
Abstract
The methylation of histone H3 at lysine 36 (H3K36me) is essential for maintaining genomic stability. Indeed, this methylation mark is essential for proper transcription, recombination, and DNA damage response. Loss- and gain-of-function mutations in H3K36 methyltransferases are closely linked to human developmental disorders and various cancers. Structural analyses suggest that nucleosomal components such as the linker DNA and a hydrophobic patch constituted by histone H2A and H3 are likely determinants of H3K36 methylation in addition to the histone H3 tail, which encompasses H3K36 and the catalytic SET domain. Interaction of H3K36 methyltransferases with the nucleosome collaborates with regulation of their auto-inhibitory changes fine-tunes the precision of H3K36me in mediating dimethylation by NSD2 and NSD3 as well as trimethylation by Set2/SETD2. The identification of specific structural features and various cis-acting factors that bind to different forms of H3K36me, particularly the di-(H3K36me2) and tri-(H3K36me3) methylated forms of H3K36, have highlighted the intricacy of H3K36me functional significance. Here, we consolidate these findings and offer structural insight to the regulation of H3K36me2 to H3K36me3 conversion. We also discuss the mechanisms that underlie the cooperation between H3K36me and other chromatin modifications (in particular, H3K27me3, H3 acetylation, DNA methylation and N6-methyladenosine in RNAs) in the physiological regulation of the epigenomic functions of chromatin.
Similar content being viewed by others
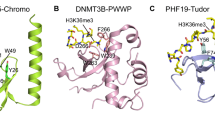

Introduction
Eukaryotic genomic DNA is packaged into structural units called nucleosomes. Each nucleosome consists of 146 base pairs (bp) of DNA that wrap around (1.6 turns) an octameric complex comprising two of each of histones H2A, H2B, H3 and H4 [1, 2]. In most eukaryotes, there exists a linker histone H1 to stabilize the chromatin structure and allow for further compaction of chromatin into more complex higher-order structures [2–5]. The amino acid residues of the core histone proteins are heavily decorated with post-translational modifications (PTMs), particularly on the unstructured histone tail domains, and these modifications help to regulate chromatin compaction and govern the availability of docking sites for chromatin-modifying factors [1, 6]. PTMs such as methylation, phosphorylation, acetylation, SUMOylation and ubiquitination occur via the transfer of chemical moieties onto specific residues of histone proteins [7] that engage in crosstalk and influence various DNA metabolic processes, including gene transcription [7–10], DNA damage repair [11, 12], specialized chromosomal loci assembly [13, 14], and cell cycle progression [15–18].
Lysine 36 of histone H3 (H3K36) can be modified by mono-, di- or trimethylation (H3K36me1, H3K36me2 and H3K36me3, respectively). In human, this is orchestrated by a redundant series of enzymes: nuclear receptor-binding SET domain (NSD) protein 3, which only catalyzes monomethylation in vivo; NSD1, NSD2,ASH1L and MYND domain-containing 2 (SMYD2), which catalyze dimethylation in vivo; and SET domain-containing 2 (SETD2) , which catalyzes trimethylation in vivo; albeit, SETD2 is capable of catalyzing all forms of methylation in vitro [19–32]. Yeasts, such as budding yeast Saccharomyces cerevisiae and fission yeast Schizosaccharomyces pombe, encode a single H3K36 methyltransferase (HMTase) in their genomes, known as Set2, which bears a high sequence similarity to SETD2 (Fig. 1A) and is responsible for generating all three forms of histone methylation in yeast [12, 33–35]. Like mammalian H3K36 methyltransferases, yeast Set2 is recruited to chromatin during transcription elongation to catalyze H3K36me in a co-transcriptional manner [33, 36–38].

Set2/SETD2 is generally conserved among fungi and metazoans. A Multiple sequence alignment (MSA) of Set2 homologs across fungi and metazoan species. MEGAX software and NCBI MSA viewer 1.13.1 [230] were used to generate an alignment of the primary amino acid sequences of Set2/SETD2 homologs from fission yeast, budding yeast, fungi Aspergillus turcosus, and metazoan species including human, rat, mouse, zebrafish, fruitfly, and nematode worm C. elegans. 35% of the human SETD2 amino acid sequence is conserved in fission yeast Set2 , even for sequences beyond the catalytic SET domain. 53% of the amino sequence in the catalytic SET and post-SET domains is identical in fission yeast Set2 and human SETD2. Residues that are generally conserved across species are indicated in red. Residues that are identical or similar in polarity across species are, respectively, highlighted in black or grey. Conserved “decision-making” residues that regulate the degree of methylation are circled in blue. Non-conserved aromatic residues that make contact with histone tails and possibly participate in methylation regulation are circled in red. Domain structure of fission yeast Set2 and human SETD2 are also shown to indicate the relative amino acid positions of the MSA sequences in the SETD2 homologues. B Conserved motifs in human SETD2 SET-domain. i Surface representation: sections of conserved motifs are highlighted (green, teal, cyan, and orange). Pymol visualization is derived from the crystallized structure database in the Protein Data Bank, entry 5V21 [74]. ii Projection of the conserved sequence of yeast Set2 and human SETD2. The SET domain crystalized structure highlights (1) a regulatory LIN loop, (2) a triangular core motif separated from the catalytic site, and (3) histone-interacting residues (refer Figs. 3 and 4). Conserved residues in the triple β-sheet in the triangular core of the SETD2 SET domain (green) endow the SET domain with its recognizable triangular shape, which maintains the structure of the domain. Conserved LIN-loop (teal, cyan and orange) and α8 (short, white α-helix region) in the closed conformation secure the histone tail in position for methylation.
Proper H3K36me epigenome function is critical for maintaining genomic stability, with defects in this process widely observed to be associated with human diseases, including prenatal developmental disorder [39, 40], and cancer [41–45]. These conditions arise presumably due to defects in processes linked with transcription, splicing [46–49], and cell cycle regulation [15–18]. In yeast cells, mutations in Set2 or H3K36 lead to a decreased lifespan, presumably by reasons of a global loss of histone methylation [44] and an inability to mount a proper response to environmental stressors like genotoxic insult [12, 16] or nutrient deprivation [50].
Here, we consolidate the structural, functional and physiological research—garnered predominantly from studies in yeast Set2 and human NSD2, NSD3, ASH1L and SETD2—pertaining to the catalysis of H3K36 methylation, and cross-reference yeast studies against those carried out using human SETD2 and other H3K36 methyltransferases.
Physiological roles of Set2/SETD2 in DNA damage repair and tumor suppression
SETD2 is a known tumor suppressor and is involved in several molecular pathways that maintain genomic stability. SETD2 mutations are associated with progression, recurrence, and survival rates, especially among patients with clear cell renal cell carcinoma (ccRCC) [45, 51–53]. We reviewed this topic in detail recently, so here we will only briefly discuss some of these findings [54]. Mutations affecting H3K36 methylation—particularly SETD2 truncation mutations—account for > 30% of pediatric high-grade gliomas [54, 55]. SETD2 loss-of-function mutations were observed in 10% of primary and 30% of metastatic ccRCC tumors, whereas H3K36 methylation is significantly reduced in ccRCC cell lines and patient samples [56]. In ccRCC, an R1625C point mutation in SETD2 destabilizes SETD2 protein, reduces its capacity for substrate binding, diminishes H3K36me3, and delays the DNA damage response, with evidence of reduced γH2A.X foci formation in an H3K36me3-dependent manner. Interestingly, introduction of the equivalent mutation in budding yeast (R195C) results in a similar attenuation of H3K36me3; albeit it affects less H3K36me1 and H3K36me2 levels, implicating a preferential role of R1625 in the formation of H3K36me3 [57]. In contrast, R2510H mutation within the SETD2 SRI domain—another common ccRCC-associated mutation—has no effect on H3K36me3. This may indicate that the interaction between SETD2 and RNA polymerase II (RNAPII) is a functionally discrete mechanism in ccRCC carcinogenesis [57]. In the case of acute lymphoblastic leukemia, H3K36me3 is enriched on most genes regulated by leukemia-associated transcription regulator MLL. Indeed, the downregulation of H3K36me3 by SETD2 mutation can attenuate leukemia cell proliferation [58].
At the molecular level, Set2/SETD2 regulates several processes that are essential for the maintenance of genomic stability, including response and repair of various types of DNA damage, alternative splicing and ensuring proper progress of the cell cycle. These three processes may compositely underlie major tumor suppression pathways regulated by SETD2 in human cancers.
H3K36 methylation has been linked to homologous recombination (HR) in response to various types of DNA lesions. For example, in the case of UV-induced DNA damage, SETD2 is involved in the recruitment of 53BP1 to damaged DNA loci via its interaction with γH2AX and H3K36me3 [59]. In contrast, at strand breaks, lens epithelium-derived growth factor (LEDGF; p75) is recruited onto DNA double-stranded breaks (DSB) via its interaction with H3K36me3 marks to generate single-stranded break overhangs with the C-terminal binding protein interacting protein (CtIP) nuclease [60, 61]; RAD51 and PALB2 are then recruited via H3K36me3 binding to stabilize and guide strand invasion for DNA repair [59, 62–65]. In fission yeast, Set2 has been reported to mediate the timely localization of the HR factor Rhp54 as well as nucleotide excision repair factor Rhp23 when the cells are exposed to DNA alkylating damage following methyl methanesulfonate (MMS) treatment [12]. These DNA damage response factors have been shown to be targeted to gene loci such as brc1+—which encodes a BRCT domain factor, similar to human BRCA1—to elicit the repair of damaged DNA [12].
Human SETD2 physically interacts with splicing factors, U2AF and SF-1, as well as heterogeneous nuclear ribonucleoproteins via SETD2-hnRNP Interaction (SHI) domain to regulate splicing [66]. Thus, not surprisingly, deregulation of alternative splicing has been shown to enhance tumorigenesis [47, 67,68,69]. Knockout of SETD2 in intestinal cells of a colorectal cancer mouse model deregulated the alternative splicing of Wnt/β-catenin signaling genes (e.g., disheveled segment polarity protein 2) to promote metastasis [47]. Primary human kidney tumors bearing SETD2 mutations and hosting a global reduction in H3K36me3 were found to have disruptions in processes concerning mRNA processing, intron retention, and splicing in ~ 25% of genes in the genome [70]. Furthermore, the knockdown of SETD2 in human gastric cancer cell lines was shown to result in the accumulation of aberrantly spliced transcripts of the mismatch repair gene hMLH1, suggesting that interfering with the alternative splicing of DNA damage repair genes may also underlie defective DNA damage responses in the disrupted function of SETD2 [67].
Fission yeast set2 null mutant showed reduced phosphorylation of the DNA damage checkpoint kinase Chk1 (similar to mammalian CHK2), suggesting that Set2 also regulates proper activation of the DNA damage checkpoint [12]. A similar effect on DNA damage checkpoint was also observed in the mammalian system, in which a leukemia-associated SETD2 F2478L point mutation caused a decrease in the activating phosphorylation of checkpoint kinases CHK1 and CHK2 and cyclin-dependent kinase (CDK)-inhibiting WEE1 kinase [18]. Transcriptomic analysis in the SETD2 F2478L leukemic cells revealed a downregulation in the genes associated with G2/M progression, DNA replication and p53 apoptotic pathways, suggesting that SETD2 may control cell cycle-related transcription programs. Indeed, both human SETD2 and fission yeast Set2 have been reported to regulate the expression of ribonucleotide reductase (RNR) complex genes to ensure the precise progression of cells through S-phase [71, 72]. In osteosarcoma cells treated with WEE1 kinase inhibitor AZD1775, SETD2 knockout induced an accumulation of cells in S-phase because of reduced RRM2 RNR subunit expression; this, in turn, induced collapse of the DNA replication fork [71]. SETD2 also regulates the cell cycle via phosphorylation of Lys-40 of α-tubulin to mediate proper mitotic spindle formation and cytokinesis [15].
Domain structure in SETD2 homologs
Set2 and SETD2 are modular enzymes bearing several defined molecular motifs that coordinate the molecular functions of the H3K36 methyltransferases (Fig. 1A and B). The primary amino acid (a.a.) sequence of mammalian SETD2 is highly similar to the sequences of fly and yeast Set2 proteins (Fig. 1A); albeit, the lengths of these proteins vary greatly from 733 a.a. in Saccharomyces cerevisiae Set2 protein to 2,537 a.a. in Mus musculus SETD2 [32, 57]. Despite the disparity in length, the function of these factors is highly conserved, owing to the dominant function of the SET domain. Indeed, the H3K36 methylation profiles on the genome are remarkably similar across Set2/SETD2 homologs, even across different orders of life [32, 73].
Set2/SETD2 homologs identified by PSI-BLAST search show similar domain architecture, consisting of post-SET, SET, Associated With SET (AWS), and Set2-Rpb1 interaction (SRI) domains [33, 57, 74] (Fig. 1A). The AWS [75], SET, and post-SET domains are necessary for the catalytic function of Set2/SETD2 in methylating H3K36 [12, 76]. In contrast, the C-terminal SRI domain drives the binding between Set2/SETD2 and the C-terminus of the Rpb1 subunit of RNAPII. The SRI motif comprises three α-helices that recognize and bind to the phosphorylated Ser-2 residue within the hepta-repeats of the hyperphosphorylated CTD of Rpb1 [36, 37, 77]. In vitro electromobility assays have been used to confirm that the SRI domain of budding yeast Set2 binds to nucleosomal linker DNA and hence determines Set2 enzyme substrate specificity [78]. Deleting the SRI domain has no effect on Set2 chromatin localization, indicating that there are other mechanisms regulating chromatin binding of Set2/SETD2 [79]. In fact, the SET domain and flanking regions are reported to bind single-stranded nucleic acids, including RNA and single-stranded DNA [80], whereas the N-terminus of Set2 interacts with histone H4 [80], thus contributing to the stability of the protein and enhancing its binding to chromatin.
In H3K36 HMTases of mammals, nematode worm and fly, the tryptophan-tryptophan (WW) and the coiled-coil (CC) domains lie distal to the post-SET domain and function in protein–protein interactions. The WW domain binds a poly-proline (Pro) stretch within itself to constitute an intramolecular auto-inhibitory module, which competes with the binding between the WW domain of SETD2 and the Pro-rich region of the Huntingtin protein [81]. The CC domain, on the other hand, promotes dimerization within another auto-inhibitory motif to regulate the extent of methylation [32, 78, 82].
Budding yeast Set2 also contains an auto-inhibitory domain (AID), which lies between the catalytic SET domain and the RNAPII-binding SRI domain. Truncation of the AID allows mutated Set2 to bind to unphosphorylated RNAPII, thereby generally promoting Set2 catalytic activity and enhancing cryptic transcription [78]. However, this domain is not conserved in fission yeast or human. In addition, the detailed regulatory mechanism of the AID remains mostly unknown [32].
Histone H3K36 di-methyl transferases
Domain structure of NSDs and ASH1L
Histone H3K36 can be dimethylated by several enzymes [27] including ASH1L, NSD2 and NSD3; these NSD proteins have been structurally studied in detail. The NSD family of proteins comprise the AWS, SET and post-SET domains, which compositely form the catalytic regulatory domain that is shared with Set2 and SETD2 trimethylase (Fig. 2). In addition, the family also contains three types of chromatin-associating domains, namely the plant homeodomain (PHD) domains: four PHD motifs are flanked by two proline-tryptophan-tryptophan-proline (PWWP) domains at the N-terminus (PWWP1 and PWWP2), with the fifth PHD motif located in the C-terminus, distal to the AWS-SET-post-SET catalytic domains. A high mobility group (HMG) motif is found solely in NSD2 (Fig. 2). The PWWP, PHD and HMG domains associate with various components of chromatin: PWWP1 in NSD2 can bind with methylated H3K36 to stabilize chromatin association of the enzymes and enable propagation of H3K36me3 [83]. PHD motifs are critical in sustaining the catalytic activity of NSD2; indeed, one study showed that ablation of the second PHD delocalized NSD2 from the nucleus to the cytoplasm, and this mislocalization correlated with complete abolishment of enzymatic activity [84]. The HMG box confers the nuclear localization of NSD2 by mediating its interaction with the DNA-binding domain of androgen receptor (AR) [85]. Overexpression of NSD2 provokes AR transcription, which in turn induces RAS signaling and promotes prostate cancer progression [86, 87].

Schematic diagram detailing the domain structure of common isoforms of NSD methyltransferases, ASH1L and SETD2. Numbers listed vertically refer to the relative amino acid positions of the domains in the different methyltransferases
ASH1L, of the Trithorax group of proteins, is another H3K36 di-methylase in fly and mammalian cells. ASH1L also shares the AWS-SET-post-SET catalytic domains, but differs to the NSD proteins, as it hosts a bromodomain and a BAH domain in its C-terminus (Fig. 2) [88, 89].
Roles of H3K36 dimethylases in oncogenesis and cancer progress
Overexpression of the NSD family proteins is shown to be oncogenic. NSD1 promotes the progression of acute myeloid leukemia, multiple myeloma, and lung cancer, as well as cancer cell migration and invasion in hepatocellular carcinoma (HCC), pediatric glioma, and breast cancer [90–95]. The carcinogenic potential of NSD1 is mediated via activation of Wnt/β-catenin signaling [90, 94].
NSD2 overexpression is linked with tumor aggressiveness [96] in cancers of the breast [97], cervix [98], lung [99], kidney [100], head and neck [101], brain [91], blood [102], colorectum [103], prostate, skin [96], and ovary [24]. Carcinogenesis associated with changes in the expression of NSD2 is also linked with cell cycle dysregulation [102], VEGF-A-mediated angiogenesis [104], hematopoietic stem cell differentiation [105], metastasis [91], chemoresistance (in osteosarcoma) [106], and the expression of various oncogenes (e.g., SYK, PTPN13 and ETV5 in multiple myeloma) [107]. Gain-of-function mutations (E1099K and T1150A) in the SET domain of NSD2 have been associated with the enhanced enzymatic activity of NSD2 in mantle cell lymphoma and pediatric acute lymphoblastic leukemia, in which they cause destabilization of the auto-inhibitory loop responsible for keeping signaling in check (refer below) [108, 109]. E1099K mutation is also found in 70% of patients with leukemia who experience relapse, and has been linked with aberrant global DNA methylation profiles in these patients [108, 110].
NSD3 is overexpressed in 58% of patients with advanced squamous cell carcinoma of the head and neck [92, 101]. NSD3 is also connected with metastatic cancers of the breast [111, 112], colorectum [113], pancreas [114] lung [115], and bone [116]. A short non-catalytic isoform of NSD3 that retains only the PWWP domain can promote oncogenesis by antagonizing proteasome-mediated degradation of the MYC oncogene [117]. Like NSD2, mutations in the SET domain of NSD3 (E1181K and T1232A), which counteract the auto-inhibition mechanism of H3K36 methylation, are associated with mantle cell lymphoma and chronic lymphocytic leukemia [118, 119].
ASH1L is involved in a global genomic nucleotide excision repair regulation and is recruited by DNA damage specific DNA-binding protein 2 (DDB2) to promote cyclobutane pyrimidine dimer excision [120]. There is evidence to show that, in acute leukemic conditions, translocation of MLL1 leads to deletion of ASH1L, followed by the transcriptional activation of various oncogenes (e.g., HOXA9) and cancer transformation [89, 121, 122]. ASH1L overexpression is associated with the progression and development of breast, liver, and thyroid cancers [123–125].
Neurodevelopmental roles of H3K36 dimethylases
NSD genes are important for pre- and postnatal neurodevelopment and their loss-of-function mutations relate to neurological syndromes, particularly Soto’s and Wolf–Hirschhorn’s syndromes [126, 127]. Soto’s syndrome, which occurs in 1/14,000 births and is associated with intellectual disability, facial deformation, and overgrowth phenotypes, is genetically mapped to NSD1 haploinsufficiency [126, 128]. Cells derived from a patient with Soto’s syndrome hosting an NSD1-inactivating mutation was shown to have a global redistribution of the DNA methyltransferase DNMT3A, along with promoter DNA hypomethylation, dysregulated synapse formation, and dysregulated neurodevelopmental gene expression [129–131]. Moreover, individuals hosting NSD1 whole-gene deletions exhibited early-onset cerebrovascular diseases [132]. This phenotype was recapitulated by deletion of an NSD1-like gene in fly, with developmental symptoms accompanied by global H3K36me2 reduction, defective motor and memory functions, and body size overgrowth [133]. Conversely, overexpression of the NSD1 homolog induced neuronal apoptosis and larval locomotive defects [134].
Mutation or deletion of NSD2 is believed to underlie Wolf–Hirschhorn’s syndrome, which is clinically characterized by pre- and postnatal neurodevelopmental disability and hypotonia [127, 135]. NSD2 point mutations at C869Y (in PHD domain), P895L (in PWWP domain), K1019R (in AWS domain), E1091K, E1099K, and S1137F (in SET domain) are associated with a global decrease in H3K36me2 levels in patients with Wolf–Hirschhorn syndrome. In particular, C869Y mutation in the PHD domain disrupts the interaction between NSD2 and its zinc cofactor, which, in turn, disrupts proper protein folding and its subsequent catalytic function. In contrast, P895L mutation in the PWWP domain causes steric clashes within the molecule, whereas K1019R mutation in the AWS domain inhibits the formation of a mobile loop in the catalytic domain that is necessary for methylation function [136].
ASH1L has a role in synaptic plasticity via H3K36me at the promoter of neurexin-1α in response to action potentials [137]. Mutations in ASH1L are clinically associated with intellectual disability [138] as well as early-onset Lewy body dementia [139]. In addition, A2780P point mutation in the BAH domain of ASH1L is associated with autism spectrum disorder [140–143], whereas Y2077F (N-terminal region) and S2200G (SET domain) point mutations are bioinformatically linked to Tourette’s syndrome (TS) [144].
Determinants of H3K36 methylation catalysis
Recent cryo-EM studies on the interactions between H3K36M and Set2, NSD2 or NSD3 have uncovered universal structural determinants of H3K36me catalysis, refining previous knowledge from X-ray diffraction and cryo-EM studies of SETD2 [75, 109, 119, 145]. Indeed, several structural features have been identified as being critical for regulating H3K36me reactions, in addition to positioning of the histone H3 tail within the catalytic pocket of the enzyme and the auto-inhibitory mechanism of human SETD2 and filamentous yeast Chaetomium thermophilum Set2 [75, 145]. Of note, the auto-inhibitory loop has been identified within the dimethylases NSD2 and NSD3 [119, 146]. Furthermore, a hydrophobic surface constructed by the C-termini of histone H2A, the αN helix of histone H3, and the linker DNA of the nucleosome plays an important role in regulating the catalytic activity of Set2/SETD2. Here, we explore each of these determinants in more detail:
Structural motifs in Set2/SETD2 in H3K36me catalysis
Set2/SETD2 is a modular enzyme that hosts several well-conserved domains. There is a 35% similarity in the a.a. sequences of the human SETD2 and both budding and fission yeast Set2, with nearly half of these conserved residues found within the catalytic pre-SET and SET domains (Fig. 1A). Projection of the conserved sequences of the SET domain of yeast Set2 onto human SETD2 SET domain (Fig. 1B) highlights three features: (I) a regulatory LIN loop, (II) a triangular core motif juxtaposing the catalytic site, and the presence of histone-interacting residues (Figs. 1B, 3, 4A and B) [145]. These features pose major impact on catalysis and the regulation of histone methylation [145, 147, 148].
-
(I) The regulatory LIN loop


Conserved histone-interacting residues in the SET domain of SETD2. Asterisk (*) and white arrow in the figure, respectively, label the position of SETD2 β15 beta sheet and α6 helix to depict relative orientation of SETD2 crystalized structure in different sub-figures. A F1589, Y1604, F1606, F1668, and Y1671 (green) interact with G33-V35 (dark red) of the histone H3 peptide (red) via hydrogen bonding. B Key catalytic residues Tyr-1578, Met-1607, Phe-1664 and Tyr-1666 (green) of the SETD2 SET domain (yellow) surround the K36/M36 residues (dark red) of the histone H3 peptide (red). C Key auto-inhibitory residue R1670 (green) of the LIN loop (brown) in SETD2 SET domain (yellow) is in proximity to K36 (dark red) on histone H3 peptide (red). D E1636 and T1637 (green) interact with Y41 (dark red) of the histone H3 peptide (red) via hydrogen bonding. Pymol visualization is derived from the crystalized structure database in the Protein Data Bank entry 5V22 [160].
Various conserved residues are found within key regions of the LIN-loop. This loop connects the SET domain with the post-SET motif and is involved in loading and positioning the H3 histone tail (Fig. 1B) [145]. Arg-1670 (SETD2 numbering) appears to serve as a key auto-inhibitory residue that dictates the opened and closed conformations of the H3K36 access pocket [145]. This residue aligns with budding yeast Arg-240 and fission yeast Arg-300, and is in close proximity to Lys-36 of the histone H3 tail (Figs. 1B, 4C) as well as other key catalytic residues critical for HMTase activity: Met-1607, Phe-1664, and Tyr-1666 in human SETD2 (Met-177, Phe-234 and Phe-236 in budding yeast; Met-237, Phe-294 and Tyr-296 in fission yeast Set2) [74, 145] (Figs. 1B, 4B).
Yang et al. (2016) observed that the LIN loop can adopt three major states—closed, partially open and open—in different stages of the auto-inhibitory model [145]. The ‘closed’ LIN loop assumes a ‘compacted conformation with Arg-1670 inserted into the space within the catalytic center that is usually occupied by Lys-36 when histone H3 is loaded into SETD2. In this way, the LIN loop acts to inhibit the enzymatic activity of SETD2 by occluding the H3K36 substrate-binding site (Fig. 4C). In the ‘partially open’ state, Arg-1670 is displaced outward to prime the catalytic pocket for entry of the histone H3 tail. The LIN loop in the ‘open’ state adopts an extended conformation with Arg-1670 shifted approximately 4.5A away to create sufficient space for loading of the histone H3 N-terminus into the substrate channel of the SETD2 catalytic site (Fig. 4C). This binding of the H3-tail peptide subsequently triggers structural organization of the residues at LIN loop and the post-SET domain into a ‘knot-like’ structure that stabilizes the docked H3-peptide [145].
Similar auto-inhibitory conformational changes were also found in the NSD2, NSD3 and ASH1L dimethylases [27, 109, 119, 149]. The compacted auto-inhibitory loop of NSD2 and NSD3 was observed to ‘open up’ upon binding of the enzymes to the nucleosome, thus making room for the K36M peptide to dock into the catalytic groove [109, 119]. Furthermore, the transition between the ‘open’ and ‘closed’ states is actively regulated via the interaction of transacting factors. Specifically, the chromatin remodeler MORF4-related gene on chromosome 15 (MRG15) binds to a FQLP motif proximal to the AWS-SET domain of ASH1L and, in so doing, displaces the auto-inhibitory loop [149]. In fly, loss-of-function mutations in MRG15 and ASH1 do not appear to impact genome-wide H3K36me2, but specifically reduce H3K36me2 levels at homeotic developmental genes, suggesting that the presence of additional factors can release the auto-inhibition on ASH1 [150].
-
(II) Residues that recognize and stabilize the H3 N-terminus in the catalytic groove of the enzyme
Various structural analyses have helped to uncover the extensive interaction between the histone H3 tail peptides and the H3K36 methyltransferases. Here, we will mainly limit our discussion to Set2/SETD2. Consistent with the view that the AWS and SET domains are functionally sufficient for associating with the histone H3 tail, an extensive array of hydrogen bonding (H-bond)—both direct and water-mediated—and hydrophobic contacts are formed between residues of the AWS-SET-post-SET domains of SETD2 (also NSD2 and NSD3) and the H3K36M peptide [75, 119, 145, 146] (Fig. 4).
SETD2 (probably also Set2, as observed from similarities in the primary sequences) is endowed with differentially charged channels to accommodate the H3 tail, which is enriched with proximal, non-charged and distal basic and bulky residues relative to the Lys-36 residue [145] (Figs. 3, 4A and B). These interactions stabilize and position the K36 residue within a triangular core bounded by three juxtaposing arrays of trans-interacting β-sheets, constituting a pseudoknot-like structure that clamps the H3 tail (S31-K37) within the catalytic site, and positions the Lys-36 residue in proximity of the methyl donor (Figs. 1B, 4A) [145, 151].
Several conserved residues within SETD2 flank the histone tail in this catalytic space, especially Tyr-1579, Met-1607, Phe-1664 and Tyr-1666 with Met-36 of the peptide (where Met-36 is used in place of Lys-36) [145]. These residues potentially stabilize the interaction between the histone tail and the HMTase (Figs. 1B, 4B) by conferring proper positioning of the histone H3 tail and accommodating steric alignment of the K36 residue within the catalytic pocket. Similar residues were also found to embrace H3M36 in NSD2, except for a leucine residue (Leu-1120) in the place of methionine residue within the FMFY motif of SETD2 (FLFY in NSD2). Future mutagenesis studies would help to reveal whether this substitution—that differentiates between a di- and trimethylase—can have an impact on the formation of di- and trimethylation [109]. Upon complex formation, the Pro-38 of the H3 tail kinks out to accommodate Met-36 in C. thermophilum Set2 [75]. Pro-38 thus appears to act as a structural hinge to confer additional control over the catalytic pocket. In line with this view, Pro-38 was pliable to conformation change via cis–trans isomerization in budding yeast [152].
Nucleosomal DNA unwrapping aids in H3K36me catalysis
NSD3 associates with the nucleosome with 12-fold higher affinity when it is wrapped with 187 bp of DNA over 147 bp of DNA, as shown through microscopic thermophoresis-based binding assays [119]. Through cryo-EM analysis, the 20-bp DNA linker termini peel away from the histone octamer surface at the dyad axis—where DNA enters and exits the nucleosome—permitting NSD3 to insert into the space between the unwrapped DNA and the core histones [119]. Similar unwrapping of DNA is also required for Set2 and NSD2 to bind H3K36 at the nucleosomal dyad axis [75, 109]. In studying NSD2, Sato et al. (2021) noted that H3K36 occupied a ‘congested’ environment co-occupied by two gyres of DNA, with unwrapping of the DNA displaces a gyre to facilitate access for NSD2 to H3K36 [109]. Thereafter, upon gaining access, the Arg-117 residue of Set2 stabilizes the displaced DNA via electrostatic interactions, which, in turn, result in rearrangement of the post-SET domain, probably to facilitate the conformation change required for the release of the auto-inhibitory LIN loop [75, 145]. Similar stabilization of the DNA backbone was also observed with several lysine residues in NSD3 (Lys-1074, Lys-1077 and Lys-1080) to sustain full catalytic activity [119].
The series of enzyme–DNA interactions allosterically stack Lys-1234 of NSD3 (and Lys-1152 of NSD2) against histone H3Y41 (Fig. 4D), which is located at the junction between the H3 N-terminal tail and the histone fold domain [153, 154]; this is perhaps to restrict the mobility of the H3 tail for docking into the catalytic site of the enzymes in vivo. Future structural studies using longer H3 tail peptides will be required to verify this possibility within the nucleosomal context. Conversely, the enzyme–DNA binding, in turn, stabilizes enzyme–nucleosome binding, as observed for C. thermophilum Set2 [75, 109].
Taken together, these findings suggest that the binding site on the nucleosome at ground state is occluded by tight winding of DNA against the histone octamers. Upon binding with linker DNA, the HMTases displace the DNA gyres from the octamer surface, possibly in cooperation with chromatin remodelers such as Asf1, which can promote H3K36 trimethylation by Set2 [155]. The enzyme then wedges into the space that become accessible at the dyad axis. This binding between the enzyme and nucleosome then allosterically activates the enzyme by releasing the auto-inhibitory LIN loop, permitting the insertion of H3 tail into the catalytic site in the formation of a stabilized enzyme–DNA-histone ternary complex.
Roles of other histones in H3K36me catalysis
Using immunoblotting to check H3K36me levels in a collection of budding yeast strains hosting mutations in histone residues, Endo and colleagues pinpointed several residues within the histone H2A C-terminus (Gly-107, Ile-112 and Leu-117) and histone H3 αN helix (Thr-45, Arg-49 and Arg-52) that are critical for H3K36me. Subsequent computational modeling led the authors to propose the presence of a structured surface on the nucleosome constituted by these histones in the regulation of H3K36me3 [156]. This hypothesis is confirmed by the cryo-EM study that revealed a hydrophobic patch constituted by Ile-111, Leu-116 and Lys-119 in histone H2A carboxyl domain and Leu-48 and Ile-51 in the αN helix of histone H3, which form hydrophobic and electrostatic interactions with Asp-146, Ile-150, and Ala-153 in the AWS domain of C. thermophilum Set2 [75]. Mutations of these histone residues abolished H3K36me, which was not observed in mutants of residues (H2A Gln-114 and Asn-115) that do not interact with Set2 [157], supporting the functional essentiality of the docking of Set2 onto the hydrophobic patch for mediating the catalysis of H3K36 [119].
Histone H2AK119 and H2BK120, which are situated within the H2A-H3 surface, can be ubiquitinated to modulate the binding of the HMTases via the AWS motif [75, 109, 119]. Artificially tethering ubiquitin to H2BK120 stimulates Set2 activity, without affecting the binding of Set2 on the nucleosome; this suggests that ubiquitin may modulate the orientation of H3K36 within the Set2 catalytic site [75]. Intriguingly, ubiquitination of H2AK119 and H2BK120 inhibited NSD3 activity, unlike that of Set2 [119], lending support to the role of ubiquitination in differentially regulating the degree of methylation on H3K36.
Set2 was also reported to bind histone H4 at Lys-44 [80]. Mutating H4K44 not only interfered with the H4–Set2 interaction, but reduced H3K36me2/3, increased H3K36ac, and enhanced transcription from cryptic promoters in gene bodies. However, these phenotypes were not observed for all H4K44 mutants: whereas K44Q and K44E disrupted H3K36me2/3, H4K44R and H4K44A did not, pointing to a more complicated role of H4. In fact, cryo-EM failed to detect a direct H4–Set2 interaction, and histone H4 is situated furthest from the dyad axis at which the enzyme (NSD3) sits on the nucleosome [119]. However, H4K44 was observed to orientate histone H2A C-terminus within the hydrophobic surface, likely to affect Set2 activity [75].
In summary, H3K36 HMTase Set2 (and also NSD2 and NSD3) interacts with DNA and the histone H2A-H3 surface to release the auto-inhibition and extended region of the H3 tail to position H3K36 for catalysis, which may bear consequences that impact the number of methyl groups that can attach to H3K36.
“Decision-making” motifs and residues in Set2 regulate the degree of methylation
Yeast Set2 catalyzes all forms of H3K36 methylation [12, 36, 158]. There have been multiple theories put forward to explain how Set2 regulates the degree of methylation (mono, di or tri). In fission yeast Set2, the SRI domain and a newly defined “domain of unknown function” (DUF) located proximal to the SRI domain [12] are two of the key motifs implicated in determining the H3K36me3 methylome. Indeed, deletion of the SRI and DUF domains from fission yeast Set2 causes a considerable reduction in total H3K36me3 levels without affecting H3K36me2 [12]. In contrast, loss of the SRI domain in budding yeast diminishes both H3K36me2 and H3K36me3 marks [79]. This discrepancy may be related to the speculative differences in binding abilities between budding yeast Set2 and fission yeast Set2 in their association with RNAPII [159]. An additional C-terminal auto-inhibitory domain located between the SRI and SET domains in both human and budding yeast Set2—but not in fission yeast—may also play a role in global methylation levels. This auto-inhibitory domain controls the DNA binding activity of the SRI domain and fine-tunes the involvement of Set2 in H3K36 methylation [78]: its deletion in budding yeast Set2 leads to a specific loss of H3K36me3, similar to that measured following deletion of the SRI domain in fission yeast Set2 [33].
H3K36me2 to H3K36me3 conversion depends on the SET domain, which encompasses the conserved residues Arg-1625 and Phe-1664 in human SETD2, Arg-315 and Phe-359 in fission yeast Set2, and Arg-195 and Phe-234 in budding yeast Set2. These residues position the H3K36me2-containing histone H3 tail [158]. The methyl group donor, S-adenosyl-L-methionine (SAM), then facilitates the transfer of an additional methyl group to the protein, resulting in trimethylation [57]. Through crystallography analysis, it was suggested that Phe-1664 of SETD2 lies near the histone H3 tail, lending further support to the importance of this residue in regulating the methylation status of H3K36 [160] (Fig. 4A). Consistently, mutation of Arg-1625 in SETD2 (R1625C) in ccRCC attenuates the level of H3K36me3 relative to that found in cells expressing the wild-type SETD2. However, the contribution of this R1625C mutation to the switch in H3K36 methylation is unclear, as the mutation causes a reduction in both SETD2 protein and mRNA levels and leads to a shortened SETD2 protein half-life [57]. The R1625C variant of SETD2 also has reduced substrate-binding capacity and H3K36 trimethylation catalytic activity, which may be linked to a disruption in the hydrogen bonding network arising from possible steric clashes near the SAM binding site [57].
Amino acid substitutions, such as Phe-to-Tyr or Tyr-to-Phe, within the catalytic domains of HMTases have an impact on histone substrate selectivity at different methylation states. Indeed, replacing Phe with Tyr results in steric clashing within the enzymatic site, thereby preventing the transfer of a third methyl group onto the dimethylated lysine residue. Conversely, a Tyr-to-Phe change creates more space, which can encourage trimethylation [161, 162]. A recent study took advantage of this principle, and mutated Phe-234 to Tyr in budding yeast Set2 (Phe-1664 in SETD2). The resultant Set2-F234Y mutant caused a selective reduction in H3K36me3 without significantly affecting the status of H3K36me1/2; conversely, a Tyr-to-Phe mutation at Tyr-149 within the SET domain of Set2 (Y149F) led to diminished H3K36me1/2 without affecting H3K36me3 levels [158].
Closer scrutiny of these conserved SET domains shows the presence of several Phe and Tyr residues, and raises the possibility that fine-contour modulation of the SET domain could influence the contact between catalytic pocket and the histone H3 tail and, in turn, affect the methylation status of H3K36. Several of these residues are only conserved in fission yeast, budding yeast, and fly (e.g., Phe-179, Phe-185, Phe-234, Tyr-244, fission yeast numbering) but not in SETD2 (Fig. 1A), which may suggest a structural basis for the enzymatic promiscuity of these Set2 homologues in catalyzing all three forms of H3K36me. Further structural and mutagenesis studies will be required to confirm these hypotheses.
As mentioned above, screening of the histone mutants that resulted in H3K36me disruption identified that residues of the hydrophobic patch on histone H2A (Gly-107, Ile-112 and Leu-117) and of the αN helix of histone H3 (Thr-45, Arg-49 and Arg-52) specifically disrupt H3K36me3 but not H3K36me2 or H3K36me [156]. It is therefore possible that Ile-150 and Ala-153 of C. thermophilum Set2 [75]—or equivalent residues in Set2/SETD2 from other species—may be involved in regulating the H3K36me2-to-me3 switch.
Interplay between H3K36me and other chromatin modifications in transcriptional regulation
Set2-mediated methylation of H3K36 depends on its physical interaction with RNAPII and its genetic interaction with other transcriptional elongation factors, including the histone chaperone, Spt6 [159]. Recent research has revealed interesting roles for the different types of H3K36me in terms of crosstalk with other chromatin PTMs, as well as with DNA and RNA templates. The interactions between H3K36me marks and the associated reader protein(s) determines the impact of H3K36me on transcription and other cellular functions. MRG15 is a multifunctional chromatin organizer that reads H3K36 marks by binding to H3K36me3 via its chromodomain to affect exon definition and splicing through the recruitment of downstream effectors (Fig. 5) [163]. Comparatively, DNA methyltransferase (DNMT) binds to H3K36me3 via its PWWP domain to control DNA methylation (Fig. 5). Eaf3, on the other hand, interacts with H3K36 via its Tudor, PWWP, and chromodomain to prevent aberrant transcription, and Phf1 and Phf19 in PRC2 interact with H3K36me3 via their Tudor domains to orchestrate H3K27 methylation (Fig. 5) [164, 165]. These interactions purportedly fine-tune the dynamic recruitment of transcription-related factors for different phases of transcription [166–168]. In the next section, we explore the recent insights into how H3K36me crosstalk with DNA methylation, histone PTMs (acetylation and H3K27 methylation), and N6-methyladenosine modifications on the RNA transcript.

Proposed crosstalk between di- and tri-methylated H3K36 and other modifications on histones, DNA, and RNA. Abbreviations: K36me: di- and/or tri-methylated H3K36, K36me2: dimethylated H3K36, K36me3: tri-methylated H3K36, ac: histone acetylation, CD: chromodomain, PWWP: proline-tryptophan-tryptophan-proline motif, m6A: N6-methyladenosine RNA modification, RNAPII: RNA polymerase II, meCpG: methylated cytosine in CpG motif on DNA. Rpd3S-K36me interaction is observed in human, budding yeast and fission yeast; Mst2-Pdp3-K36me interaction is reported solely from fission yeast system thus far, while interactions of DNMTs and MTTL14-K36me are studies from human
Antagonism between H3K36me and H3K27me
Trimethylated Lys-27 on histone H3 (H3K27me3) confers silencing, particularly on developmentally regulated genes. H3K27me3 and H3K36me2/3 are mutually exclusive on the same histone H3 and, consequently, do not co-occur across most of the genome [169–171]. Such H3K27me/H3K36me antagonism plays important role in setting up developmental programs [169, 172]. Indeed, disruption of H3K36 HMTase activities in worm, fly and Neurospora crassa (N. crassa) results in aberrant genome-wide H3K27me3 distribution, leading consequently to developmental defects that arise from transcriptional de-repression of genes that are typically repressed by downstream effectors of H3K27me3 silencing, such as Polycomb group proteins [173–175]. Studies performed in human glioma cells further point to the formation of a boundary by H3K36me2/3 to prohibit the spread of H3K27me3 on chromatin [176]. Consistent with these observations, myeloma cells overexpressing the H3K36 di-methylating MMSET HMTase exhibit hypersensitivity to an inhibitor of the Polycomb repressive complex 2 (PRC2) H3K27 HMTase [177].
In HeLa cells and in the human genome, when histone H3 is unmethylated on H3K36 it tends to be mostly methylated on H3K27; this is except for any newly synthesized histone H3. Furthermore, the enzymatic activity of PRC2 is inhibited on H3K36 pre-methylated nucleosomal substrates [171]. This inhibition requires the presence of H3K36me2/3 (or H3K4me3) in cis on the same or opposite histone tail hosting the H3K27 substrate [169, 171, 178]. H3K36me2/3 does not counteract docking of PRC2 onto histone H3 but, rather, inhibits its catalytic activity [170, 179]. Through mutagenesis analysis, a H3K36me-sensing motif—D630PVQK634—was uncovered on the catalytic EZH2 subunit of PRC2. This motif forms a solvent-accessible pocket for the insertion of an unmodified H3 tail [170]. Using cryo-EM, H3K36 was shown to be strategically situated at a critical site that permits the correct alignment of the histone H3 tail along the EZH2 interface to ensure H3K27 methylation [179].
The antagonism between H3K36 and H3K27 methylation is envisioned to stabilize epigenetic states in vivo, which is essential to target chromatin modifiers for enforcing transcriptional specificity in key developmental genes [172]. In line with this view, sequencing of cancer patient genomes revealed mutually exclusive occurrence of K27M and K36M mutations in histone H3.3 genes in several pediatric cancers, with K27M alteration found in brain tumors (pediatric diffuse pontine glioma and pediatric glioblastoma) [180, 181] and K36M mutation in bone tumors (chondroblastoma, chondrosarcoma and giant cell tumor of bone) [182, 183]. Loss of H3K36me in undifferentiated sarcoma cells is correlated with upregulation of H3K27me preferentially at intergenic regions. These ectopically induced H3K27me is proposed to compete for binding of reader proteins with H3K27me-enriched gene loci, leading to the transcriptional de-repression of these genes [183]. Various mutants of H3.3-Gly-34 were also identified in these pediatric cancers [180, 182]. Study of ectopically expressed G34L and G34W-mutated histone H3.3 proteins in HeLa cells revealed reduced H3K36me3 and increased H3K27me3 in cis, which correlated with augmentation of H3K27me3-interacting PRC1 and PRC2 complexes and obstruction of H3K36me3-reader ZMYND11 binding at the incorporation sites of these Gly-34-mutated histone H3.3. These observations show that H3K27me–H3K36me balance fine-tune localization of effector proteins to enforce epigenetic programs at specific genomic loci [172, 184].
Even though there is a general exclusivity of H3K27me and H3K36me on the genome, a small proportion of nucleosomes was, however, detected via mass spectrometry to contain both H3K36me2 and H3K27me1/2/3. Computational modeling supported the co-occurrence of these usually mutually exclusive marks in the same nucleosome if Lys-27 is first methylated on the histone H3 tail before Lys-36. This prediction is consistent with the reported evidence that H3K36me3 directly inhibits EZH2 methylation of H3K27, whereas similar evidence has not been reported for the reverse scenario [169–172, 178].
Regulation of histone acetylation by H3K36me
H3K36me2 and H3K36me3 tend to show a higher enrichment within gene coding sequences [185–189] with H3K36me2 preferentially enriched closer to the transcriptional start site than H3 K36me3. The level of H3K36me2 appears to be less correlated with transcription than H3K36me3 [190]. Set2-mediated methylation of H3K36 acts at the end of the coding sequence in a negative feedback mechanism to slow RNAPII transcription; this action counteracts histone acetylation, thereby compacting chromatin to limit RNAPII accessibility, via the recruitment of the Rpd3S histone deacetylase (HDAC) complex [191]. The Rpd3S HDAC complex employs two of its subunits—Rco1 and Eaf3—to engage H3K36-methylated nucleosomes [192]. The recruitment of Rpd3S also depends on elongation factor Spt4/5 (yeast equivalence of human DSIF) and association with phosphorylated RNAPII CTD (Fig. 4) [193]. The targeting of HDAC complexes to localities of H3K36me counters histone acetylation, which promotes the formation of a less compacted chromatin conformation besides serving as the binding sites for recruiting transcriptional activators [194, 195]. Deacetylation of chromatin represses transcription of antisense and aberrant non-coding transcripts that arise from cryptic promoters in the wake of the elongating RNAPII, especially in genes with long coding sequences [196, 197, 198].
Eaf3–H3K36me interaction is essential for keeping the Rpd3S complex active (Fig. 5) [193]. Eaf3 binds H3K36me at a shallow histone binding surface cleft comprising a C-terminal α-helix and a β-barrel core, in which the conserved Tyr and Trp residues (Tyr-23, Tyr-81, Trp-84 and Trp-88 for budding yeast Eaf3) confer selective recognition of H3K36me3 [192]. This selectivity is imposed by a conformational switch that occurs when the Rpd3S complex contacts chromatin: this contact leads to allosteric enhancement of the binding strength of the CD on H3K36me2/3 and is thought to constitute one of the strongest chromatin–protein interactions documented to date [32, 35, 192, 199]. H3K36me3 and the yeast Eaf3 homolog , Alp13, also co-localize on heterochromatin in the fission yeast genome during S-phase, when the heterochromatic repeat sequences are preferentially transcribing [200, 201]. Deletion of alp13 or set2 in budding and fission yeast increases antisense transcription in euchromatin and heterochromatin regions of the genome [33, 200, 202–204], indicating a conserved interaction among Eaf3/Alp13, Set2, and H3K36me across different species; albeit, a direct interaction between Alp13 and H3K36me is yet to be structurally determined.
The transcription spike is accompanied by the momentary de-repression of the non-coding repeats at constitutive heterochromatic loci induces the activation of RNA interference during S-phase of the cell cycle, along with high levels of histone acetylation [200]. These events suggest the possibility that HAT, including the Mst2 complex, may be targeted to heterochromatin in normal cell cycling. H3K36me, which functions to coordinate non-coding sequence transcription during S-phase, will temporarily co-localize with the HAT and HDAC effectors, which possess opposing enzymatic activities on histone acetylation [200, 205]. It is possible that H3K36me can maintain a dynamic equilibrium wherein transcriptional activation is fine-tuned in conjunction with chromatin packaging to accommodate the needs of chromatin unraveling during DNA replication and transcription [154, 200]. During S-phase, although heterochromatin becomes less compacted, it is rarely completely disrupted (as in mutants of heterochromatin factors, such as that of H3K9 HMTase). H3K36me may facilitate the maintenance of the partially opened state of heterochromatin during the DNA replicative phase by regulating a balance between silencing and anti-silencing activities in conjunction with other chromatin factors and histone modifications. For instance, crosstalk is likely to be established with H3Y41 phosphorylation (H3Y41p), which functions to differentially modulate chromatin binding of CD-containing proteins. The release of Swi6/HP1 and the recruitment of RITS component Chp1—both CD-containing proteins—by H3Y41p extends the duration in which centromeric heterochromatin becomes accessible to RNAPII to transcribe underlying non-coding repetitive sequences [154, 206].
Apart from recruiting the Rpd3S HDAC complex, H3K36me3 also anchors the Mst2 H3K14 histone acetyltransferase (HAT) complex to euchromatin to prevent HAT from being mistargeted (Fig. 5); mistargeted HAT purportedly antagonizes silenced chromatin at the heterochromatic loci in fission yeast [201]. The budding yeast Sas3-dependent NuA3 complex and fission yeast Mst2 complex (equivalent to human MYST3 complex) is localized to H3K36me3 via its PWWP domain within the Pdp3 subunit and Yng1 subunit, respectively [207, 208]. A mutation at residue Phe-109 of the PWWP domain (F109A) abolishes the interaction between the Mst2 complex and H3K36me3; this allows HAT to be misplaced from the euchromatic gene loci and leads to aberrant activation and transcription of heterochromatic sequences, along with a dissolution of transcriptional silencing [207].
Interactions between H3K36me and DNA methylation
DNA methylation is important for heterochromatin formation not only in mammals, but also in some yeast species [209, 210]; consistently, DNA methylation occurs on 0.3%-1% and 3%-8% of total cytosine (C) residues in yeast and mammalian (of total C) genomes, respectively [211, 212]. DNA methyltransferase 3A (DNMT3A) and DNMT3B in mammals and Pmt1 in fission yeast are capable of catalyzing methylation at the 5’-position of cytosine (C) residues on the CpG nucleotide pair [212, 213].
A specific interaction occurs between H3K36me2 and the N-terminus of the PWWP domain of DNMT3A (Fig. 5): DNMT3A is recruited onto nucleosomal or linker DNA where its enzymatic activity is stimulated [40, 210, 214]. Studies show that mutation of the conserved Lys-295 residue on the DNA-interacting surface of the PWWP domain in DMNT3A disrupts its binding to both DNA and H3K36me2, leading to an aberrant sub-nuclear localization of the DNA methyltransferase [210]. Yet, unlike the recognition of H3K36me2 by DNMT3A, the PWWP domain of DNMT3B preferentially binds to SETD2-induced H3K36me3 to anchor itself onto gene bodies in CpG-rich regions [210, 215]. Indeed, a genome-wide increase in H3K36me2 levels correlates with an increase in nucleosomal 5-methylcytosine levels; this, in turn, is correlated with an overexpression of the NSD2 (Nuclear Receptor Binding SET Domain Protein 2) cancer-driver and an upregulation in genes enriched in oncogenic pathways [24]. However, removal of genomic H3K36me2 via the concurrent knocking down of NSD1 and NSD2 results in a genome-wide reduction in DNA methylation at intergenic loci, along with hypersensitivity to the DNA-hypomethylating agent decitabine. In contrast, Setd2-knockdown cells, which become depleted of H3K36me3, show no decitabine sensitivity, providing evidence for the differential regulation of H3K36me2 and H3K36me3 on DNA methylation [216].
Interaction of H3K36me with RNA N6-methyladenosine (m6A) modification
N6-Methyladenosine (m6A) is an abundant modification imposed on messenger RNA (mRNA), ribosomal RNA (rRNA), and small nuclear RNA (snRNA) of higher eukaryotes [217, 218]. A stable and accurate level of m6A modification ensures proper nuclear RNA export and splicing, mRNA stability, circular RNA translation, microRNA biogenesis, and long non-coding RNA metabolism. This type of methylation is also physiologically implicated in counteracting obesity, immune dysregulation, the generation of meiotic defects and carcinogenesis [219–221].
The METTL14 methyltransferase, responsible for m6A modifications, directly binds H3K36me3 (Fig. 5) and methylates adjacent nascent RNAPII transcripts; in the human transcriptome, this was observed at more than 12,000 loci of consensus RRACH sequences ([G/A][G > A]m6AC[U > A > C]) [222–225]. Most m6A peaks are close to stop codons or 3’ untranslated regions, with the methylation status fluctuating along with meiosis events and cellular stress [226]. m6A marks on mRNA transcripts can be significantly depleted by SETD2 knockout, R1625C mutation in the SET domain, or a disruption in the RNAPII-interacting SRI domain of SETD2 [227]. It remains unknown how METTL14 recognizes H3K36me3 for specific mRNA methylation, as no known H3K36me3-interacting domains have been identified from the METTL3–METTL14 complex crystal [228]. Yeast Set2 is also believed to interact with a defined group of nascent RNAPII transcripts, and this interaction likely affects chromatin occupancy in a manner similar to the related protein, Set1. It has been hypothesized that RNA binding structurally stabilizes Set2 chromatin binding [229].
Conclusions and future questions
The structural and physiological studies reviewed here show that HMTases, including NSD2, NSD3, Set2 and SETD2, possess structural features across multiple domains that confer the catalytic specificity of di- and tri-methylated histone H3K36. These HMTases act in conjunction with various nucleosomal determinants and via crosstalk with other modifications and transacting factors. The selectively of H3K36me2/3 elicits specific physiological outcomes in the chromatin context in conjunction with co-occurring modifications on histones, DNA, and RNA, as well as various transacting factors. However, many questions remain to be answered. For example: (1) What is the sequence of events concerning the interactions of the enzymes with DNA and histone H2A/H3 hydrophobic patches in terms of stimulating the release of the auto-inhibitory loop, and how the latter can further potentiate stronger binding? (2) How is (1) reversed after catalysis is completed? (3) How does the ubiquitination of histone H2A and H2B contribute to the selectivity of di-to-trimethylation? (4) What are the critical residues within the full-length enzyme(s) that influence di-to-trimethylation within the catalytic AWS-SET-post-SET domains and beyond (such as DUF, AID and SRI)? (5) What is the impact of these structural mechanisms on various H3K36me-regulated cellular processes? And, finally, (6) Does the disruption of any of these regulatory pathways underpin the cancer-driving capability of the mutated HMTases in various cancers? Future structural studies and in vivo analyses are expected to provide the answers to these questions.
Availability of data and materials
Data are depicted in the submitted figures. No material was involved in this work.
References
Luger K, Dechassa ML, Tremethick DJ (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13:436–447
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260
Woodcock CL, Skoultchi AI, Fan Y (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 14:17–25
Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A et al (2017) Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol Cell 66:384-397.e8
Healton SE, Pinto HD, Mishra LN, Hamilton GA, Wheat JC, Swist-Rosowska K et al (2020) H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Natl Acad Sci 117:14251–14258
Shahid Z, Simpson B, Singh G (2019) Genetics, histone code. StatPearls Publishing, St. Petersburg
Shilatifard A (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 75:243–269
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Prakash K, Fournier D (2018) Evidence for the implication of the histone code in building the genome structure. BioSystems 164:49–59
O’Hagan HM (2014) Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environ Mol Mutagen 55:278–291
Lim KK, Nguyen TTT, Li AY, Yeo YP, Chen ES (2018) Histone H3 lysine 36 methyltransferase mobilizes NER factors to regulate tolerance against alkylation damage in fission yeast. Nucleic Acids Res 46:5061–5074
Bergmann JH, Rodríguez MG, Martins NMC, Kimura H, Kelly DA, Masumoto H et al (2011) Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J 30:328–340
Alper BJ, Lowe BR, Partridge JF (2012) Centromeric heterochromatin assembly in fission yeast-balancing transcription, RNA interference and chromatin modification. Chromosome Res 20:521–534
Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL et al (2016) Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166:950–962
Pai CC, Kishkevich A, Deegan RS, Keszthelyi A, Folkes L, Kearsey SE et al (2017) Set2 methyltransferase facilitates DNA replication and promotes genotoxic stress responses through MBF-dependent transcription. Cell Rep 20:2693–2705
Dronamraju R, Jha DK, Eser U, Adams AT, Dominguez D, Choudhury R et al (2018) Set2 methyltransferase facilitates cell cycle progression by maintaining transcriptional fidelity. Nucleic Acids Res 46:1331–1344
Dong Y, Zhao X, Feng X, Zhou Y, Yan X, Zhang Y et al (2019) SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia 33:2585–2598
Rayasam GV, Wendling O, Angrand P-O, Mark M, Niederreither K, Song L et al (2003) NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J 22:3153–3163
Brown MA, Sims RJ, Gottlieb PD, Tucker PW (2006) Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer 5:26
Tanaka Y, Katagiri Z, Kawahashi K, Kioussis D, Kitajima S (2007) Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene 397:161–168
Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R et al (2009) Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci 106:21830–21835
Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ et al (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 284:34283–34295
Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J et al (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44:609–620
Yuan W, Xie J, Long C, Erdjument-Bromage H, Ding X, Zheng Y et al (2009) Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem 284:15701–15707
Eom GH, Kim KB, Kim JH, Kim JY, Kim JR, Kee HJ et al (2011) Histone methyltransferase SETD3 regulates muscle differentiation. J Biol Chem 286:34733–34742
Wagner EJ, Carpenter PB (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13:115–126
Sun Z, Zhang Y, Jia J, Fang Y, Tang Y, Wu H et al (2020) H3K36me3, message from chromatin to DNA damage repair. Cell Biosci 10:1–9
Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM (2011) The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem 286:8361–8368
Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB (2010) Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci 107:16952–16957
Gregory GD, Vakoc CR, Rozovskaia T, Zheng X, Patel S, Nakamura T et al (2007) Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol 27:8466–8479
McDaniel SL, Strahl BD (2017) Shaping the cellular landscape with Set2/SETD2 methylation. Cell Mol Life Sci 74:3317–3334
Suzuki S, Kato H, Suzuki Y, Chikashige Y, Hiraoka Y, Kimura H et al (2016) Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res 44:4147–4162
Strahl BD, Grant PA, Briggs SD, Sun Z-W, Bone JR, Caldwell JA et al (2002) Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol 22:1298–1306
Venkatesh S, Workman JL (2013) Set2 mediated H3 lysine 36 methylation: regulation of transcription elongation and implications in organismal development. Wiley Interdiscip Rev Dev Biol 2:685–700
Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD (2005) A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 25:3305–3316
Vojnic E, Simon B, Strahl BD, Sattler M, Cramer P (2006) Structure and carboxyl-terminal domain (CTD) binding of the Set2 SRI domain that couples histone H3 Lys36 methylation to transcription. J Biol Chem 281:13–15
Cramer P (2019) Organization and regulation of gene transcription. Nature 573:45–54
Li C, Diao F, Qiu D, Jiang M, Li X, Han L et al (2018) Histone methyltransferase SETD2 is required for meiotic maturation in mouse oocyte. J Cell Physiol 234:661–668
Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB et al (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51:844–856
Fahey CC, Davis IJ (2017) SETting the stage for cancer development: SETD2 and the consequences of lost methylation. Cold Spring Harb Perspect Med 7:1–16
Kim IK, McCutcheon JN, Rao G, Liu S, Pommier Y, Skrzypski M et al (2019) Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene 38:180–193
Chen R, Zhao W-Q, Fang C, Yang X, Ji M (2020) Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer 11:3349–3356
Hu M, Hu M, Zhang Q, Lai J, Liu X (2020) SETD2, an epigenetic tumor suppressor: a focused review on GI tumor. Front Biosci 25:781–797
González-Rodríguez P, Engskog-Vlachos P, Zhang H, Murgoci AN, Zerdes I, Joseph B (2020) SETD2 mutation in renal clear cell carcinoma suppress autophagy via regulation of ATG12. Cell Death Dis 11:69
Sorenson MR, Jha DK, Ucles SA, Flood DM, Strahl BD, Stevens SW et al (2016) Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae RNA Biol 13:412–426
Yuan H, Li N, Fu D, Ren J, Hui J, Peng J et al (2017) Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Investig 127:3375–3391
Pajoro A, Severing E, Angenent GC, Immink RGH (2017) Histone H3 lysine 36 methylation affects temperature-induced alternative splicing and flowering in plants. Genome Biol 18:1–12
Zhu K, Lei PJ, Ju LG, Wang X, Huang K, Yang B et al (2017) SPOP-containing complex regulates SETD2 stability and H3K36me3-coupled alternative splicing. Nucleic Acids Res 45:92–105
McDaniel SL, Hepperla AJ, Huang J, Dronamraju R, Adams AT, Kulkarni VG et al (2017) H3K36 methylation regulates nutrient stress response in Saccharomyces cerevisiae by enforcing transcriptional fidelity. Cell Rep 19:2371–2382
Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen Y-B, Gonen M et al (2013) Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 19:3259–3267
Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K (2016) SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget 7:50719–50734
Liu L, Guo R, Zhang X, Liang Y, Kong F, Wang J et al (2017) Loss of SETD2, but not H3K36me3, correlates with aggressive clinicopathological features of clear cell renal cell carcinoma patients. Biosci Trends 11:214–220
Lam UTF, Chen ES (2022) Molecular mechanisms in governing genomic stability and tumor suppression by the SETD2 H3K36 methyltransferase. Intl J Biochem Cell Biol. 144:106155
Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, Liu X-Y, Sturm D, Korshunov A et al (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 125:659–669
Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RMW et al (2010) Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res 70:4287–4291
Hacker KE, Fahey CC, Shinsky SA, Chiang YCJ, DiFiore J, Jha DK et al (2016) Structure/function analysis of recurrent mutations in SETD2 protein reveals a critical and conserved role for a SET domain residue in maintaining protein stability and histone H3 Lys-36 trimethylation. J Biol Chem 291:21283–21295
Skucha A, Ebner J, Schmöllerl J, Roth M, Eder T, César-Razquin A et al (2018) MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat Commun 9:1983
Carvalho S, Vítor AC, Sridhara SC, Filipa BM, Ana CR, Desterro JMP et al (2014) SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 2014:e02482
Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sørensen CS et al (2012) LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 19:803–810
Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA (2012) Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 8:e1002717
Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S et al (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21:366–374
House NCM, Koch MR, Freudenreich CH (2014) Chromatin modifications and DNA repair: beyond double-strand breaks. Front Genet 5:296
Jha DK, Pfister SX, Humphrey TC, Strahl BD (2014) SET-ting the stage for DNA repair. Nat Struct Mol Biol 21:655–657
Bleuyard JY, Fournier M, Nakato R, Couturier AM, Katou Y, Ralf C et al (2017) MRG15-mediated tethering of PALB2 to unperturbed chromatin protects active genes from genotoxic stress. Proc Natl Acad Sci 114:7671–7676
Bhattacharya S, Levy MJ, Zhang N, Li H, Florens L, Washburn MP et al (2021) The methyltransferase SETD2 couples transcription and splicing by engaging mRNA processing factors through its SHI domain. Nat Commun 12:1443
Zhao J-X, Li X-W, Shi B-Y, Wang F, Xu Z-R, Meng H-L et al (2017) Effect of histone modifications on hMLH1 alternative splicing in gastric cancer. Tumour Biol 39:1010428317697546
Gonçalves Vânia, Pereira Joana, Jordan Peter (2018) Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes 9(1):9. https://doi.org/10.3390/genes9010009
Bonnal SC, López-Oreja I, Valcárcel J (2020) Roles and mechanisms of alternative splicing in cancer — implications for care. Nature Reviews Clinical Oncology 17(8):457–474. https://doi.org/10.1038/s41571-020-0350-x
Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M et al (2014) Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res 24:241–250
Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G et al (2015) Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 28:557–568
Pai C-C, Hsu K-F, Durley SC, Keszthelyi A, Kearsey SE, Rallis C et al (2019) An essential role for dNTP homeostasis following CDK-induced replication stress. J Cell Sci 132:jcs226969
Wozniak GG, Strahl BD (2014) Hitting the “mark”: interpreting lysine methylation in the context of active transcription. Biochem Biophys Acta 1839:1353–1361
Zheng W, Ibáñez G, Wu H, Blum G, Zeng H, Dong A et al (2012) Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J Am Chem Soc 134:18004–18014
Bilokapic S, Halic M (2019) Nucleosome and ubiquitin position Set2 to methylate H3K36. Nat Commun 10:1–9
Adhvaryu KK, Morris SA, Strahl BD, Selker EU (2005) Methylation of histone H3 lysine 36 is required for normal development in Neurospora crassa Eukaryot Cell 4:1455–1464
Li M, Phatnani HP, Guan Z, Sage H, Greenleaf AL, Zhou P (2005) Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc Natl Acad Sci 102:17636–17641
Wang Y, Niu Y, Li B (2015) Balancing acts of SRI and an auto-inhibitory domain specify Set2 function at transcribed chromatin. Nucleic Acids Res 43:4881–4892
Youdell ML, Kizer KO, Kisseleva-Romanova E, Fuchs SM, Duro E, Strahl BD et al (2008) Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol Cel Biol 28:4915–4926
Du H-N, Fingerman IM, Briggs SD (2008) Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Gene Dev 22:2786–2798
Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME (1998) Huntingtin interacts with a family of WW domain proteins. Human Mol Genet 7:1463–1474
Poon BPK, Mekhail K (2011) Cohesin and related coiled-coil domain-containing complexes physically and functionally connect the dots across the genome. Cell Cycle 10:2669–2682
Sankaran SM, Wilkinson AW, Elias JE, Gozani O (2016) A PWWP domain of histone-lysine N-methyltransferase NSD2 binds to dimethylated Lys-36 of histone H3 and regulates NSD2 function at chromatin. J Biol Chem 291:8465–8474
Huang Z, Wu H, Chuai S, Xu F, Yan F, Englund N et al (2013) NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res 73:6277–6288
Kang H-B, Choi Y, Lee JM, Choi K-C, Kim H-C, Yoo J-Y et al (2009) The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett 583:1880–1886
Aytes A, Giacobbe A, Mitrofanova A, Ruggero K, Cyrta J, Arriaga J et al (2018) NSD2 is a conserved driver of metastatic prostate cancer progression. Nat Commun 9:5201
Stangl-Kremser J, Lemberger U, Hassler MR, Garstka N, Grubmüller B, Haitel A et al (2020) The prognostic impact of tumour NSD2 expression in advanced prostate cancer. Biomarkers 25:268–273
Nakamura T, Blechman J, Tada S, Rozovskaia T, Itoyama T, Bullrich F et al (2000) huASH1 protein, a putative transcription factor encoded by a human homologue of the Drosophila ash1 gene, localizes to both nuclei and cell-cell tight junctions. Proc Natl Acad Sci 97:7284–7289
Rogawski DS, Grembecka J, Cierpicki T (2016) H3K36 methyltransferases as cancer drug targets: rationale and perspectives for inhibitor development. Future Med Chem 8:1589–1607
Zhang S, Zhang F, Chen Q, Wan C, Xiong J, Xu J (2019) CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J Exp Clin Cancer Res 38:467
Yu J-R, Gary L, Devin B, Frenster JD, Ricardo S-M, Ying J et al (2022) The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci Adv 7:eabg7444
Gameiro SF, Ghasemi F, Zeng PYF, Mundi N, Howlett CJ, Plantinga P et al (2021) Low expression of NSD1, NSD2, and NSD3 define a subset of human papillomavirus-positive oral squamous carcinomas with unfavorable prognosis. Infect Agents Cancer 16:13
Chen Y, Tang W, Zhu X, Zhang L, Zhu Y, Xiao H et al (2021) Nuclear receptor binding SET domain protein 1 promotes epithelial-mesenchymal transition in paclitaxel-resistant breast cancer cells via regulating nuclear factor kappa B and F-box and leucine-rich repeat protein 11. Bioengineered 12:11506–11519
Zhang S, Xu J, Cao H, Jiang M, Xiong J (2021) KB-68A7.1 inhibits hepatocellular carcinoma development through binding to NSD1 and suppressing Wnt/β-catenin signalling. Frontiers Oncol. 11:808291.1
Wang GG, Cai L, Pasillas MP, Kamps MP (2007) NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol 9:804–812
Kassambara A, Klein B, Moreaux J (2009) MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res Commun 379:840–845
Gao B, Liu X, Li Z, Zhao L, Pan Y (2021) Overexpression of EZH2/NSD2 histone methyltransferase axis predicts poor prognosis and accelerates tumor progression in triple-negative breast cancer. Front Oncol 10:600514
Zhu L, Yu C-L, Zheng Y (2019) NSD2 inhibition suppresses metastasis in cervical cancer by promoting TGF-β/TGF-βRI/SMADs signaling. Biochem Biophys Res Commun 519:489–496
Sengupta D, Zeng L, Li Y, Hausmann S, Ghosh D, Yuan G et al (2021) NSD2 dimethylation at H3K36 promotes lung adenocarcinoma pathogenesis. Mol Cell 81:4481-4492.e9
Han X, Piao L, Xu X, Luo F, Liu Z, He X (2020) NSD2 promotes renal cancer progression through stimulating Akt/Erk signaling. Cancer Manag Res 12:375–383
Saloura V, Cho H-S, Kiyotani K, Alachkar H, Zuo Z, Nakakido M et al (2015) WHSC1 promotes oncogenesis through regulation of NIMA-related kinase-7 in squamous cell carcinoma of the head and neck. Mol Cancer Res 13:293–304
Brito JLR, Walker B, Jenner M, Dickens NJ, Brown NJM, Ross FM et al (2009) MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 94:78–86
Zhao L, Li Q, Huang Z-J, Sun M-X, Lu J, Zhang X et al (2021) Identification of histone methyltransferase NSD2 as an important oncogenic gene in colorectal cancer. Cell Death Dis 12:974
Song D, Lan J, Chen Y, Liu A, Wu Q, Zhao C et al (2021) NSD2 promotes tumor angiogenesis through methylating and activating STAT3 protein. Oncogene 40:2952–2967
Park JW, Kang J-Y, Hahm JY, Kim HJ, Seo S-B (2020) Proteosomal degradation of NSD2 by BRCA1 promotes leukemia cell differentiation. Commun Biol 3:462
He C, Liu C, Wang L, Sun Y, Jiang Y, Hao Y (2019) Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis 10:65
Lhoumaud P, Badri S, Rodriguez-Hernaez J, Sakellaropoulos T, Sethia G, Kloetgen A et al (2019) NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat Commun 10:4843
Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z et al (2014) Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28:198–201
Sato K, Kumar A, Hamada K, Okada C, Oguni A, Machiyama A et al (2021) Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat Commun 12:6605
Pierro J, Saliba J, Narang S, Sethia G, Saint Fleur-Lominy S, Chowdhury A et al (2020) The NSD2 p.E1099K mutation is enriched at relapse and confers drug resistance in a cell context-dependent manner in pediatric acute lymphoblastic leukemia. Mol Cancer Res 18:1153–1165
Jeong G-Y, Park MK, Choi H-J, An HW, Park Y-U, Choi H-J et al (2021) NSD3-induced methylation of H3K36 activates NOTCH signaling to drive breast tumor initiation and metastatic progression. Cancer Res 81:77–90
Turner-Ivey B, Smith EL, Rutkovsky AC, Spruill LS, Mills JN, Ethier SP (2017) Development of mammary hyperplasia, dysplasia, and invasive ductal carcinoma in transgenic mice expressing the 8p11 amplicon oncogene NSD3. Breast Cancer Res Treat 164:349–358
Yi L, Yi L, Liu Q, Li C (2019) Downregulation of NSD3 (WHSC1L1) inhibits cell proliferation and migration via ERK1/2 deactivation and decreasing CAPG expression in colorectal cancer cells. OncoTargets Ther 12:3933–3943
Sun Y, Xie J, Cai S, Wang Q, Feng Z, Li Y et al (2021) Elevated expression of nuclear receptor-binding SET domain 3 promotes pancreatic cancer cell growth. Cell Death Dis 12:913
Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M et al (2021) Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 590:504–508
Liu Z, Piao L, Zhuang M, Qiu X, Xu X, Zhang D et al (2017) Silencing of histone methyltransferase NSD3 reduces cell viability in osteosarcoma with induction of apoptosis. Oncol Rep 38:2796–2802
Gonzalez-Pecchi V, Kwan AK, Doyle S, Ivanov AA, Du Y, Fu H (2020) NSD3S stabilizes MYC through hindering its interaction with FBXW7. J Mol Cel Biol 12:438–447
Beà S, Valdés-Mas R, Navarro A, Salaverria I, Martín-Garcia D, Jares P et al (2013) Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci 110:18250–18255
Li W, Tian W, Yuan G, Deng P, Sengupta D, Cheng Z et al (2021) Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590:498–503
Balbo Pogliano C, Gatti M, Rüthemann P, Garajovà Z, Penengo L, Naegeli H (2017) ASH1L histone methyltransferase regulates the handoff between damage recognition factors in global-genome nucleotide excision repair. Nat Commun 8:1333
Aljazi MB, Gao Y, Wu Y, Mias GI, He J (2021) Histone H3K36me2-specific methyltransferase ASH1L promotes MLL-AF9-induced leukemogenesis. Front Oncol 11:754093
Zhu L, Li Q, Wong SHK, Huang M, Klein BJ, Shen J et al (2016) ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Dis 6:770–783
Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y et al (2016) Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 48:500–509
Colamaio M, Puca F, Ragozzino E, Gemei M, Decaussin-Petrucci M, Aiello C et al (2015) miR-142–3p down-regulation contributes to thyroid follicular tumorigenesis by targeting ASH1L and MLL1. J Clin Endocrinol Metab 100:E59–E69
Liu L, Kimball S, Liu H, Holowatyj A, Yang Z-Q (2015) Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 6:2466–2482
Choufani S, Cytrynbaum C, Chung BHY, Turinsky AL, Grafodatskaya D, Chen YA et al (2015) NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun 6:10207
Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF et al (1998) WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma Hum. Mol Genet 7:1071–1082
Türkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, Hesse V et al (2003) Mutations in NSD1 are responsible for Sotos syndrome but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 11:858–865
Li Y, Chen X, Lu C (2021) The interplay between DNA and histone methylation: molecular mechanisms and disease implications. EMBO Rep 22:e51803
Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN et al (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573:281–286
Brennan K, Zheng H, Fahrner JA, Shin JH, Gentles AJ, Schaefer B et al (2022) NSD1 mutations deregulate transcription and DNA methylation of bivalent developmental genes in Sotos syndrome. Hum Mol Genet. https://doi.org/10.1093/hmg/ddac026
Itai T, Miyatake S, Hatano T, Hattori N, Ohno A, Aoki Y et al (2021) Cerebrovascular diseases in two patients with entire NSD1 deletion. Hum Genome Var 8:20
Choi S, Song B, Shin H, Won C, Kim T, Yoshida H et al (2021) Drosophila NSD deletion induces developmental anomalies similar to those seen in Sotos syndrome 1 patients. Genes Genomics 43:737–748
Kim T, Shin H, Song B, Won C, Yoshida H, Yamaguchi M et al (2020) Overexpression of H3K36 methyltransferase NSD in glial cells affects brain development in Drosophila. Glia 68:2503–2516
Barrie ES, Alfaro MP, Pfau RB, Goff MJ, McBride KL, Manickam K et al (2019) De novo loss-of-function variants in NSD2 (WHSC1) associate with a subset of Wolf-Hirschhorn syndrome. Cold Spring Harb Mol Case Stud. 5:a004044
Zanoni P, Steindl K, Sengupta D, Joset P, Bahr A, Sticht H et al (2021) Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet Med 23:1474–1483
Zhu Τ, Liang C, Li D, Tian M, Liu S, Gao G et al (2016) Histone methyltransferase Ash1L mediates activity-dependent repression of neurexin-1α. Sci Rep 6:26597
Xi H, Peng Y, Xie W, Pang J, Ma N, Yang S et al (2020) A chromosome 1q22 microdeletion including ASH1L is associated with intellectual disability in a Chinese family. Mol Cytogenet 13:20
Rongve A, Witoelar A, Ruiz A, Athanasiu L, Abdelnour C, Clarimon J et al (2019) GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci Rep 9:7013
Dhaliwal J, Qiao Y, Calli K, Martell S, Race S, Chijiwa C et al (2021) Contribution of multiple inherited variants to autism spectrum disorder (ASD) in a family with 3 affected siblings. Genes 12:1053
Okamoto N, Miya F, Tsunoda T, Kato M, Saitoh S, Yamasaki M et al (2017) Novel MCA/ID syndrome with ASH1L mutation. Am J Med Genet 173:1644–1648
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, de Rubeis S, An J-Y et al (2020) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180:568-584.e23
Shen W, Krautscheid P, Rutz AM, Bayrak-Toydemir P, Dugan SL (2019) De novo loss-of-function variants of ASH1L are associated with an emergent neurodevelopmental disorder. Eur J Med Genet 62:55–60
Liu S, Tian M, He F, Li J, Xie H, Liu W et al (2020) Spontaneous hyperactivity in Ash1l mutant mice, a new model for Tourette syndrome. Mol Psychiatry 25:241–242
Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H (2016) Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 30:1611–1616
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T et al (2013) Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 45:860–867
Schuhmacher MK, Beldar S, Khella MS, Bröhm A, Ludwig J, Tempel W et al (2020) Sequence specificity analysis of the SETD2 protein lysine methyltransferase and discovery of a SETD2 super-substrate. Commun Biol 3:511
Liu Y, Zhang Y, Xue H, Cao M, Bai G, Mu Z et al (2021) Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discovery 7:32
Lee Y, Yoon E, Cho S, Schmähling S, Müller J, Song J-J (2019) Structural basis of MRG15-mediated activation of the ASH1L histone methyltransferase by releasing an autoinhibitory loop. Structure 27:846-852.e3
Schmähling S, Meiler A, Lee Y, Mohammed A, Finkl K, Tauscher K et al (2018) Regulation and function of H3K36 di-methylation by the trithorax-group protein complex AMC. Development 145:dev163808
Dillon SC, Zhang X, Trievel RC, Cheng X (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6:227
Nelson CJ, Santos-Rosa H, Kouzarides T (2006) Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126:905–916
Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR et al (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 461:819–822
Ren B, Tan HL, Nguyen TTT, Sayed AMM, Li Y, Mok Y-K et al (2018) Regulation of transcriptional silencing and chromodomain protein localization at centromeric heterochromatin by histone H3 tyrosine 41 phosphorylation in fission yeast. Nucleic Acids Res 46:189–202
Lin L-J, Minard LV, Johnston GC, Singer RA, Schultz MC (2010) Asf1 can promote trimethylation of H3 K36 by Set2. Mol Cel Biol 30:1116–1129
Endo H, Nakabayashi Y, Kawashima S, Enomoto T, Seki M, Horikoshi M (2012) Nucleosome surface containing nucleosomal DNA entry/exit site regulates H3–K36me3 via association with RNA polymerase II and Set2. Genes Cells 17:65–81
Du H-N, Briggs SD (2010) A nucleosome surface formed by histone H4, H2A, and H3 residues is needed for proper histone H3 Lys36 methylation, histone acetylation, and repression of cryptic transcription. J Biol Chem 285:11704–11713
DiFiore JV, Ptacek TS, Wang Y, Li B, Simon JM, Strahl BD (2020) Unique and shared roles for histone H3K36 methylation states in transcription regulation functions. Cell Rep 31:107751
Fuchs SM, Kizer KO, Braberg H, Krogan NJ, Strahl BD (2012) RNA polymerase II carboxyl-terminal domain phosphorylation regulates protein stability of the set2 methyltransferase and histone H3 di- and trimethylation at lysine 36. J Biol Chem 287:3249–3256
Zhang Y, Shan C-M, Wang J, Bao K, Tong L, Jia S (2017) Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep 7:43906
Cheng X, Collins RE, Zhang X (2005) Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct 34:267–294
Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X et al (2005) In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem 280:5563–5570
Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T (2010) Regulation of alternative splicing by histone modifications. Science 327:996–1000
Sun B, Hong J, Zhang P, Dong X, Shen X, Lin D et al (2008) Molecular basis of the interaction of Saccharomyces cerevisiae Eaf3 chromo domain with methylated H3K36. J Biol Chem 283:36504–36512
Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM et al (2012) Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol 19:1273–1281
Filtz TM, Vogel WK, Leid M (2014) Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol Sci 35:76–85
Benayoun BA, Veitia RA (2009) A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol 19:189–197
Ruthenburg AJ, Li H, Patel DJ, Allis CD (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8:983–994
Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J et al (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42:330–341
Jani KS, Jain SU, Ge EJ, Diehl KL, Lundgren SM, Müller MM et al (2019) Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc Natl Acad Sci 116:8295–8300
Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B (2011) H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 286:7983–7989
Alabert C, Loos C, Voelker-Albert M, Graziano S, Forné I, Reveron-Gomez N et al (2020) Domain model explains propagation dynamics and stability of histone H3K27 and H3K36 methylation landscapes. Cell Rep 30:1223-1234.e8
Bicocca VT, Ormsby T, Adhvaryu KK, Honda S, Selker EU (2018) ASH1-catalyzed H3K36 methylation drives gene repression and marks H3K27me2/3-competent chromatin. Elife 7:e41497
Patel T, Tursun B, Rahe DP, Hobert O (2012) Removal of Polycomb repressive complex 2 makes C. elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep 2:1178–1186
Dorighi KM, Tamkun JW (2013) The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila Development 140:4182–4192
Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, Krug B et al (2020) H3K27M in gliomas causes a one-step decrease in h3k27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep 33:108390
Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T et al (2014) Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 10:e1004566
Voigt P, LeRoy G, Drury WJ 3rd, Zee BM, Son J, Beck DB et al (2012) Asymmetrically modified nucleosomes. Cell 151:181–193
Finogenova K, Bonnet J, Poepsel S, Schäfer IB, Finkl K, Schmid K et al (2020) Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. Elife 9:e61964
Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231
Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA et al (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340:857–861
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, van Loo P et al (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45:1479–1482
Lu Y-W, Zhang H-F, Liang R, Xie Z-R, Luo H-Y, Zeng Y-J et al (2016) Colorectal cancer genetic heterogeneity delineated by multi-region sequencing. PLoS ONE 11:e0152673
Shi L, Shi J, Shi X, Li W, Wen H (2018) Histone H3.3 G34 mutations alter histone H3K36 and H3K27 methylation in cis. J Mol Biol 430:1562–1565
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Tong IL et al (2005) Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122:517–527
Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S et al (2012) Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489:452–455
Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N et al (2005) Single-nucleosome mapping of histone modifications in S. cerevisiae PLoS Biol 3:e328
Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ et al (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181
Rao B, Shibata Y, Strahl BD, Lieb JD (2005) Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol 25:9447–9459
Kim JH, Lee BB, Oh YM, Zhu C, Steinmetz LM, Lee Y et al (2016) Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nat Commun 7:1–12
Ruan C, Lee CH, Cui H, Li S, Li B (2015) Nucleosome contact triggers conformational changes of Rpd3S driving high-affinity H3K36me nucleosome engagement. Cell Rep 10:204–215
Drouin S, Laramée L, Jacques P-É, Forest A, Bergeron M, Robert F (2010) DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet 6:e1001173
Sathianathan A, Ravichandran P, Lippi JM, Cohen L, Messina A, Shaju S et al (2016) The Eaf3/5/7 subcomplex stimulates nua4 interaction with methylated histone H3 Lys-36 and RNA polymerase II. J Biol Chem 291:21195–21207
Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL (2006) Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science 311:844–847
Nakayama J-I, Xiao G, Noma K-I, Malikzay A, Bjerling P, Ekwall K et al (2003) Alp13, an MRG family protein, is a component of fission yeast Clr6 histone deacetylase required for genomic integrity. EMBO J 22:2776–2787
Shevchenko A, Roguev A, Schaft D, Buchanan L, Habermann B, Sakalar C et al (2008) Chromatin central: towards the comparative proteome by accurate mapping of the yeast proteomic environment. Genome Biol 9:R167
Venkatesh S, Li H, Gogol MM, Workman JL (2016) Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat Commun 7:1–14
Huh J-W, Wu J, Lee C-H, Yun M, Gilada D, Brautigam CA et al (2012) Multivalent di-nucleosome recognition enables the Rpd3S histone deacetylase complex to tolerate decreased H3K36 methylation levels. EMBO J 31:3564–3574
Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SIS (2008) Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451:734–737
Georgescu PR, Capella M, Fischer-Burkart S, Braun S (2020) The euchromatic histone mark H3K36me3 preserves heterochromatin through sequestration of an acetyltransferase complex in fission yeast. Microb Cell 7:80–92
Nicolas E, Yamada T, Cam HP, FitzGerald PC, Kobayashi R, Grewal SIS (2007) Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 14:372–380
DeGennaro CM, Alver BH, Marguerat S, Stepanova E, Davis CP, Bahler J et al (2013) Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol Cell Biol 33:4779–4792
Hennig BP, Bendrin K, Zhou Y, Fischer T (2012) Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep 13:997–1003
Cam HP, Chen ES, Grewal SIS (2009) Transcriptional scaffolds for heterochromatin assembly. Cell 136:610–614
Ren B, Chen ES (2019) Regulation of centromeric heterochromatin in the cell cycle by phosphorylation of histone H3 tyrosine 41. Curr Genet 65:829–836
Flury V, Georgescu PR, Iesmantavicius V, Shimada Y, Kuzdere T, Braun S et al (2017) The histone acetyltransferase Mst2 protects active chromatin from epigenetic silencing by acetylating the ubiquitin ligase Brl1. Mol Cell 67:294-307.e9
Martin BJE, McBurney KL, Maltby VE, Jensen KN, Brind’Amour J, Howe LJ (2017) Histone H3K4 and H3K36 methylation independently recruit the NuA3 histone acetyltransferase in Saccharomyces cerevisiae Genetics 205:1113–1123
Rountree MR, Selker EU (2010) DNA methylation and the formation of heterochromatin in Neurospora crassa Heredity 105:38–44
Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P et al (2019) H3K36me2/3 binding and DNA binding of the DNA methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J Mol Biol 431:5063–5074
Hattman S, Kenny C, Berger L, Pratt K (1978) Comparative study of DNA methylation in three unicellular eucaryotes. J Bacteriol 135:1156–1157
Tang Y, Gao X-D, Wang Y, Yuan B-F, Feng Y-Q (2012) Widespread existence of cytosine methylation in yeast DNA measured by gas chromatography/mass spectrometry. Anal Chem 84:7249–7255
Wilkinson CR, Bartlett R, Nurse P, Bird AP (1995) The fission yeast gene pmt1+ encodes a DNA methyltransferase homologue. Nucleic Acids Res 23:203–210
Rondelet G, Dal Maso T, Willems L, Wouters J (2016) Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J Struct Biol 194:357–367
Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR et al (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520:243–247
Rajagopalan KN, Chen X, Weinberg DN, Chen H, Majewski J, Allis CD et al (2021) Depletion of H3K36me2 recapitulates epigenomic and phenotypic changes induced by the H3.3K36M oncohistone mutation. Proc Natl Acad Sci 118:e2021795118
Harper JE, Miceli SM, Roberts RJ, Manley JL (1990) Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res 18:5735–5741
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F (1994) Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem 269:17697–17704
Shafik A, Schumann U, Evers M, Sibbritt T, Preiss T (2016) The emerging epitranscriptomics of long noncoding RNAs. Biochem Biophys Acta 1859:59–70
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y et al (2017) Extensive translation of circular RNAs driven by N 6-methyladenosine. Cell Res 27:626–641
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W et al (2017) Roles of RNA methylation by means of N6-methyladenosine (m6A) in human cancers. Cancer Lett 408:112–120
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G et al (2019) The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol 12:121
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L et al (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10:93–95
Lee M, Kim B, Kim VN (2014) Emerging roles of RNA modification: M6A and U-Tail. Cell 158:980–987
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M et al (2019) Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567:414–419
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z et al (2016) Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 534:575–578
Sayou C, Millán-Zambrano G, Santos-Rosa H, Petfalski E, Robson S, Houseley J et al (2017) RNA Binding by histone methyltransferases Set1 and Set2. Mol CellBiol. 37:e00165
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Acknowledgements
We thank Rebecca Jackson, Meng Jie Ho, and Kim Kiat Lim for editing a draft of this manuscript.
Funding
This work was supported by the Singapore Ministry of Education Academic Research Funds MOE2018-T2-1-100 and MOE2016-T2-2-063, and National University Health System fund NUHSRO/2021/062/NUSMed/04/LOA.
Author information
Authors and Affiliations
Contributions
ESC, UTFL, and JJXP wrote the manuscript; ESC secured the funding; BKYT and UTFL performed the computer modeling. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to the publication.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lam, U.T.F., Tan, B.K.Y., Poh, J.J.X. et al. Structural and functional specificity of H3K36 methylation. Epigenetics & Chromatin 15, 17 (2022). https://doi.org/10.1186/s13072-022-00446-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13072-022-00446-7