIKV outbreak in Puerto Rico from blood donors [15], and were de-identified prior to inclusion in the study. Total nucleic acid was extracted from 400 μL of a CHIKV-positive plasma sample (Chik1) inactivated in a 1:3 ratio of TRIzol LS (Life Technologies, Carlsbad, CA, USA) at the American Red Cross prior to shipping to UCSF. The Direct-zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA) was used for nucleic acid extraction, including on-column treatment with Turbo DNAse (Life Technologies) for 30 min at 37 °C to deplete human host genomic DNA.
For the EBOV samples, total nucleic acid was extracted using the QIAamp Viral RNA kit (Qiagen, Valencia, CA, USA) from 140 μL of whole blood from two patients with suspected Ebola hemorrhagic fever during a 2014 outbreak in the DRC (Ebola1 and Ebola2). RNA was extracted at Institut National de Recherche Biomédicale in Kinshasa, DRC, preserved using RNAstable (Biomatrica, San Diego, CA, USA), and shipped at room temperature to UCSF. Upon receipt, the extracted RNA sample was treated with 1 μL Turbo DNase (Life Technologies), followed by cleanup using the Direct-zol RNA MiniPrep Kit (Zymo Research).
For the HCV sample, an HCV-positive serum sample at a titer of 1.6 × 107 copies/mL (HepC1) was diluted to 1 × 105 copies/mL using pooled negative serum. Total nucleic acid was then extracted from 400 μL of serum using the EZ1 Viral RNA kit, followed by treatment with Turbo DNase for 30 min at 37 °C and cleanup using the RNA Clean and Concentrator Kit (Zymo Research).
Molecular confirmation of viral infection
A previously reported TaqMan quantitative reverse-transcription polymerase chain reaction (qRT-PCR) assay targeting the EBOV NP gene was used for detection of EBOV and determination of viral load [18]. The assay was run on a Stratagene MX300P real-time PCR instrument and performed using the TaqMan Fast Virus 1-Step Master Mix (Life Technologies) in 20 μL total reaction volume (5 μL 4× TaqMan mix, 1 μL sample extract), with 0.75 μM of each primer (F565 5′-TCTGACATGGATTACCACAAGATC-3′, R640 5′-GGATGACTCTTTGCCGAACAATC-3′) and 0.6 μM of the probe (p597S 6FAM-AGGTCTGTCCGTTCAA-MGBNFQ). Conditions for the qRT-PCR were modified as follows: 50 °C for 10 min and 95 °C for 20 s followed by 45 cycles of 95 °C for 3 s plus 60 °C for 30 s. Viral copy number was calculated by standard curve analysis using a plasmid vector containing the EBOV amplicon. The first EBOV sample analyzed by nanopore sequencing (Ebola1) corresponded to the Ebola virus/H.sapiens-wt/COD/2014/Lomela-Lokolia16 strain, while the second Ebola sample (Ebola2) corresponded to the Ebola virus/H.sapiens-wt/COD/2014/Lomela-LokoliaB11 strain. The CHIKV-positive sample was identified and quantified using a transcription-mediated amplification assay (Hologic, Bedford, MA, USA) as previously described [15]. HCV was quantified using the Abbott RealTime RT-PCR assay, approved by the Food and Drug Administration, as performed in the UCSF Clinical Microbiology Laboratory on the Abbott Molecular m2000 system.
Construction of metagenomic amplified cDNA libraries
To obtain ≥1 μg of metagenomic complementary DNA (cDNA) for the library required for the nanopore sequencing protocol, randomly amplified cDNA was generated using a primer-extension pre-amplification method (Round A/B) as described previously [19–21]. Of note, this protocol has been extensively tested on clinical samples for metagenomic pan-pathogen detection of DNA and RNA viruses, bacteria, fungi, and parasites [4, 6, 19, 21, 22]. Briefly, in Round A, RNA was reverse-transcribed with SuperScript III Reverse Transcriptase (Life Technologies,) using Sol-PrimerA (5′-GTTTCCCACTGGAGGATA-N9-3′), followed by second-strand DNA synthesis with Sequenase DNA polymerase (Affymetrix, Santa Clara, CA, USA). Reaction conditions for Round A were as follows: 1 μL of Sol-PrimerA (40 pmol/μL) was added to 4 μL of sample RNA, heated at 65 °C for 5 min, then cooled at room temperature for 5 min. Then 5 μL of SuperScript Master Mix (2 μl 5× First-Strand Buffer, 1 μL water, 1 μL 12.5 mM dNTP mix, 0.5 μL 0.1 M DTT, 0.5 μL SS III RT) was added and incubated at 42 °C for 60 min. For second strand synthesis, 5 μL of Sequenase Mix #1 (1 μL 5× Sequenase Buffer, 3.85 μL ddH2O, 0.15 μL Sequenase enzyme) was added to the reaction mix and incubated at 37 °C for 8 min, followed by addition of Sequenase Mix #2 (0.45 μl Sequenase Dilution Buffer, 0.15 μl Sequenase Enzyme) and there was a second incubation at 37 °C for 8 min. Round B reaction conditions were as follows: 5 μL of Round A-labeled cDNA was added to 45 μL of KlenTaq master mix per sample (5 μL 10× KlenTaq PCR buffer, 1 μL 12.5 mM dNTP, 1 μL 100 pmol/μL Sol-PrimerB (5′-GTTTCCCACTGGAGGATA-3′), 1 μL KlenTaq LA (Sigma-Aldrich, St Louis, MO), 37 μL ddH2O). Reaction conditions for the PCR were as follows: 94 °C for 2 min; 25 cycles of 94 °C for 30 s, 50 °C for 45 s, and 72 °C for 60 s, followed by 72 °C for 5 min.
Preparation of nanopore sequencing libraries
Amplified cDNA from Round B was purified using AMPure XP beads (Beckman Coulter, Brea, CA), and 1 μg DNA was used as input into Oxford Nanopore Genomic DNA MAP-003 Kits (Chik1, Ebola1) or MAP-004 Kits (HepC1, Ebola2) for generation of MinION Oxford Nanopore-compatible libraries [9, 11]. Briefly, the steps include: (1) addition of control lambda phage DNA, (2) end-repair with the NEBNext End Repair Module, (3) 1× AMPure purification, (4) dA-tailing with the NEBNext dA-tailing Module, (5) ligation to protein-linked adapters HP/AMP (Oxford Nanopore Technologies, Oxford, UK) using the NEBNext QuickLigation Module for 10 min at room temperature, (6) purification of ligated libraries using magnetic His-Tag Dynabeads (Life Technologies), and (7) elution in 25 μL buffer (Oxford Nanopore Technologies). Lambda phage DNA was not added during preparation of the Ebola2 sample library.
Nanopore sequencing
Nanopore libraries were run on an Oxford Nanopore MinION flow cell after loading 150 μL sequencing mix (6 μL library, 3 μL fuel mix, 141 μL buffer) per the manufacturer’s instructions. The Chik1 and Ebola1 samples were run consecutively on the same flow cell, with an interim wash performed using Wash-Kit-001 (Oxford Nanopore).
Illumina sequencing
For the Chik1 and Ebola1 samples, amplified Round B cDNA were purified using AMPure XP beads (Beckman Coulter) and 2 ng used as input into the Nextera XT Kit (Illumina). After 13 cycles of amplification, Illumina library concentration and average fragment size were determined using the Agilent Bioanalyzer. Sequencing was performed on an Illumina MiSeq using 150 nucleotide (nt) single-end runs and analyzed for viruses using either the MetaPORE or SURPI computational pipeline (UCSF) [19].
MetaPORE bioinformatics pipeline
We developed a custom bioinformatics pipeline for real-time pathogen identification and visualization from nanopore sequencing data (MetaPORE) (Fig. 1b), available under license from UCSF at [23]. The MetaPORE pipeline consists of a set of Linux shell scripts, Python programs, and JavaScript/HTML code, and was tested and run on an Ubuntu 14.10 computational server with 64 cores and 512 GB memory. In addition, MetaPORE was tested and run on a laptop (Ubuntu 14.10, eight hyper-threaded cores, 32 GB RAM). On the laptop, to maximize sensitivity while still retaining the speed necessary for real-time analysis and web-based visualization, MetaPORE can either (1) restrict the reference database for nucleotide BLAST (BLASTn) alignment to viral sequences or (2) use the faster MegaBLAST instead of the BLASTn algorithm at word sizes ranging from 11 to 28 to align nanopore reads to all of the National Center for Biotechnology Information (NCBI) nucleotide collection database (NT database). Running MegaBLAST to NT at a word size of 16 was found to detect ~85 % of nanopore CHIKV reads (n = 196) with an ~8× speedup in processing time relative to BLASTn, or 100 % of EBOV reads (n = 98) with an ~5× speedup (Additional file 1: Table S1). Overall, speeds of MegaBLAST to NT alignment at a word size of 16 versus BLASTn to the viral database were slower but comparable (Additional file 2: Table S2).
Raw FAST5/HDF files from the MinION instrument are base-called using the Metrichor 2D Basecalling v1.14 pipeline (Metrichor). The MetaPORE pipeline continually scans the Metrichor download directory for batch analysis of downloaded sequence reads. For each batch of files (collected every time 200 reads are downloaded in the download directory, or ≥2 min of elapsed time, whichever comes first), the 2D read or either the template or complement read, depending on which is of higher quality, is converted into a FASTQ file using HDF5 Tools [24]. The cutadapt program is then used to trim Sol-PrimerB adapter sequences from the ends of the reads [25]. Next, the BLASTn aligner is used to subtract host reads computationally [19, 26], aligning to the human fraction of the NT database at word size 11 and e-value cutoff of 10-5. The remaining, non-human reads are then aligned by BLASTn (on a 64-core server) or MegaBLAST (on a laptop) to the entire NT database, using the same parameters. Alternatively, the remaining reads can be aligned on a laptop using BLASTn to just the viral fraction of the NT database, followed by BLASTn alignment of the viral reads to the NT database to verify that they are correctly identified. For each read, the single best match by e-value is retained, and the NCBI GenBank gene identifier assigned to the best match is then annotated by taxonomic lookup of the corresponding lineage, family, genus, and species [19].
It has been reported that the LAST alignment algorithm [27] may be more sensitive for nanopore read identification [12, 28]. However, LAST was originally developed for genome-scale alignments, and not for huge databases such as the NT database. To date, it has only been used to align nanopore reads to individual reference sequences [12, 28]. We attempted to use the LAST software to align nanopore reads to the NT database (June 2014, ~60 Gb in size). LAST automatically created multiple formatted database volumes (n > 20), each approximately 24 Gb, to encompass all of the NT database. As the run time for loading each volume into memory was just under 2 minutes, resulting in a >40 minutes overhead time, LAST was considered to be impractical for real-time metagenomic sequencing analysis on a single server or laptop.
For real-time visualization of results, a graphical user interface was developed for the MetaPORE pipeline. A live taxonomic count table is displayed as a donut chart using the CanvasJS graphics suite [29], with the chart refreshing every 30 s (Additional file 3). For each viral species detected, the top hit is chosen to be the reference sequence (GenBank identifier) in the NT database assigned to that species with the highest number of aligned reads, with priority given to reference sequences in the following order: (1) complete genomes, (2) complete sequence, or (3) partial sequences or individual genes. Coverage maps are generated by mapping all aligned viral species reads to the top hit reference sequence using LASTZ v1.02 [30], with interactive visualization provided using a custom web program that accesses the HighCharts JavaScript library [31]. A corresponding interactive pairwise identity plot is generated using SAMtools [32] to calculate the consensus FASTA sequence from the coverage map, followed by pairwise 100-bp sliding-window comparisons of the consensus to the reference sequence using the BioPython implementation of the Needleman–Wunsch algorithm [33, 34]. For comparison, the MetaPORE pipeline was also run on a subset of 100,000 reads from parallel Illumina MiSeq data corresponding to the Chik1, Ebola1, and Ebola2 samples.
Phylogenetic analysis
The overall CHIKV phylogeny consisted of all 188 near-complete or complete genome CHIKV sequences available in the NT database as of March 2015. A subphylogeny, including the MiSeq- and nanopore-sequenced Puerto Rico strain PR-S6 presented here and previously [15], as well as additional Caribbean CHIKV strains and other representative members of the Asian-Pacific clade, was also analyzed. The EBOV phylogeny consisted of the newly MiSeq- and nanopore-sequenced Ebola strain Lomela-LokoliaB11 from the 2014 DRC outbreak [17], as well as other representative EBOV strains, including strains from the 2014–2015 West African outbreak [8, 35]. Sequences were aligned using the MAFFT algorithm [36], and phylogenetic trees were constructed using the MrBayes algorithm [37] in the Geneious software package [38].
Data availability
Nanopore and MiSeq sequencing data corresponding to non-human reads identified by MetaPORE, along with sample metadata, have been submitted to NCBI under the following GenBank Sequence Read Archive (SRA) accession numbers: Ebola virus/H.sapiens-wt/COD/2014/Lomela-Lokolia16 [SRA:SRP057409], Ebola virus/H.sapiens-wt/COD/2014/Lomela-LokoliaB11 [SRA:SRS933322], Chik1 [SRA:SRP057410] and HepC1 [SRA:SRP057418]. Sequence reads were additionally filtered for exclusion of human sequences by both BLASTn alignment at an e-value cutoff of 10-5 and Bowtie2 high-sensitivity local alignment to the human hg38 reference database.
Results
Example 1: Nanopore sequencing of high-titer chikungunya virus (Flow cell #1)
To test the ability of nanopore sequencing to identify metagenomic reads from a clinical sample, we first analyzed a plasma sample harboring high-titer CHIKV and previously sequenced on an Illumina MiSeq platform (Fig. 2a) [15]. The plasma sample corresponded to an asymptomatic blood donor who had screened positive for CHIKV infection during the 2014 outbreak in Puerto Rico (strain PR-S6), with a calculated viral titer of 9.1 × 107 copies/mL.

Metagenomic identification of CHIKV and EBOV from clinical blood samples by nanopore sequencing. a Time line of sequencing runs on flow cell #1 with sample reloading, plotted as a function of elapsed time in hours since the start of flow cell sequencing. b Cumulative numbers of all sequenced reads (black line) and target viral reads (red line) from the Chik1 run (left panel) and Ebola1 run (right panel), plotted as a function of individual sequencing run time in minutes. c Taxonomic donut charts generated using the MetaPORE bioinformatics analysis pipeline from the Chik1 run (left panel) and Ebola1 run (right panel). The total number of reads analyzed is shown in the center of the donut. d Coverage plots generated in MetaPORE by mapping reads aligning to CHIKV (left, Chik1 run) or EBOV (right, Ebola1 run) to the closest matching reference genome ((e), asterisk). A corresponding pairwise identity plot is also shown for CHIKV, for which there is sufficient coverage. e Whole-genome phylogeny of CHIKV. Representative CHIKV genome sequences from the Asian-Pacific clade, including the Puerto Rico PR-S6 strain recovered by nanopore and MiSeq sequencing, or all available 188 near-complete or complete CHIKV genomes (inset), are included. Branch lengths are drawn proportionally to the number of nucleotide substitutions per position, and support values are shown for each node. were was analyzed in MetaPORE on a 64-core Ubuntu Linux server using the June 2014 and January 2015 NT databases as the reference databases for the CHIKV and EBOV samples, respectively
A read aligning to CHIKV, the 96th read, was sequenced within 6 min (Fig. 2b, left panel) and detected by BLASTn alignment to the NT database within 8 min of data acquisition, demonstrating an overall sample-to-detection turnaround time of <6 hr (Fig. 1). After early termination of the sequencing run at the 2 hr 15 min time point, 556 of 19,452 total reads (2.8 %) were found to align to CHIKV (Fig. 2b, c, left panels). The individual CHIKV nanopore reads had an average length of 455 bp (range 126–1477 bp) and average percentage identity of 79.4 % to the most closely matched reference strain, a CHIKV strain from the neighboring British Virgin Islands (KJ451624), corresponding to an average nanopore read error rate of 20.6 % (range 8–49 %) (Table 1). When only high-quality 2D pass reads were included, 346 of 5139 (6.7 %) reads aligned to CHIKV, comparable to the proportion of CHIKV reads identified by corresponding metagenomic sequencing on the Illumina MiSeq (7.6 % by MetaPORE analysis of 100,000 reads; Fig. 3a, left panel).

MetaPORE analysis of Illumina MiSeq data from samples containing CHIKV and EBOV. Taxonomic donut charts were generated from Illumina MiSeq data corresponding to the Chik1 run (a) and Ebola1 run (b) using the MetaPORE bioinformatics analysis pipeline. The total number of MiSeq reads analyzed is shown in the center of the donut. Note that given computational time constraints, only a subset of reads (n = 100,000) was analyzed using MetaPORE. Coverage and pairwise identity plots were generated from MiSeq CHIKV reads from the Chik1 sample (248,677 of 3,235,099 reads, 7.7 %) (c), or EBOV reads from the Ebola1 sample (20,820 of 2,743,589 reads, 0.76 %) (d), identified using SURPI analysis and LASTZ mapping {Harris, 2007 #34} at an e-value of 10-5 to the closest matching reference genome. Data were analyzed in MetaPORE on a 64-core Ubuntu Linux server using the June 2014 and January 2015 NT databases as the reference databases for the CHIKV and EBOV samples, respectively.
Mapping of the 556 nanopore reads aligning to CHIKV to the assigned reference genome (KJ451624) showed recovery of 90 % of the genome at 3× coverage and 98 % at 1× coverage (Fig. 2d, left panel). Notably, despite high individual read error rates, 97–99 % identity to the reference genome (KJ451624) was achieved across contiguous regions with at least 3× coverage. Furthermore, phylogenetic analysis revealed co-clustering of the CHIKV genomes independently assembled from MinION nanopore or Illumina MiSeq reads (Fig. 2d, left panel and Fig. 3b, left panel) on the same branch within the Caribbean subclade (Fig. 2e). Overall, a large proportion of reads (55 %) in the error-prone nanopore data remained unidentifiable, while other aligning reads aside from CHIKV corresponded to human, lambda phage control spike-in, uncultured bacterial, or other eukaryotic sequences (Fig. 2c, left panel).
Example 2: Nanopore sequencing of high-titer Ebola virus (Flow cell #1)
We next attempted to replicate our metagenomic detection result on the nanopore sequencer with a different virus by testing a whole blood sample from a patient with Ebola hemorrhagic fever during the August 2014 outbreak in the DRC (Ebola1, strain Lomela-Lokolia16) [17]. To conserve flow cells, the same nanopore flow cell used to run the Chik1 sample was washed and stored overnight at 4 °C, followed by nanopore sequencing of the Ebola1 sample (viral titer of 1.0 × 107 copies/mL by real-time qRT-PCR) (Fig. 2b, right panel). Only 41 of 13,090 nanopore reads (0.31 %) aligned to EBOV (Fig. 2c, right panel), comparable to the percentage of reads obtained for Illumina MiSeq (0.84 % by MetaPORE analysis of 100,000 reads; Fig. 3a, right panel). The decrease in relative number and percentage of target viral nanopore reads in the Ebola1 sample relative to the Chik1 sample is consistent with the lower levels of viremia (1.0 × 107 versus 9.1 × 107 copies/mL) and higher host background (whole blood versus plasma). Nonetheless, the first read aligning to EBOV was detected in a similar timeframe as in the Chik1 sample, sequenced within 8 min and detected within 10 min of data acquisition. EBOV nanopore reads were 359 bp in length on average (range 220–672 nt), with an average error rate of 22 % (range 12–43 %) (Table 1). However, despite these error rates, the majority of Ebola nanopore sequences (31 of 41, 76 %) were found to align to the correct strain, Lomela-Lokolia16, as confirmed by MiSeq sequencing (Fig. 2d, right panel and Fig. 3b, right panel).
Despite washing the flow cell between the two successive runs, seven CHIKV reads were recovered during the Ebola1 library sequencing, suggesting the potential for carryover contamination. CHIKV reads were not present in the corresponding Illumina MiSeq Ebola1 run (Fig. 3a, right panel), confirming that the source of the contamination originated from the Chik1 nanopore library, which was run on the same flow cell as and just prior to the Ebola1 library.
Example 3: Nanopore sequencing of moderate-titer hepatitis C virus (Flow cell #2)
Our previous experiments revealed both the total number of metagenomic reads and proportion of target viral reads at a given titer that could be obtained from a single MinION flow cell, and showed that the proportion of viral reads obtained by metagenomic nanopore and MiSeq sequencing was comparable. Thus, we projected that the minimum concentration of virus that could be reproducibly detected using our current metagenomic protocol would be 1 × 105 copies/mL. An HCV-positive clinical sample (HepC1) was diluted in negative control serum matrix to a titer of 1 × 105 copies/mL and processed for nanopore sequencing using an upgraded library preparation kit (MAP-004). After four consecutive runs on the same flow cell with repeat loading of the same metagenomic HepC1 library (Fig. 4a), a total of 85,647 reads were generated, of which only six (0.0070 %) aligned to HCV (Fig. 4b). Although the entire series of flow cell runs lasted for >12 hr, the first HCV read was sequenced within 34 min, enabling detection within 36 min of data acquisition. Given the low titer of HCV in the HepC1 sample and hence low corresponding fraction of HCV reads in the nanopore data, the vast majority (96 %) of viral sequences identified corresponded to the background lambda phage spike-in (Fig. 4c). Importantly, although nanopore sequencing identified only six HCV reads, all six reads aligned to the correct genotype, genotype 1b (Fig. 4d).

Metagenomic identification of HCV from a clinical serum sample by nanopore sequencing. a Time line of sequencing runs on flow cell #2 with HepC1 sample reloading, plotted as a function of elapsed time in hours since the start of flow cell sequencing. b Cumulative number of all sequenced reads (black line) and HCV viral reads (red line), plotted as a function of individual sequencing run time in minutes. c Taxonomic donut charts generated using the MetaPORE bioinformatics analysis pipeline. The total number of reads analyzed is shown in the center of the donut. d Coverage and pairwise identity plots generated in MetaPORE by mapping reads aligning to HCV to the closest matching reference genome. Data were analyzed in MetaPORE on a 64-core Ubuntu Linux server using the January 2015 NT reference database
Example 4: Nanopore sequencing of high-titer Ebola virus with real-time MetaPORE analysis (Flow cell #3)
To enable real-time analysis of nanopore sequencing data, we combined pathogen identification with monitoring and user-friendly web visualization into a real-time bioinformatics pipeline named MetaPORE. We tested MetaPORE by sequencing a nanopore library (Ebola2) constructed using the upgraded MAP-004 kit and corresponding to a whole blood sample from a patient with suspected Ebola hemorrhagic fever during the 2014 DRC outbreak. Four consecutive runs of the Ebola2 library on the same flow cell over 34 hr (Fig. 5a) yielded a total of 335,308 reads, of which 609 (0.18 %) aligned to EBOV (141 of 6009 or 2.3 %, of 2D pass reads), comparable to the 0.91 % achieved by Illumina MiSeq sequencing (Fig. 5c).

Metagenomic identification of EBOV from a clinical blood sample by nanopore sequencing and MetaPORE real-time bioinformatics analysis. Nanopore data generated from the Ebola2 library and sequenced on flow cell #3 were analyzed in real time using the MetaPORE bioinformatics analysis pipeline, and compared to corresponding Illumina MiSeq data. a Time line of nanopore sequencing runs on flow cell #3 with sample reloading, plotted as a function of elapsed time in hours since the start of flow cell sequencing. b Cumulative numbers of all sequenced reads (black line) and target viral reads (red line) from the nanopore run (left panel) or MiSeq run (right panel), plotted as a function of individual sequencing run time in minutes. c Taxonomic donut charts generated by real-time MetaPORE analysis of the nanopore reads (left panel) and post-run analysis of the MiSeq reads (right panel). The total number of reads analyzed is shown in the center of the donut. Note that given computational time constraints, only a subset of MiSeq reads (n = 100,000) was analyzed using MetaPORE. d Coverage and pairwise identity plots generated from nanopore (left panel) or MiSeq data (right panel) by mapping reads aligning to EBOV to the closest matching reference genome ((e), asterisk). e Whole-genome phylogeny of EBOV. Representative EBOV genome sequences, including those from the 2014-2015 West Africa outbreak (tan) and 2014 DRC outbreak (pink), are included. Branch lengths are drawn proportionally to the number of nucleotide substitutions per position, and support values are shown for each node. Data were analyzed in MetaPORE on a 64-core Ubuntu Linux server using the January 2015 NT reference database.
Notably, the first EBOV read was sequenced 44 s after data acquisition and correctly detected in ~3 min by MetaPORE (Fig. 5b, right panel; Additional file 3). The mapping of nanopore reads across the EBOV genome was relatively uniform with at least one read mapping to >88 % of the genome and areas of zero coverage also seen with much higher-coverage Illumina MiSeq data (Fig. 5d). The detection of EBOV by real-time metagenomic nanopore sequencing was confirmed by qRT-PCR testing of the clinical blood sample, which was positive for EBOV at an estimated titer of 7.64 × 107 copies/mL. Phylogenetic analysis of the Ebola2 genome independently recovered by MinION nanopore and Illumina MiSeq sequencing revealed that nanopore sequencing alone was capable of pinpointing the correct EBOV outbreak strain and country of origin (Fig. 5e).
Discussion
Unbiased point-of-care testing for pathogens by rapid metagenomic sequencing has the potential to transform radically infectious disease diagnosis in clinical and public health settings. In this study, we sought to demonstrate the potential of the nanopore instrument for metagenomic pathogen identification in clinical samples by coupling an established assay protocol with a new real-time sequence analysis pipeline. To date, high reported error rates (10–30 %) and relatively low throughput (<100,000 reads per flow cell) have hindered the utility of nanopore sequencing for analysis of metagenomic clinical samples [9, 11]. Prior work on infectious disease diagnostics using nanopore has focused on rapid PCR amplicon sequencing of viruses and bacteria [11], or real-time sequencing of pure bacterial isolates in culture, such as Salmonella in a hospital outbreak [12]. To our knowledge, this is the first time that nanopore sequencing has been used for real-time metagenomic detection of pathogens in complex, high-background clinical samples in the setting of human infections. Here, we also sequenced a near-complete viral genome to high accuracy (97–99 % identity) directly from a primary clinical sample and not from culture. As also demonstrated previously for the bacterium Escherichia coli K-12 [13], the CHIKV genome was assembled using only multiple overlapping, albeit error-prone, nanopore reads and without resorting to the use of a secondary platform such as an Illumina MiSeq for sequence correction (Fig. 2d).
Real-time sequence analysis is necessary for time-critical applications such as outbreak investigation [7] and metagenomic diagnosis of life-threatening infections in hospitalized patients [3, 4, 6]. NGS analysis for clinical diagnostics is currently performed after sequencing is completed, analogous to how PCR products were analyzed by agarose gel electrophoresis in the 1990s. Most clinical PCR assays to date have since been converted to a real-time format that reduces hands-on laboratory technician time and effort and decreases overall sample-to-answer turnaround times. Importantly, our nanopore data suggest that very few reads are needed to provide an unambiguous diagnostic identification, despite high individual per read error rates of 10–30 %. The ability of nanopore sequence analysis to identify viruses accurately to the species and even strain or genotype level is facilitated by the high specificity of viral sequence data, especially with the longer reads achievable by nanopore versus second-generation sequencing (Table 1, 452 bp; range 126–1477 bp).
Although the overall turnaround time for metagenomic sample-to-detection has now been reduced to <6 hr with nanopore sequencing, many challenges remain for routine implementation of this technology in clinical and public health settings. Improvements to make library preparation faster and more robust are critical, including automation and optimization of each step in the protocol. Standardized external and internal spike-in controls run in parallel will be needed to control for laboratory and carryover contamination. Here we looked only at clinical samples at moderate to high titers of 105–108 copies/mL, and the sensitivity of metagenomic nanopore sequencing at lower titers remains unclear at current achievable sequencing depths. Standard wash protocols also appear inadequate to prevent carryover contamination when reusing the same flow cell, as CHIKV reads were identified in the downstream Ebola1 sample sequence run. One solution may be to perform only one nanopore sequencing run per flow cell for clinical diagnostic purposes, akin to how individual disposable cartridges are used for clinical quantitative PCR testing on a Cepheid GenXpert instrument to prevent cross-contamination [39]. Another potential solution is to give unique barcodes to individual samples as part of a multiplexed sequencing run at the cost of added time and effort.
A key challenge with microbial identification by metagenomic nanopore sequencing is that the current accuracy of sparse nanopore reads is insufficient to allow confident species identification of bacteria, fungi, or parasites, which have much larger genomes and share more conserved genes than viruses. Indeed, distinct bacterial species are often defined by as little as 5 % genomic divergence and 1 % sequence divergence in highly conserved housekeeping genes such as 16S ribosomal RNA [40]. Of note, the majority of nanopore reads aligning to bacteria in this study likely originated from the inclusion of lambda phage DNA in the sequencing library, reagent contamination, or, for the Ebola virus samples, environmental contamination from sample collection in a rural hospital setting (Additional file 4: Table S3). Accurate identification of eukaryotic pathogens from sparse, error-prone nanopore reads also appears to be challenging (Additional file 4: Table S3). In addition, single-nucleotide resolution will likely be required for detection of antimicrobial resistance markers [41], which is difficult to achieve from relatively low-coverage metagenomic data [42]. These limitations can potentially be overcome in the future by target enrichment methods such as capture probes to increase coverage, improvements in nanopore sequencing technology, or more accurate base-calling and alignment algorithms for nanopore data [43, 44].
Conclusions
Our results indicate that unbiased metagenomic detection of viral pathogens from clinical samples with a sample-to-answer turnaround time of <6 hr and real-time bioinformatics analysis is feasible with nanopore sequencing. We demonstrate unbiased, diagnostic identification of EBOV within ~3 min of sequence acquisition. This technology will be particularly desirable for enabling point-of-care genomic analyses in the developing world, where critical resources, including reliable electric power, laboratory space, and computational server capacity, are often severely limited. Importantly, MetaPORE, the real-time sequencing analysis platform developed here, is web-based and can be run on a laptop. As sequencing yield, quality, and turnaround times continue to improve, we anticipate that third-generation technologies such as nanopore sequencing will challenge clinical diagnostic mainstays such as PCR and transcription-mediated amplification testing, fulfilling the dream of an unbiased, point-of-care test for infectious diseases.
Abbreviations
- bp:
-
base pair
- cDNA:
-
complementary DNA
- Chik1:
-
chikungunya virus, strain PR-S6 sample
- CHIKV:
-
chikungunya virus
- DNA:
-
deoxyribonucleic acid
- DRC:
-
Democratic Republic of the Congo
- Ebola1:
-
Ebola virus, strain Lomela-Lokolia16 sample
- Ebola2:
-
Ebola virus, strain Lomela-LokoliaB11 sample
- EBOV:
-
Ebola virus
- Gb:
-
gigabase pair
- HCV:
-
hepatitis C virus
- HepC1:
-
hepatitis C virus, genotype 1b sample
- HTML:
-
hypertext markup language
- kb:
-
kilobase pair
- MAP:
-
MinION Access Program
- MetaPORE:
-
a bioinformatics analysis pipeline for real-time pathogen identification and visualization from nanopore NGS data
- MinION:
-
nanopore sequencing platform developed by Oxford Nanopore, Inc
- NCBI:
-
National Center for Biotechnology Information
- NGS:
-
next-generation sequencing
- nt:
-
nucleotide
- NT database:
-
NCBI nucleotide collection database
- qRT-PCR:
-
quantitative reverse transcription polymerase chain reaction
- RNA:
-
ribonucleic acid
- SURPI:
-
sequence-based ultra-rapid pathogen identification, a bioinformatics analysis pipeline for pathogen identification from NGS data developed at UCSF
- UCSF:
-
University of California, San Francisco
- dNTP:
-
deoxynucleotide triphosphate
- DTT:
-
Dithiothreitol
- SS III RT:
-
Superscript III reverse transcriptase
References
Pallen MJ. Diagnostic metagenomics: potential applications to bacterial, viral and parasitic infections. Parasitology. 2014;141:1856–62.
Miller RR, Montoya V, Gardy JL, Patrick DM, Tang P. Metagenomics for pathogen detection in public health. Genome Med. 2013;5:81.
Brown JR, Morfopoulou S, Hubb J, Emmett WA, Ip W, Shah D, et al. Astrovirus VA1/HMO-C: an increasingly recognized neurotropic pathogen in immunocompromised patients. Clin Infect Dis. 2015;60:881–8.
Naccache SN, Peggs KS, Mattes FM, Phadke R, Garson JA, Grant P, et al. Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin Infect Dis. 2015;60:919–23.
Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–8.
Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med. 2014;370:2408–17.
Briese T, Paweska JT, McMullan LK, Hutchison SK, Street C, Palacios G, et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009;5:e1000455.
Gire SK, Goba A, Andersen KG, Sealfon RS, Park DJ, Kanneh L, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–72.
Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2015;33:296–300.
Goodwin S, Gurtowski J, Ethe-Sayers S, Despande P, Schatz M, McCombie WR. Oxford Nanopore Sequencing and de novo assembly of a eukaryotic genome. bioRxiv 2015. http://dx.doi.org/10.1101/013490.
Kilianski A, Haas JL, Corriveau EJ, Liem AT, Willis KL, Kadavy DR, et al. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. Gigascience. 2015;4:12.
Quick J, Ashton P, Calus S, Chatt C, Gossain S, Hawker J, et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015;16:114.
Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12:733–5.
The MinION(TM) Access Programme - Community - Oxford Nanopore Technologies. http://nanoporetech.com/community/the-minion-access-programme/. AccessedSeptember 2015.
Chiu CY, Bres V, Yu G, Krysztof D, Naccache SN, Lee D, et al. Emerging genomic assays for identification of chikungunya virus infection in blood donors from Puerto Rico, 2014. Emerg Infect Dis. 2015;21:1409–13.
Epelboin A, Formenty P, Anoko J, Allarangar Y, B J-M. Humanisation and informed consent for people and populations during responses to VHF in central Africa (2003–2008). In: Biquet JM, editor, Humanitarian stakes No 1. Geneva, Switzerland: Médecins Sans Frontières (MSF) Switzerland; 2008. p. 25–38.
Maganga GD, Kapetshi J, Berthet N, Kebela Ilunga B, Kabange F, Mbala Kingebeni P, et al. Ebola virus disease in the Democratic Republic of Congo. N Engl J Med. 2014;371:2083–91.
Trombley AR, Wachter L, Garrison J, Buckley-Beason VA, Jahrling J, Hensley LE, et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am J Trop Med Hyg. 2010;82:954–60.
Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–92.
Chen EC, Miller SA, DeRisi JL, Chiu CY. Using a pan-viral microarray assay (Virochip) to screen clinical samples for viral pathogens. J Vis Exp 2011;50.
Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Yu G, et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One. 2010;5:e13381.
Greninger AL, Naccache SN, Messacar K, Clayton A, Yu G, Somasekar S, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012–14): a retrospective cohort study. Lancet Infect Dis. 2015;15:671.
MetaPORE – Chiu laboratory, University of California, San Francisco. http://github.com/chiulab/MetaPORE. Accessed September 2015.
HDF5/Tools API Specification. http://www.hdfgroup.org/HDF5/doc/RM/Tools.html. Accessed September 2015.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–2.
Chiu CY. Viral pathogen discovery. Curr Opin Microbiol. 2013;16:468–78.
Frith MC, Hamada M, Horton P. Parameters for accurate genome alignment. BMC Bioinformatics. 2010;11:80.
Quick J, Quinlan AR, Loman NJ. A reference bacterial genome dataset generated on the MinION portable single-molecule nanopore sequencer. Gigascience. 2014;3:22.
Beautiful HTML5 Javascript Charts | Canvas JS. http://canvasjs.com. Accessed September 2015.
Harris R: Improved pairwise alignment of genomic DNA. PhD thesis. PA: Pennsylvania State University, University Park; 2007.
Interactive Javascript charts for your webpage | Highcharts. http://www.highcharts.com. Accessed September 2015.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–3.
Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–53.
Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N, et al. Emergence of Zaire Ebola virus disease in Guinea. N Engl J Med. 2014;371:1418–25.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Blakemore R, Story E, Helb D, Kop J, Banada P, Owens MR, et al. Evaluation of the analytical performance of the Xpert MTB/RIF assay. J Clin Microbiol. 2010;48:2495–501.
Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–31.
Fournier PE, Dubourg G, Raoult D. Clinical detection and characterization of bacterial pathogens in the genomics era. Genome Med. 2014;6:114.
Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P. A bioinformatician’s guide to metagenomics. Microbiol Mol Biol Rev. 2008;72:557–78.
Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12:351–6.
Loman NJ, Watson M. Successful test launch for nanopore sequencing. Nat Methods. 2015;12:303–4.
Acknowledgements
This study is supported in part by a grant from the National Institutes of Health (R01-HL105704) (CYC) and an UCSF-Abbott Viral Discovery Award (CYC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CYC is the director of the UCSF-Abbott Viral Diagnostics and Discovery Center and receives research support in pathogen discovery from Abbott Laboratories, Inc. JML and VB are employees of Hologic, Inc. P. Mbala, P. Mulembakani, and BS are employees of Metabiota, Inc. The other authors declare that they have no competing interests.
Authors’ contributions
ALG and CYC conceived and designed the study. ALG performed the nanopore experiments. SNN, GY, SS, and JB provided cDNA libraries and generated Illumina libraries. SF, ALG, and CYC developed the MetaPORE pipeline. SF, DS, and CYC generated the real-time visualization software for the nanopore data. ALG, SF, SNN, and CYC validated the MetaPORE pipeline using Oxford nanopore and Illumina data. VB, JML, RD, and SS provided the chikungunya samples. P.Mbala, P.Mulembakani, BSS, and JJM provided the Ebola samples. ALG, SNN, and CYC wrote the manuscript. All authors read and approved the final manuscript.
Samia N. Naccache and Scot Federman contributed equally to this work.
Additional files
Additional file 1: Table S1.
Optimization of word count for MegaBLAST alignment to the NT reference database. (XLSX 12 kb)
Additional file 2: Table S2.
MetaPORE performance according to system configuration and alignment mode. (XLSX 18 kb)
Additional file 3:
Movie. MetaPORE real-time bioinformatics analysis and visualization (clip 0:19–10:13, 9.8 min). Detection of EBOV by metagenomic nanopore sequencing and real-time MetaPORE bioinformatics analysis. Raw FAST5 files are uploaded to the Metrichor cloud-based analytics platform for 2D base-calling (clip, right panel). After downloading from Metrichor, base-called FAST5 reads are collected in batches of 200 reads and automatically processed in real time by MetaPORE. Read counts corresponding to detected organisms (e.g. humans, viruses, bacteria, non-human eukaryotes) and viral species are displayed in donut plots that are updated each minute in real time (left panel). Note that the first EBOV read from the Ebola2 sequencing run is detected 3 min 6 s (3:26) after the start of sequence acquisition (0:19). (clip 10:21–11:51, 1.5 min). Web-based, interactive coverage map and pairwise identity plots, generated in real time by MetaPORE, enable zooming, highlighting of individual values, outputting of relevant statistical data, and exporting of the graphs in various formats. The plots shown in the movie correspond to the analyzed data after completion of nanopore sequencing. Data were analyzed in MetaPORE on a 64-core Ubuntu Linux server using the January 2015 NT reference database. (MP4 20493 kb)
Additional file 4: Table S3.
Taxonomic classification of non-human nanopore reads identified using MetaPORE. (XLSX 117 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Greninger, A.L., Naccache, S.N., Federman, S. et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med 7, 99 (2015). https://doi.org/10.1186/s13073-015-0220-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-015-0220-9