
Abstract
Background
Anopheles melas is an understudied malaria vector with a potential role in malaria transmission on the Bijagós Archipelago of Guinea-Bissau. This study presents the first whole-genome sequencing and population genetic analysis for this species from the Bijagós. To our knowledge, this also represents the largest population genetic analysis using WGS data from non-pooled An. melas mosquitoes.
Methods
WGS was conducted for 30 individual An. melas collected during the peak malaria transmission season in 2019 from six different islands on the Bijagós Archipelago. Bioinformatics tools were used to investigate the population structure and prevalence of insecticide resistance markers in this mosquito population.
Results
Insecticide resistance mutations associated with pyrethroid resistance in Anopheles gambiae s.s. from the Bijagós were absent in the An. melas population, and no signatures of selective sweeps were identified in insecticide resistance-associated genes. Analysis of structural variants identified a large duplication encompassing the cytochrome-P450 gene cyp9k1. Phylogenetic analysis using publicly available mitochondrial genomes indicated that An. melas from the Bijagós split into two phylogenetic groups because of differentiation on the mitochondrial genome attributed to the cytochrome C oxidase subunits COX I and COX II and the NADH dehydrogenase subunits 1, 4, 4L and 5.
Conclusions
This study identified an absence of insecticide-resistant SNPs common to An. gambiae in the An. melas population, but did identify structural variation over insecticide resistance-associated genes. Furthermore, this study presents novel insights into the population structure of this malaria vector using WGS analysis. Additional studies are required to further understand the role of this vector in malaria transmission.
Graphical abstract

Similar content being viewed by others

Background
Malaria is a persistent public health problem in Guinea-Bissau, West Africa, where the population of 2.1 million people experienced an estimated 225,200 malaria cases and 1023 malaria deaths in 2022 [1]. The Bijagós Archipelago (Bijagós) is a group of 88 islands and islets located approximately 50 km off the coast of Guinea-Bissau and is a designated UNESCO Biosphere Reserve [2]. The Archipelago is home to approximately 25,000 people, who live on 19 permanently inhabited islands [3]. Malaria transmission on the Bijagós is highly seasonal and stable [4], and Plasmodium falciparum prevalence on the Archipelago can peak at up to 15% at the end of the rainy season in November [5].
A survey in 2017 on Bubaque Island, the most populous island of the Archipelago, identified all Anopheles species on Bubaque to be members of the Anopheles gambiae sensu lato (s.l.) complex. Of the species present, Anopheles gambiae sensu stricto (s.s.) was identified as the primary malaria vector and Anopheles melas as having a role in low level transmission during the dry season [4]. Anopheles melas is a saltwater-tolerant species able to sustain population numbers during the dry season by laying eggs in brackish water, giving this species a particular advantage during the dry season when freshwater oviposition sites have dried up [6]. The Bijagós Archipelago has an abundance of mangroves and mud flats [2], which are commonly associated with the presence of An. melas [7, 8]. A larger survey conducted on 16 of the inhabited islands of the Archipelago was conducted between October and December 2019 during the peak malaria transmission season. This survey used indoor and outdoor light-traps and identified 85.2% of trapped mosquitoes to be An. melas (Pretorius et al. 2024, in review). A sub-sample of mosquitoes were investigated for Plasmodium falciparum sporozoite positivity using circumsporozoite protein (CSP) ELISA, which calculated a sporozoite rate of 0.86% and identified all CSP-positive specimens collected as An. melas (Pretorius et al. 2024, in review). This study indicates that An. melas may be important in malaria transmission on the Bijagós, particularly regarding residual transmission during the dry season (Pretorius et al. 2024, in review). This is supported by previous studies which have identified An. melas to have a role in malaria transmission, including in Senegal [9], The Gambia [10, 11] and Equatorial Guinea [12].
Vector control on the Bijagós relies on the use of insecticide-treated nets (ITNs) impregnated with pyrethroid insecticides, which are distributed every 3 years and have high estimated coverage and usage of around 90% [13, 14]. Pyrethroid ITNs are the most successful vector control intervention developed to date, having prevented approximately 68% of malaria deaths in Africa between 2000 and 2015 [15]. Alarmingly, resistance to pyrethroids is highly prevalent worldwide [16]. Of all countries that reported resistance data to WHO between 2010 and 2020, 87% declared pyrethroid resistance in at least one malaria vector [1]. Resistance to pyrethroids has been associated with single-nucleotide polymorphisms (SNPs) in the voltage-gated sodium channel gene (vgsc) of An. gambiae, including L995F and L995S, also known as the kdr west and kdr east alleles [17, 18], and the N1570Y mutation [19]. In addition, target site mutations in the gste2 gene have been associated with pyrethroid resistance, including L119V and I114T [20]. Resistance to pyrethroids has been associated with copy number variants (CNVs) encompassing genes in three major enzyme families: cytochrome-P450s, esterases and glutathione-S-transferases (GSTs) [21,22,23,24].
Insecticide resistance is a growing threat to the control of malaria. This threat has propelled the understanding of Anopheles genetic variation through international collaborations such as the Anopheles Gambiae 1000 Genomes Project [25]. Genomics research has focused on the key malaria vectors An. gambiae s.s. and Anopheles coluzzii, but few studies have investigated An. melas. On the Bijagós, a previous study on Bubaque Island investigated the presence of the kdr east and west alleles in An. gambiae s.l. mosquitoes using targeted PCR sequencing [4], and a subsequent study across 13 islands investigated the prevalence of known insecticide resistance mutations using high-throughput multiplex-amplicon sequencing [26]. This study identified four mutations associated with insecticide resistance in An. melas at low prevalence. This included three mutations in the vgsc gene, L995F, N1570Y and A1746S, one mutation in the rdl gene, A296G, and no known insecticide resistance mutations in the ace1 or gste2 genes [26]. However, no previous studies using whole-genome sequence (WGS) data from An. melas on the Bijagós Archipelago have been conducted, and population structure and signatures of selection have not previously been investigated. Furthermore, to our knowledge, only two studies analysing WGS data from An. melas have previously been published. This includes a study of Pool-seq WGS data from An. melas in The Gambia, Cameroon and Equatorial Guinea, where the DNA from several individual mosquitoes was pooled prior to sequencing [27] and the Anopheles 16 Genomes Project, which produced the reference genome assembly for An. melas [28]. The previous study using Pool-seq WGS data identified three genetically distinct clusters of An. melas on the West African coast, An. melas West, An. melas South and An. melas Bioko, which ranged respectively from The Gambia to Northwest Cameroon, Southeast Cameroon to Angola and Bioko Island in Equatorial Guinea [27, 29]. Genetic differentiation between these clusters was high and mostly distributed evenly across the genome, with elevated differentiation along the X chromosome, indicative of allopatric divergence [27].
Here, we generate and analyse WGS data from 30 individual mosquitoes of this little-known species, collected during the 2019 vector survey on the Bijagós Archipelago, combined with WGS data from seven An. melas specimens made available through the Anopheles 16 Genomes Project [28]. WGS data are a valuable resource which we utilise to investigate the genetic diversity of this mosquito population, the presence of SNPs associated with insecticide resistance and genomic signatures of selection in this species.
Methods
Mosquito sampling
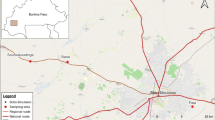
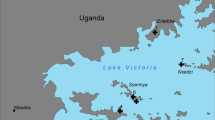
Mosquitoes were collected from the Bijagós Archipelago during October and November 2019 using CDC indoor and outdoor light traps (Model 512; John W. Hock Co., Gainesville, FL, USA) using previously described methodology [30]. This included mosquitoes from six different islands across the Archipelago highlighted with purple triangles in Fig. 1: Soga, Bubaque, Tchedega (Maio), Uno, Caravela and Uracane. Collected mosquitoes were separated by genus, and Anopheles mosquitoes were morphologically identified using previously described keys [31]. All mosquitoes were identified as belonging to the An. gambiae s.l. complex.

Mosquito sample collection sites. A Location of Bijagós Archipelago, created using ArcGIS ArcMap 10.8.1. B Mosquitoes were collected from the six islands labelled with purple triangles. Map from OpenStreetMap 2023‑05‑06
DNA extraction
DNA extraction was conducted following the manufacturer’s instructions, using the QIAamp® 96 DNA QIAcube® HT Kit (Qiagen) with the QIAcube Extraction Robot. DNA was eluted in 80 µl AE buffer and stored at – 20 ℃. No additional processing of DNA, for example whole-genome amplification or selective genome amplification, was conducted prior to sequencing.
Species identification
Female mosquitoes were then identified to species at the Medical Research Council Unit The Gambia at London School of Hygiene & Tropical Medicine using PCR-RFLP to distinguish between members of the An. gambiae s.l. complex, based on the protocol of Fanello et al. [32]. This used primers to amplify the intergenic spacer (IGS) region, which differs in size between members of the An. gambiae complex. The following primers were used: Universal F: GTGTGCCCCTTCCTCGATGT; Anopheles gambiae R: CTGGTTTGGTCGGCACGTTT; An. arabiensis R: AAGTGTCCTTCTCCATCCTA; An. melas R: TGACCAACCCACTCCCTTGA. Amplified PCR products were then digested using the HhaI enzyme to differentiate An. gambiae s.s. and An. coluzzii specimens. The band sizes of the PCR products were visualised using electrophoresis with the QIAxcel capillary electrophoresis system (Qiagen). The band sizes of PCR product were analysed to distinguish species: An. gambiae s.s. (257 and 110 bp), An. arabiensis (292 bp), An. melas (435 bp), An. coluzzii (367 bp) and An. coluzzii/An. gambiae s.s. hybrid (257 bp, 110 bp and 367 bp).
Whole-genome sequencing and bioinformatic analysis
The DNA of 30 An. melas mosquitoes from six different islands across the archipelago was whole-genome sequenced at Eurofins Genomics using the Illumina Novaseq 6000 (2 × 150 bp configuration). This included 11 An. melas from Soga, 7 from Bubaque, 7 from Tchedega (Maio), 3 from Uno, 1 from Caravela and 1 from Uracane. Average read depth for all samples across the whole genome was 32.61. Over 55% of the genome for each sample had a read depth ≥ 20. Publicly available An. melas data were included for population analyses. This included one sample from The Gambia and six samples from Cameroon, made available through the Anopheles 16 Genomes Project [28, 33]. Mosquitoes were phenotypically identified as female during sampling, and this was called again using the modal coverage ratio between chromosome 3R and chromosome X (with a ratio between 0.4 and 0.6 = male and ratio between 0.8 and 1.2 = female, with other ratios leading to sample exclusion, as previously described [34]).
Raw WGS data were trimmed using trimmomatic (version 0.39) [35] before aligning to the Anopheles gambiae (AgamP4) reference genome and the Anopheles melas (AmelC2) reference genome, using bwa-mem software (default parameters)[36]. The AgamP4 alignment was taken forward as the AgamP4 reference genome is of better quality than the An. melas reference genome, and the percentage of reads which mapped to AgamP4 was higher (average 92.8%) than the percentage of reads which mapped to AmelC2 (average 79.3%). Furthermore, the AmelC2 reference genome is not a chromosomal level assembly, consisting of over 22,000 scaffolds with a mean N50 value of 18,103, compared to a AgamP4, which is a chromosomal level assembly with mean N50 value of 49,364,325. Therefore, mapping of An. melas WGS data to the AgamP4 reference genome also allowed interpretation of genetic variation in the context of chromosome location. Finally, mapping of An. melas data to the An. gambiae reference genome has been conducted in previously published studies with amplicon [26] and WGS data [27] and is an accepted method. Mapping and coverage statistics from the resulting bam files were calculated using samtools [37]. Variants were called for each sample using GATK’s HaplotypeCaller (v 4.1.4.1) to generate a VCF for each sample [38]. A combined, genotyped VCF was created for the An. melas samples from the Bijagós, The Gambia and Cameroon using GATK’s GenomicsDBImport and GenotypeGVCFs function [38]. The multi-sample VCF was filtered using bcftools (v 1.17) and GATK’s VariantFiltration to include chromosomal variants with the following parameters: QD > 5.0, QUAL > 30.0, SOR < 3.0, FS < 60.0, MQ > 40.0, MQRankSum > − 12.5, ReadPosRankSum > -8.0. Reads were subsequently filtered to remove reads with DP < 5.0 or GQ < 20.0, and variants were filtered to remove those with > 20.0% missing genotypes or MAF < 0.01. The final filtered VCF contained 6,767,012 variants.
Population genetic analysis
A distance matrix was generated using PLINK (v 1.90b6.21), and principal component analyses were computed in R (v 4.3.1). A maximum likelihood tree was made using RAxML-NG (v 1.2.0) with the mitochondrial FASTA sequences for the An. melas samples and 43 An. gambiae samples from the Bijagós Archipelago [26]. The resulting maximum likelihood tree was visualised using iTOL [39]. Admixture analysis was computed using ADMIXTURE (v 1.3.0) [40]. The estimated number of ancestral populations (optimum K-value) was computed through cross-validation of 1–10 dimensions of eigenvalue decay (k = 4).
Nucleotide diversity (π) was computed for the N = 30 Bijagós An. melas mosquitoes in 20,000-bp windows on chromosome 3 L using phased filtered variants and the scikit-allel function allel.windowed_diversity. Tajima’s D was calculated using the function allel.windowed_tajima_d. Analysis was conducted with chromosome 3 L only because of the presence of large chromosomal inversions in the other chromosomes in the An. gambiae s.l. complex [41].
Genetic divergence between An. melas from the Bijagós and Cameroon was investigated using the fixation index, FST. The windowed_weir_cockerham_fst function in scikit-allel was used to compute FST in 1-kbp windows over each chromosome (https://scikit-allel.readthedocs.io/en/stable/). Signatures of selection were investigated using three different complementary statistics: H12 [42], iHS and XP-EHH [43, 44]. Garud’s H12 was computed using the moving_garud_h function in scikit-allel, using phased biallelic SNPs in windows of 1000 SNPs. Two hundred iterations of H12 were calculated, and the mean value for each window was plotted. iHS was computed using phased biallelic SNPs using the allel.ihs function in scikit-allel (https://scikit-allel.readthedocs.io/en/stable/). Raw iHS scores were standardized using the allel.standardize_by_allele_count function, and p-values were computed and plotted. XP-EHH was calculated using phased biallelic SNPs using the allel.xpehh function in scikit-allel (https://scikit-allel.readthedocs.io/en/stable/). XP-EHH scores were standardised using the allel.standardize_by_allele_count function and plotted.
Identification of SNPs associated with insecticide resistance
The filtered VCF was analysed to identify SNPs in four genes previously associated with resistance: vgsc, rdl, gste2 and ace1. Identified variants were annotated using SnpEff (v 5.1d). The vgsc G2042C non-synonymous (NS) SNP was identified in 100% of allele calls in An. melas from all locations. This mutation has not been reported in this study as a novel NS SNP with possible association with resistance, as the identified presence in 100% of An. melas may have resulted from alignment to the AgamP4 genome, reflecting a species-specific mutation between our An. melas samples and the reference An. gambiae.
Identification of structural variants
DELLY software was used to identify large structural variants (SVs) [45]. Individual bcf files were created for each sample from their bam files using DELLY [45], which were then merged and filtered to remove samples with average genome read depth of < 20 × and SVs with > 20% missingness. Filtered SVs were retained for analysis.
Results
Whole-genome sequence data and genetic diversity
WGS data from 30 An. melas from the Bijagós Archipelago were combined with an additional seven An. melas mosquitoes with publicly available WGS data, which were downloaded and incorporated for analysis. This included An. melas from Cameroon (N = 6) and The Gambia (N = 1). The combined dataset (N = 37) contained 6,767,012 high-quality SNPs after filtering. Average sequencing depth across the core genome ranged from 23.1 to 55.61-fold coverage, with an average of 32.61-fold coverage across all samples. The mitochondrial genome was sequenced to very high depth, averaging 2600-fold coverage and ranging from a minimum of 420- up to 5930-fold coverage.
Nucleotide diversity (π) and Tajima’s D were computed in 20,000-bp windows across chromosome 3L. This resulted in mean π = 0.003 (SD = 0.001), indicating that for any pair of mosquitoes, 0.3% of nucleotides would differ, implying low nucleotide diversity in the population. Mean Tajima’s D for chromosome 3L was calculated as D = − 1.531 (SD = 0.710), indicating an excess of rare alleles. Tajima’s D was much higher at the start of the chromosome, with mean D = 1.518 (SD = 1.334) for the first 50 windows (1Mbp) of the chromosome, suggesting balancing selection in this region (Supplementary Data 1).
Population genetic and ancestry analysis
A maximum likelihood tree was generated using mitochondrial sequences from the Bijagós, combined with publicly available mitochondrial sequences from additional Anopheles mosquitoes from across Africa in the Anopheles gambiae s.l. complex: An. gambiae s.s., An. melas, An. merus, An. arabiensis and An. coluzzii (Fig. 2). As expected, An. melas from the Bijagós group with the other An. melas specimens from The Gambia and Cameroon.

Maximum likelihood tree of whole mitochondrial sequences: Anopheles melas and An. gambiae from the Bijagós Archipelago with other species from the An. gambiae sensu lato species complex
A second maximum likelihood tree was computed using whole mitochondrial sequences of An. melas (N = 37) and previously published An. gambiae samples from the Bijagós (N = 43) ([26], in review) (Fig. 3). The An. melas samples from the Bijagós group with the An. melas individual from The Gambia, and are situated next to the An. melas samples from Cameroon. No clustering of An. melas from different islands on the Bijagós was identified in the maximum likelihood tree. However, the Bijagós An. melas appear to form two distinct clusters, labelled A and B (Fig. 3). These clusters do not correspond to different islands of the Bijagós Archipelago, with mosquito specimens from several islands appearing in each cluster.

Maximum likelihood tree using Anopheles melas mitochondrial sequences from the Bijagós Archipelago, The Gambia and Cameroon and An. gambiae mitochondrial sequences from the Bijagós Archipelago. An. melas from the Bijagós split into two groups, labelled A and B. Support values can be seen in Supplementary Data 1
Principal component analysis (PCA) was conducted per chromosome to further investigate the relationship between the Bijagós An. melas mosquitoes (Fig. 4). For all chromosomes, PCA indicated that An. melas from the Bijagós is genetically distinct from An. melas samples from Cameroon and The Gambia. Comparisons with the Gambia should be treated with caution as this analysis includes only one sample. This geographic separation depicted by PC1 explains a large amount of variation for all chromosomes, ranging from 28.7% in chromosome 2 L to 59.8% in the mitochondrial genome. The clustering of An. melas from the Bijagós into two phylogenetic groups (A and B in Fig. 3) is supported by the PCA analysis for the mitochondrial genome. However, PCA analysis of the chromosomes 2 L, 2R and 3 L shows An. melas from the Bijagós clustering into one group, and chromosome X and 3R indicate some divergence but not clearly between groups A and B. Furthermore, little variance was explained by PC2 for the X and 3R chromosomes compared to the mitochondrial genome, 1.8% for chromosome X and 5.5% for chromosome 3R, compared with 15.7% for the mitochondrial genome (Fig. 4).

Principal component analysis for An. melas from the Bijagós Archipelago, Guinea-Bissau (N = 30), Cameroon (N = 6) and The Gambia (N = 1)
The clustering of Bijagós An. melas into groups A and B was further investigated using fixation index (FST) analysis across 100-bp and 1000-bp windows. The highest FST values were on the mitochondrial genome, with a mean FST value of 0.476, compared to a mean FST value between 0.038 and 0.040 on the other chromosomes (Table 1). This indicates that most of the population differentiation is due to genetic differentiation between An. melas groups A and B in regions on the mitochondrial genome.
Genes underlying peaks in FST were identified. There were seven genomic windows with an FST ≥ 0.5. These windows of the genome include the protein-coding genes detailed in Table 2. This includes genes encoding the cytochrome C oxidase subunits COX I and COX II and the NADH dehydrogenase subunits 1, 4, 4L and 5.
Additional PCA was conducted to investigate the relationship between the genomes of the An. melas specimens from the Bijagós, Cameroon and The Gambia and An. gambiae s.s. mosquitoes from the Bijagós to investigate the possibility of hybridisation between An. melas and An. gambiae s.s. (Fig. 5). This mitochondrial PCA indicates that An. melas from the Bijagós separates from Anopheles gambiae s.s. from the Bijagós, and this relationship is also reflected in the PCA analyses of all other individual chromosomes (Supplementary Data 1). This gives no indication of hybridization between the An. melas and An. gambiae s.s. from the Archipelago.

Principal components analysis comparing Anopheles melas and An. gambiae s.s. from the Bijagós Archipelago. Includes additional An. melas samples from Cameroon and The Gambia
Admixture analysis was conducted with WGS data from the combined sample set of N = 37 An. melas available globally (Fig. 6). This admixture analysis indicated an optimum of K = 4 ancestral groups. Anopheles melas from the Bijagós and The Gambia had a mixture of three different K ancestries (K = 1, 2 and 4), whilst An. melas from Cameroon had a clearly separate ancestry (K = 3).

Admixture based on geographic location, K = 4. N = 1 The Gambia, N = 6 Cameroon, N = 30 Bijagós [N = 11 from Soga, 7 from Bubaque, 7 from Tchedega (Maio), 3 from Uno, 1 from Caravela and 1 from Uracane]
Population differentiation between An. melas from the Bijagós (N = 30) and An. melas from Cameroon (N = 6) was investigated using the fixation index (FST) statistic (Table 3).
Fifty-nine protein coding genes were identified as overlapping windows with FST ≥ 0.9 on chromosome 2L, 72 on chromosome 2R, 51 on chromosome 3L, 56 on chromosome 3R and 56 on chromosome X (Supplementary Data 1). No genomic windows with FST ≥ 0.9 were identified on the mitochondrial genome. Consequently, windows with FST ≥ 0.6 were investigated, and 15 protein coding genes were identified (Supplementary Data 1). This included cytochrome C oxidase subunits II (cox2) and III (cox3) and NADH dehydrogenase subunits ND2 (nadh2), ND3 (nadh3), ND4 (nadh4), ND4L (nadh4l) and ND5 (nadh5).
Selection analysis identified signals of selection within An. melas populations
Genome-wide selection scans of filtered variants were performed to identify signals of directional selection within and between populations of An. melas. Three different selection metrics were calculated: integrated haplotype score (iHS) was used to identify regions of the genome under selection within the An. melas Bijagós population. The cross-population haplotype homozygosity metric (XP-EHH) was used to identify regions of the genome under selection when comparing the Bijagós and Cameroon An. melas populations. Finally, Garud’s H12 was computed as an additional method to identify both hard and soft selective sweeps within the Bijagós An. melas population. In a single population analysis of An. melas from the Bijagós using the iHS metric [44], 194 loci were identified as having significant iHS scores (iHS ≥ 4) (Fig. 7). This included 102 SNPs in 29 different protein coding genes: 3 in chromosome 2L, 13 in chromosome 2R, 7 in chromosome 3L, 4 in chromosome 3R and 2 in chromosome X (Table 4). None of the protein coding genes identified with significant iHS scores have previously been implicated in insecticide resistance.

Within population selection analysis using iHS scores for Anopheles melas from the Bijagós Archipelago
Cross-population analysis using XP-EHH was conducted between An. melas from the Bijagós and Cameroon (Fig. 8). More positive scores indicate positive selection in the Bijagós An. melas population (significant at XP-EHH ≥ 5), whereas more negative scores indicate positive selection in the Cameroon An. melas population (significant at XP-EHH ≤ − 5). Protein coding genes containing SNPs under positive selection in the Bijagós population (Table 5) and the Cameroon population (Table 6) are detailed.

Cross population selection analysis using XP-EHH metric between Anopheles melas from the Bijagós and Cameroon
Garud’s H12 was used to identify signatures of recent positive selection in the Bijagós An. melas population [42]. H12 was computed per chromosome, and no clear peaks of selection were identified on any chromosome (Supplementary Data 1).
Detection of structural variants
Structural variants (SVs) were identified in the Bijagós An. melas mosquito population using DELLY software [45] and were discovered in relation to the AgamP4 (An. gambiae) reference genome. A total of 113,121 SVs were identified across the whole genome following quality control filtering. Of these 113,121 filtered SVs, 116 were identified in genes associated with insecticide resistance or in gene families associated with insecticide metabolism. This included 48 deletions, 38 inversions, 22 insertions and 8 duplications.
SVs were annotated using SnpEff [46]. Of the 48 identified deletions, four were annotated as having high impact. This included one deletion (40 bp) in chromosome 2R, which was found at 3% allelic frequency and resulted in a frameshift in cyp6p15p and the up- or downstream modification of cyp6aa2, coeae60, cyp6p3 and cyp6p5. Two high-impact deletions on chromosome 2 L (766,776 bp) and 3R (714,267 bp) resulted in feature ablation of multiple genes and were found in all An. melas samples from the Bijagós and Cameroon, indicating that these deletions may be species specific to An. melas and could have been identified as a result of aligning to the AgamP4 reference genome. The fourth high-impact deletion was identified in chromosome X (505,172 bp) at 47.9% allelic frequency in the Bijagós population and 28.6% in the Cameroon population, which resulted in the deletion of multiple genes including AGAP000817, AGAP000816 and AGAP013474 (protein coding genes with unspecified products). Two deletions were annotated as having moderate impact. The first was found at 1.7% allelic frequency in the cyp6m4 gene on chromosome 3R (575 bp). The second was on chromosome 2R (38 bp) in AGAP013202 and was identified in all An. melas samples from the Bijagós and Cameroon so may be An. melas specific.
Of the 22 insertions identified, 5 were in introns, 8 were in intergenic regions, 4 were downstream gene variants, and 5 were upstream gene variants. None of these insertions were identified as having high impact, and 14 were identified in all of the Bijagós An. melas isolates, indicating that they may be species specific insertions.
Of the eight identified duplications, five were annotated as having high impact. Two of these duplications were identified on chromosome 2R at 2% allelic frequency. The first leads to bidirectional gene fusion between cyp6p4 and the solute carrier family 8 sodium/calcium exchanger (AGAP002859), and the second leads to bidirectional gene fusion between AGAP002876 (encoding a DNA glycosylase) and AGAP002877 (encoding a tetratricopeptide repeat protein). Two other high-impact duplications were identified on chromosome 3R. The first was found at 2% allelic frequency and leads to gene fusion between cyp6m2 and cyp6m3, whereas the second occurred at 4% allelic frequency and results in a premature stop codon in gste1. Finally, a large duplication on chromosome X between genomic positions 14,939,828 and 15,316,733 (376,905 bp) was identified to have high impact. All Bijagós An. melas and six of the seven Cameroon An. melas were heterozygous for this duplication, which spans multiple protein coding genes including the cytochrome P450 cyp9k1. In addition, one moderate impact duplication was identified at 1.7% allelic frequency in cyp9k1 on chromosome X.
Of the 38 identified inversions, 8 were annotated as having high impact. This included three different inversions in the vgsc gene on chromosome 2L found at 10%, 4% and 2% allelic frequency, respectively, two different inversions in the AGAP009189 gene on chromosome 3R found at 2% allelic frequency, one inversion in gste8 on chromosome 3R found at 4% allelic frequency and one inversion in the zf-C3Hc3H domain-containing protein (AGAP029190) gene in chromosome 2R found at 6% frequency. One large, high-impact, inversion in chromosome 2L spanned from genomic positions 2,161,433 to 2,666,161 and led to bidirectional gene fusion between AGAP004717 and AGAP029667 (protein coding genes with unspecified products). All An. melas Bijagós and Cameroon mosquitoes were homozygous alternate for this inversion, indicating that this could be a species-specific inversion in An. melas identified through alignment to the AgamP4 reference genome. One additional moderate impact inversion was identified on chromosome 3L at 15% allelic frequency, impacting the DNA-directed RNA polymerase III subunit RPC1 (AGAP004703), compass component SPP1 (AGAP004704), arginyl-tRNA synthetase (AGAP004708) and five protein coding genes with unspecified products.
Identification of non-synonymous SNPs in resistance genes
We investigated the presence of target site mutations in the vgsc, gste2, rdl and ace1 genes, which have previously been associated with insecticide resistance (Supplementary Data 1). In total, we identified 28 non-synonymous mutations in these resistance genes (Table 7). Of these, the only mutation that has been reported previously is vgsc M490I, which we identified at an allelic frequency of 2% and is highlighted with a † symbol in Table 7. This mutation was previously identified in An. gambiae samples from Kenya, where it was found to potentially be under selection [47]. To our knowledge, none of the other missense mutations identified here have previously been reported in the An. gambiae s.l. complex. The vgsc G2046S mutation was fixed in the Bijagós An. melas population (100% allelic frequency) and was identified in one An. melas sample from The Gambia (100% allelic frequency) but was not found in any An. melas samples from Cameroon (0% allelic frequency).
Discussion
Anopheles melas is highly abundant on the Bijagós Archipelago of Guinea-Bissau and may have a role in malaria transmission [4] (Pretorius et al. 2024, in review). However, the population structure and insecticide resistance status of this malaria vector are not well understood. This study used WGS data from 30 An. melas from across the Archipelago to investigate genetic diversity, population structure and signatures of selection in insecticide resistance genes within this vector population.
Maximum likelihood trees generated using the whole mitochondrial genome showed that An. melas from the Bijagós split into two groups. Mosquito samples were collected from six different islands in the Archipelago, but this split was not associated with sampling island, and analyses indicated that the clustering of An. melas into two groups was due to genetic differentiation on the mitochondrial genome. The protein coding genes underlying the peaks in FST on the mitochondrial genome included the cytochrome C oxidase subunits cox1 and cox2 and the NADH dehydrogenase subunits nadh1, nadh4, nadh4L and nadh5. Both cox1 and nadh4 have previously been used to investigate phylogenetic relationships within multiple vector complexes and between cryptic species, including the An. gambiae s.l. complex [48,49,50]. Further investigation with a larger sample size of An. gambiae s.l. complex mosquitoes from the Bijagós Archipelago will help to understand the clusters observed.
The computed maximum likelihood trees indicate that An. melas from the Bijagós are closely related to An. melas from The Gambia and Cameroon. The level of genetic differentiation between An. melas from the Bijagós and Cameroon was higher than previously identified for other species of An. gambiae. Average FST across chromosomes 3L and 3R between the Bijagós and Cameroon An. melas was 0.27. However, FST across these chromosomes was previously identified at a tenfold lower score of 0.028 between mainland Guinea-Bissau and Cameroon An. gambiae [25]. Higher than expected levels of genetic differentiation have previously been identified between An. melas populations using whole-genome data. A previous study by Deitz et al. found that divergence between large An. melas clusters along the West African coast were due to high levels of differentiation across the whole genome, indicative of allopatric separation [27]. Nucleotide diversity was similar to that previously identified for An. melas from Bioko Island, Equatorial Guinea (π = 0.0034) [27], and was lower than the average nucleotide diversity calculated for An. gambiae sampled from 15 locations across Africa (π = 0.015) [25]. Ancestry analysis using a combined database of these An. melas samples indicated K = 4 ancestral populations, with An. melas samples from the Bijagós and The Gambia sharing ancestries K = 1, 2 and 4 and Cameroon An. melas samples sharing a distinct K = 3 ancestry. There was no clear distinction between the ancestries of An. melas from different islands on the Archipelago, indicating historical gene flow between the islands, despite the geographical distance between islands being greater than the distance An. gambiae s.l. are known to disperse [51]. This is supported by a previous study, which identified extensive gene flow between Anopheles s.s. on the Bijagós Archipelago and mainland Guinea-Bissau [52]. Fixation index analysis identified 59 protein coding genes with high genetic differentiation between An. melas from the Bijagós and Cameroon, including the cyp307a1 gene (AGAP001309) on chromosome X, which is a member of the cytochrome P450 gene family associated with metabolic resistance to insecticides [53, 54]. This analysis was conducted with a large sample size disparity, with 30 samples from the Bijagós vs. 6 samples from Cameroon. Additional WGS data are required to further investigate this genetic differentiation.
Genome-wide selection scans were computed to identify signatures of selection across the genome. Within-population analysis of Bijagós An. melas using the iHS statistic identified signatures of directional selection in 29 protein coding genes, none of which have previously been associated with insecticide resistance. Cross-population analysis between An. melas from the Bijagós and Cameroon using the XP-EHH metric identified 15 protein coding genes within the Bijagós population undergoing positive selection compared to the Cameroon population. This included the gene encoding tep6, a thioester-containing protein in the same family as tep1, which is implicated in An. gambiae resistance to parasite infection [55]. This analysis also identified 11 protein coding genes undergoing positive selection in the Cameroon population compared to the Bijagós population, none of which have previously been associated with insecticide resistance. Genome-wide selection scans using the H12 statistic did not identify any clear selective sweeps in the Bijagós An. melas genome, in contrast to our previous study of An. gambiae s.s. collected during 2022 from Bubaque Island, where H12 analysis identified two distinct selective sweeps on chromosomes X and 2R spanning multiple cytochrome-P450 genes involved in insecticide metabolism ([26], in review). The absence of selective sweeps in insecticide resistance-associated genes in the An. melas genome suggests that this species may be under less selective pressure from insecticides than An. gambiae on these islands.
Structural variants (SVs) were analysed in the Bijagós An. melas population in relation to the An. gambiae AgamP4 reference genome. One deletion identified in chromosome 2R at 3% allelic frequency resulted in a frameshift in cyp6p15p and modification of cyp6aa2, coeae60, cyp6p3 and cyp6p5. These cyp6p genes are associated with metabolic insecticide resistance in mosquitoes [53, 54, 56, 57]. Another variant resulted in the duplication of multiple protein coding genes including the cytochrome P450 cyp9k1, which metabolises deltamethrin and has been associated with pyrethroid resistance in An. coluzzii populations following vector control interventions [21, 22]. Furthermore, cyp9k1 duplications have previously been identified in An. gambiae s.l. complex mosquitoes from mainland Guinea-Bissau ([23], Supplementary S7) [23]. Copy number variants resulting in gene duplication are under positive selection in the An. gambiae s.l. complex [23], and CNVs leading to increased expression of metabolic genes have been shown to increase insecticide metabolism, leading to insecticide resistance [56, 58,59,60]. Eight high-impact inversions were identified, including three inversions in the vgsc gene associated with resistance to DDT and pyrethroids [17, 18, 56, 61], and one inversion in the gste8 gene, which is in the same gene family as gste2, which encodes a DDT-detoxifying enzyme [62].
Analysis of non-synonymous SNPs in insecticide resistance genes identified the vgsc M490I mutation in the Bijagós An. melas population at low frequency, which has previously been reported in An. gambiae in Kenya as under possible directional selection [47]. No other SNPs previously associated with insecticide resistance were found. In our previous study of amplicon data from An. melas from the Archipelago collected in the same year, three SNPs previously associated with insecticide resistance were identified. These were vgsc L995F (2.14% allelic frequency), N1570Y (1.12% allelic frequency) and A1746S (0.76% allelic frequency) [26]. However, these SNPs were identified at very low frequency and found at significantly lower frequency in An. melas than in An. gambiae s.s. Therefore, the absence of these SNPs in our dataset is not unexpected.
The absence of insecticide resistance-associated SNPs in An. melas further suggests that this species is under less insecticide resistance selection pressure than An. gambiae on the Bijagós Archipelago. This is supported by a previous study in Equatorial Guinea, where no insecticide resistance mutations were identified in An. melas [12]. This may be because An. melas is biting people outdoors, circumventing exposure to the insecticides in ITNs, or because An. melas may be feeding mostly on non-human hosts [6]. In an entomological survey on the Bijagós in 2019, a greater proportion of Plasmodium-positive An. melas were caught in the outdoor than indoor traps. However, Plasmodium-positive An. melas were also caught in indoor traps, indicating human-host seeking indoors and outdoors (Pretorius et al. 2024, in review). As ITNs are the only vector control method used in the Bijagós, outdoor biting would reduce selection pressure for resistance evolution in this species. Whilst maintaining susceptibility to insecticides is positive, preferential outdoor biting by An. melas may present further issues for vector control on the islands as conventional ITNs and IRS may not be as effective. This is supported by studies in Equatorial Guinea which identified high levels of outdoor biting by An. melas [63]. Alternatively, the absence of insecticide resistance-associated mutations and selective sweeps in An. melas found in this study may be because this species is evolving separate molecular mechanisms of resistance to An. gambiae s.s. Notably, Anopheles mosquitoes are among the most genetically diverse eukaryotic organisms known [25], and it is plausible that different species may evolve unique molecular pathways for resistance. Further investigation using additional sampling, phenotypic bioassays, synergist-insecticide bioassays and ‘omics studies should be undertaken to understand the resistance status and molecular mechanisms of resistance in this understudied malaria vector.
The main limitations of this study are the absence of phenotypic insecticide resistance data for An. melas and that the current An. melas reference genome necessitated aligning our An. melas WGS data to the An. gambiae (AgamP4) reference genome. The AgamP4 reference genome is of higher quality, and 92.8% of An. melas reads mapped successfully to AgamP4 compared to 79.3% mapping to the poorer quality AmelC2 reference genome, giving us confidence in our approach. However, future analyses would benefit from a chromosome-level reference genome assembly for An. melas, particularly as all structural variants were discovered in relation to the AgamP4 reference. Furthermore, the genes discussed in this study have been annotated from the An. gambiae reference genome, and though these genes are likely to have very similar functions in An. melas, they could play different roles.
Conclusions
In conclusion, using WGS data, this study identifies two separate phylogenetic clusters of An. melas on the Bijagós Archipelago of Guinea-Bissau because of genetic differentiation on the mitochondrial genome. Structural variants encompassing genes that could be involved in metabolic insecticide resistance were identified. However, common SNPs associated with insecticide resistance in An. gambiae s.s. were absent in the An. melas population, and there were no clear signatures of selection in known insecticide-resistance genes. This suggests that An. melas may experience less selective pressure for insecticide resistance evolution than An. gambiae, potentially through biting outdoors and circumventing selection pressure from ITNs or because they are feeding primarily on non-human hosts. Further investigations using larger data sets and phenotypic bioassays are required.
Availability of data and materials
The raw sequence data generated and analysed during this study are available in the European Nucleotide Archive (project ID: PRJEB75927, accession numbers ERS20101387 to ERS20101416).
References
World Health Organization. World Malaria Report 2023. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023. Accessed 15 Jan 2024.
UNESCO. Boloma Bijagós Biosphere Reserve, Guinea-Bissau. https://www.unesco.org/en/mab/bolama-bijagos. Accessed 28 Mar 2024
National Institute of Statistics, Guinea-Bissau. Guinea-Bissau 2009 Census. https://guinebissau.opendataforafrica.org/amthtjd/guinea-bissau-2009-census. Accessed 28 Mar 2024
Ant T, Foley E, Tytheridge S, Johnston C, Goncalves A, Ceesay S, et al. A survey of Anopheles species composition and insecticide resistance on the island of Bubaque, Bijagós Archipelago, Guinea-Bissau. Malar J. 2020;19:1–9.
Hutchins H, Bradley J, Pretorius E, Teixeira da Silva E, Vasileva H, Jones RT, et al. Protocol for a cluster randomised placebo-controlled trial of adjunctive ivermectin mass drug administration for malaria control on the Bijagós Archipelago of Guinea-Bissau: the MATAMAL trial. BMJ Open. 2023;13:e072347.
Sinka ME, Bangs MJ, Manguin S, Coetzee M, Mbogo CM, Hemingway J, et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: occurrence data, distribution maps and bionomic précis. Parasit Vectors. 2010;3:117.
Thomson RCM. Studies on the breeding places and control of Anopheles gambiae and A. gambiae var. melas in Coastal Districts of Sierra Leone. Bull Entom Res. 1946;36:185–252.
Giglioli ME. Oviposition by Anopheles melas and its effect on egg survival during the dry season in the Gambia. West Africa Ann Entomol Soc Am. 1965;58:885–91.
Sy O, Sarr PC, Assogba BS, Nourdine MA, Ndiaye A, Konaté L, et al. Residual malaria transmission and the role of Anopheles arabiensis and Anopheles melas in central Senegal. J Med Entomol. 2023;60:546–53.
Caputo B, Nwakanma D, Jawara M, Adiamoh M, Dia I, Konate L, et al. Anopheles gambiae complex along The Gambia river, with particular reference to the molecular forms of An. gambiae s.s. Malar J. 2008;7:182.
Bryan JH, Petrarca V, Di Deco MA, Coluzzi M. Adult behaviour of members of the Anopheles gambiae complex in the Gambia with special reference to An. melas and its chromosomal variants. Parassitologia. 1987;29:221–49.
Overgaard HJ, Reddy VP, Abaga S, Matias A, Reddy MR, Kulkarni V, et al. Malaria transmission after five years of vector control on Bioko Island, Equatorial Guinea. Parasit Vectors. 2012;5:253.
McGregor D, da Silva ET, Grignard L, Goncalves A, Vasileva H, Mabey D, et al. The epidemiology of Plasmodium falciparum malaria in the Bijagós islands of Guinea-Bissau. Am J Trop Med and Hyg. 2021;104:2117–22.
Hutchins H, Power G, Ant T, Teixeira E, Goncalves A, Rodrigues A, et al. A survey of knowledge, attitudes and practices regarding malaria and bed nets on Bubaque Island, Guinea—Bissau. Malar J. 2020;19:1–15.
Bhatt S, Weiss DJ, Cameron E, Bisanzio D, Mappin B, Dalrymple U, et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature. 2015;526:207.
World Health Organization. Malaria Threat Map. https://apps.who.int/malaria/maps/threats/. Accessed 1 Mar 2024.
Ranson H, Jensen B, Vulule JM, Wang X, Hemingway J, Collins FH. Identification of a point mutation in the voltage-gated sodium channel gene of Kenyan Anopheles gambiae associated with resistance to DDT and pyrethroids. Insect Mol Biol. 2000;9:491–7.
Martinez-Torres D, Chandre F, Williamson MS, Darriet F, Bergé JB, Devonshire AL, et al. Molecular characterization of pyrethroid knockdown resistance (kdr) in the major malaria vector Anopheles gambiae s.s. Insect Mol Biol. 1998;7:179–84.
Jones CM, Liyanapathirana M, Agossa FR, Weetman D, Ranson H, Donnelly MJ, et al. Footprints of positive selection associated with a mutation (N1575Y) in the voltage-gated sodium channel of Anopheles gambiae. Proc Natl Acad Sci U S A. 2012;109:6614–9.
Lucas ER, Rockett KA, Lynd A, Essandoh J, Grisales N, Kemei B, et al. A high throughput multi-locus insecticide resistance marker panel for tracking resistance emergence and spread in Anopheles gambiae. Sci Rep. 2019;9:13335.
Vontas J, Grigoraki L, Morgan J, Tsakireli D, Fuseini G, Segura L, et al. Rapid selection of a pyrethroid metabolic enzyme CYP9K1 by operational malaria control activities. PNAS. 2018;115:4619–24.
Hearn J, Djoko Tagne CS, Ibrahim SS, Tene-Fossog B, Mugenzi LMJ, Irving H, et al. Multi-omics analysis identifies a CYP9K1 haplotype conferring pyrethroid resistance in the malaria vector Anopheles funestus in East Africa. Mol Ecol. 2022;31:3642–57.
Lucas ER, Miles A, Harding NJ, Clarkson CS, Lawniczak MKN, Kwiatkowski DP, et al. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 2019;29:1250–61.
Ibrahim SS, Amvongo-Adjia N, Wondji MJ, Irving H, Riveron JM, Wondji CS. Pyrethroid resistance in the major malaria vector Anopheles funestus is exacerbated by overexpression and overactivity of the P450 CYP6AA1 across Africa. Genes. 2018;9:140.
Anopheles gambiae 1000 Genomes Consortium. Genetic diversity of the African malaria vector Anopheles gambiae. Nature. 2017;552:96–100.
Moss S, Pretorius E, Ceesay S, Hutchins H, da Silva ET, Ndiath MO, et al. Genomic surveillance of Anopheles mosquitoes on the Bijagós Archipelago using custom targeted amplicon sequencing identifies mutations associated with insecticide resistance. Parasit Vectors. 2024;17:10.
Deitz KC, Athrey GA, Jawara M, Overgaard HJ, Matias A, Slotman MA. Genome-wide divergence in the West-African malaria vector Anopheles melas. G3. 2016;6:2867–79.
Neafsey DE, Waterhouse RM, Abai MR, Aganezov SS, Alekseyev MA, Allen JE, et al. Mosquito genomics. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science. 2015;347:1258522.
Deitz KC, Athrey G, Reddy MR, Overgaard HJ, Matias A, Jawara M, et al. Genetic isolation within the malaria mosquito Anopheles melas. Mol Ecol. 2012;21:4498–513.
Mboera LE, Kihonda J, Braks MA, Knols BG. Short report: Influence of Centers for Disease Control light trap position, relative to a human-baited bed net, on catches of Anopheles gambiae and Culex quinquefasciatus in Tanzania. Am J Trop Med Hyg. 1998;59:595–6.
Coetzee M. Key to the females of Afrotropical Anopheles mosquitoes (Diptera: Culicidae). Malar J. 2020;19:70.
Fanello C, Santolamazza F, Della TA. Simultaneous identification of species and molecular forms of the Anopheles gambiae complex by PCR-RFLP. Med Vet Entomol. 2002;16:461–4.
Neafsey DE, Christophides GK, Collins FH, Emrich SJ, Fontaine MC, Gelbart W, et al. The evolution of the Anopheles 16 genomes project. G3. 2013;3:1191–4.
Lucas ER, Nagi SC, Egyir-Yawson A, Essandoh J, Dadzie S, Chabi J, et al. Genome-wide association studies reveal novel loci associated with pyrethroid and organophosphate resistance in Anopheles gambiae and Anopheles coluzzii. Nat Commun. 2023;14:4946.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv. 2013;16:1303.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021. https://doi.org/10.1093/gigascience/giab008.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–8.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Anopheles gambiae 1000 Genomes Consortium. Genome variation and population structure among 1142 mosquitoes of the African malaria vector species Anopheles gambiae and Anopheles coluzzii. Genome Res. 2020;30:1533–46.
Garud NR, Rosenberg NA. Enhancing the mathematical properties of new haplotype homozygosity statistics for the detection of selective sweeps. Theor Popul Biol. 2015;102:94–101.
Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007;5:e171.
Gautier M, Klassmann A, Vitalis R. rehh 2.0: a reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol Ecol Resour. 2017;17:78–90.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Clarkson CS, Miles A, Harding NJ, O’Reilly AO, Weetman D, Kwiatkowski D, et al. The genetic architecture of target-site resistance to pyrethroid insecticides in the African malaria vectors Anopheles gambiae and Anopheles coluzzii. Mol Eco. 2021;30:5303–17.
Rubinoff D, Holland BS. Between two extremes: mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst Biol. 2005;54:952–61.
Campos M, Phelan J, Spadar A, Collins E, Gonçalves A, Pelloquin B, et al. High-throughput barcoding method for the genetic surveillance of insecticide resistance and species identification in Anopheles gambiae complex malaria vectors. Sci Rep. 2022;12:13893.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Kaufmann C, Briegel H. Flight performance of the malaria vectors Anopheles gambiae and Anopheles atroparvus. J Vector Ecol. 2004;29:140–53.
Marsden CD, Cornel A, Lee Y, Sanford MR, Norris LC, Goodell PB, et al. An analysis of two island groups as potential sites for trials of transgenic mosquitoes for malaria control. Evol Appl. 2013;6:706–20.
Scott JG. Cytochromes P450 and insecticide resistance. Insect Biochem Mol Biol. 1999;29:757–77.
Vontas J, Katsavou E, Mavridis K. Cytochrome P450-based metabolic insecticide resistance in Anopheles and Aedes mosquito vectors: muddying the waters. Pestic Biochem Physiol. 2020;170:104666.
Hamid-Adiamoh M, Jabang AMJ, Opondo KO, Ndiath MO, Assogba BS, Amambua-Ngwa A. Distribution of Anopheles gambiae thioester-containing protein 1 alleles along malaria transmission gradients in The Gambia. Malar J. 2023;22:89.
Edi CV, Djogbénou L, Jenkins AM, Regna K, Muskavitch MAT, Poupardin R, et al. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS Genet. 2014;10:e1004236.
Müller P, Warr E, Stevenson BJ, Pignatelli PM, Morgan JC, Steven A, et al. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS Genet. 2008;4:e1000286.
Grigoraki L, Lagnel J, Kioulos I, Kampouraki A, Morou E, Labbé P, et al. Transcriptome profiling and genetic study reveal amplified carboxylesterase genes implicated in temephos resistance, in the Asian tiger mosquito Aedes albopictus. PLoS Negl Trop Dis. 2015;9:e0003771.
Schmidt JM, Good RT, Appleton B, Sherrard J, Raymant GC, Bogwitz MR, et al. Copy number variation and transposable elements feature in recent, ongoing adaptation at the Cyp6g1 locus. PLoS Gen. 2010;6:e1000998.
Itokawa K, Komagata O, Kasai S, Masada M, Tomita T. Cis-acting mutation and duplication: history of molecular evolution in a P450 haplotype responsible for insecticide resistance in Culex quinquefasciatus. Insect Biochem Mol Biol. 2011;41:503–12.
Williams J, Cowlishaw R, Sanou A, Ranson H, Grigoraki L. In vivo functional validation of the V402L voltage gated sodium channel mutation in the malaria vector Anopheles gambiae. Pest Manag Sci. 2022;8:1155–63.
Mitchell SN, Rigden DJ, Dowd AJ, Lu F, Wilding CS, Weetman D, et al. Metabolic and target-site mechanisms combine to confer strong DDT resistance in Anopheles gambiae. PLoS ONE. 2014;9:e92662.
Reddy MR, Overgaard HJ, Abaga S, Reddy VP, Caccone A, Kiszewski AE, et al. Outdoor host seeking behaviour of Anopheles gambiae mosquitoes following initiation of malaria vector control on Bioko Island, Equatorial Guinea. Malar J. 2011;10:184.
Acknowledgements
We acknowledge the communities of the Bijagós Archipelago, where these mosquito samples were collected, and all who took part or assisted in the cross-sectional survey.
Funding
SM and EC are funded by Medical Research Council UK (grant no. MR/N013638/1). TGC, SC, HAP and MH are funded by Medical Research Council UK grants (ref. no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, MR/R020973/1 and MR/X005895/1). AL, EP, ETS, SC, RTJ and HV are funded by Joint Global Health Trials Scheme (MRC, Wellcome Trust, UKRI, NIHR, grant no. MR/S005013/1).
Author information
Authors and Affiliations
Contributions
EP coordinated mosquito collection on the Bijagós Archipelago during a cross‑sectional survey with HH, ES and RTJ. The cross‑sectional survey was designed by AL, EP and RTJ. SCe conducted DNA extraction and species identification at the Medical Research Council, The Gambia (MRCG), under the supervision of MON. SM designed this study and conducted all bioinformatic analysis of sequence data under the supervision of TGC and SCa, with guidance from HAF, EC, MH and JP. HV assisted with sample logistics and transport. SM wrote the first draft of this manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Moss, S., Pretorius, E., Ceesay, S. et al. Whole genome sequence analysis of population structure and insecticide resistance markers in Anopheles melas from the Bijagós Archipelago, Guinea-Bissau. Parasites Vectors 17, 396 (2024). https://doi.org/10.1186/s13071-024-06476-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06476-2