
Abstract
Background
Ixodes ricinus is an important vector of several pathogens, primarily in Europe. Recently, Ixodes inopinatus was described from Spain, Portugal, and North Africa and then reported from several European countries. In this study, a multiplex polymerase chain reaction (PCR) was developed to distinguish I. ricinus from I. inopinatus and used in the surveillance of I. inopinatus in Algeria (ALG) and three regions in the Czech Republic (CZ).
Methods
A multiplex PCR on TROSPA and sequencing of several mitochondrial (16S rDNA, COI) and nuclear markers (TROSPA, ITS2, calreticulin) were used to differentiate these two species and for a subsequent phylogenetic analysis.
Results
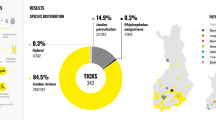
Sequencing of TROSPA, COI, and ITS2 separated these two species into two subclades, while 16S rDNA and calreticulin could not distinguish I. ricinus from I. inopinatus. Interestingly, 23 nucleotide positions in the TROSPA gene had consistently double peaks in a subset of ticks from CZ. Cloning of these PCR products led to a clear separation of I. ricinus and I. inopinatus indicating hybridization and introgression between these two tick taxa. Based on a multiplex PCR of TROSPA and analysis of sequences of TROSPA, COI, and ITS2, the majority of ticks in CZ were I. ricinus, no I. inopinatus ticks were found, and 10 specimens showed signs of hybridization. In contrast, most ticks in ALG were I. inopinatus, four ticks were I. ricinus, and no signs of hybridization and introgression were detected.
Conclusions
We developed a multiplex PCR method based on the TROSPA gene to differentiate I. ricinus and I. inopinatus. We demonstrate the lack of evidence for the presence of I. inopinatus in Central Europe and propose that previous studies be re-examined. Mitochondrial markers are not suitable for distinguishing I. inopinatus from I. ricinus. Furthermore, our data indicate that I. inopinatus and I. ricinus can hybridize, and the hybrids can survive in Europe.
Graphical abstract

Similar content being viewed by others


Background
Ixodes ricinus (Acari, Ixodidae) is an important vector of many pathogens of medical and veterinary importance with a wide host and distribution range across the entire West Palearctic region, from British Islands to Russian Ural and from North Africa to Scandinavia [1].
Despite the complexity of the genetic structure of I. ricinus populations, two patterns have been consistently shown, regardless of the approach used: the genetic distance of ticks on the British Islands compared with that of populations in North Africa [2,3,4] and the genetic dissimilarity between the North African tick population and that on continental Europe [5, 6]. In 2014, the latter pattern resulted in a description of the new species Ixodes inopinatus, based on the morphological characteristics and the partial sequence of the 16S rRNA gene of ticks from Spain (type locality), Portugal, and North Africa (Algeria, Tunisia, Morocco) [7].
Since then, ticks referred to as I. inopinatus have been reported in several European countries including Germany, Austria, Switzerland, Romania, and Turkey. These reports are based solely on morphology [8,9,10,11,12], morphology combined with sequencing of 16S ribosomal DNA (rDNA) [13,14,15,16,17,18], and sometimes using other genetic markers [2, 19,20,21]. However, with the rising number of available sequences, the number of reports of a failure to distinguish the two species has been growing, resulting in the use of the term I. ricinus/inopinatus complex [22,23,24,25]. In our surveillance of I. inopinatus in the Czech Republic, we also encountered difficulties in differentiating I. ricinus from I. inopinatus based on the morphology and sequencing of 16S rDNA. This resulted in our search for other genetic and easy-to-use markers that would differentiate these two sympatric and morphologically close to identical species.
So far, no consensus has been reached on the selection of a single molecular marker to differentiate ixodid tick species. The short fragment of mitochondrial 16S rRNA is often the first choice for tick identification together with the morphological description [26,27,28], followed by sequencing of the cytochrome c oxidase I subunit (COI) [29,30,31]. As for nuclear markers, the internal transcribed spacer 2 (ITS2) was used not only for species delineation [32,33,34] but also for the detection of natural hybrids between Ixodes persulcatus and I. ricinus as well as between I. ricinus and I. pavlovskyi [35, 36]. Use of other genes is scarce and in the context of I. ricinus/inopinatus, only the tick receptor for the OspA (TROSPA) and defensin genes showed discrimination power [5, 19,20,21].
In this study, we aimed for an easy and fast method for differentiation of the closely related and possibly sympatric species, I. ricinus and I. inopinatus, and searched for I. inopinatus in the Czech Republic. During the validation of a multiplex polymerase chain reaction (PCR) targeting the TROSPA gene, we were driven to a deeper study of the genetic diversity of these two species using mitochondrial and nuclear markers.
Methods
Adult ticks were collected by flagging in the Czech Republic (CZ) and Algeria (ALG) between 2015 and 2020. Ticks were collected in three different regions in CZ: Libava, northern Moravia (n = 114), Prostredni Porici, central Moravia (n = 110), and Podyji, southern Moravia (n = 103), and from one locality in ALG: El-Tarf province (n = 47). Based on the morphological characteristics according to Estrada-Peña et al. [7], all ticks were identified as I. ricinus or I. inopinatus. For phylogenetic purposes, another five Ixodes spp. were used: I. frontalis (Italy, 2021), I. gibbosus (Italy, 2021), I. hexagonus (Czech Republic, 2021), I. persulcatus (Russia, 2019), and I. ventalloi (Italy, 2013). These ticks were collected by our team as part of other ongoing projects with DNA of I. ventalloi obtained from colleagues from Italy [37]. Ticks were identified by BLASTn (Nucleotide Basic Local Alignment Search Tool) analysis of their 16S sequences. All samples were stored in 70% EtOH at −20 °C until further analysis.
Genomic DNA was isolated from a longitudinal half of each tick using the Exgene Cell SV mini 250p Kit (GeneAll, Seoul, Korea) according to the standard protocol for animal tissues with 100 µl of elution buffer added in the final step. The other half of the tick was stored for potential reanalysis.
For an easy and fast way to distinguish the two main variants of the TROSPA gene sequences reported as I. ricinus and I. inopinatus, we designed a multiplex PCR. A specific pair of primers for each variant was designed within the intron region based on the alignment of available sequences in GenBank (Table 1). The resulting amplicons differed by 126 base pairs (bp) for an easy on-gel identification. PCR was performed in a total volume of 25.0 µl using 2× PCRBIO Taq Mix Red (PCR Biosystems, UK), 0.4 µM of each of the four primers and 2.0 µl of template DNA. Reaction conditions followed manufacturer instructions with the annealing temperature of 52 °C and the elongation time of 15 s for 40 cycles.
For validation of the multiplex PCR and for subsequent sequence analysis, the 824 bp long fragment of the TROSPA gene including the entire intron was amplified and sequenced. To assess the genetic variability in more detail, fragments of two nuclear (ITS2 and calreticulin) and two mitochondrial genes (16S rRNA and cytochrome C oxidase subunit I—COI) were also amplified and sequenced. Primer sequences and PCR conditions are shown in Table 1.
Amplification of TROSPA, 16S rDNA, and ITS2 was performed in a total volume of 25 µl using 2× PCRBIO Taq Mix Red (PCR Biosystems, UK), 0.4 µM of each primer and 2 µl of template DNA. Reaction conditions followed manufacturer instructions for 40 cycles. COI and calreticulin genes were amplified in the total volume of 20 µl using Phusion Green Hot Start II High-Fidelity PCR Master Mix (Thermo Fisher Scientific, USA), 0.5 µM of each primer, and 2 µl of template DNA. Reaction conditions followed manufacturer instructions for 40 cycles. All PCR reactions were visualized on 1.5% agarose gel with the Midori Green Advance system (Nippon Genetics Europe, Germany). All products of expected length were cut from the gel, purified by the Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech Ltd., Taiwan), and sequenced by the Macrogen capillary sequencing services (Macrogen Europe, Netherlands) in both directions using the amplification primers.
Obtained sequences were assembled and visually inspected using Geneious R11.1.5 [38]. The identity of the amplicons was confirmed by BLASTn analysis (NCBI GenBank). Due to the appearance of double peaks in otherwise high-quality sequences (TROSPA and calreticulin genes), the detection of heterozygotes was performed using the Geneious plugin Find Heterozygotes followed by visual inspection and assigning ambiguous bases in positions with double peaks detected in both strands (with settings of peak similarity 30% and peak detection height 50%).
Representative samples with a high number of double peaks detected in the TROSPA gene as well as samples with a sudden loss of sequencing signal followed by an apparently mixed product chromatogram in the ITS2 region were cloned using pGEM®-T Easy Vector Systems (Promega Corporation, USA). Acquired plasmid DNA was purified from the bacterial culture using the GenElute™ Plasmid Miniprep Kit (Sigma-Aldrich, USA) and sequenced by the Macrogen capillary sequencing services (Macrogen Europe, The Netherlands) using universal T7/SP6 primers.
For phylogenetic analyses, sequences representing 16S rDNA, COI, TROSPA, ITS2, and calreticulin from various tick species within the genus Ixodes, preferably from different studies and geographical origins, were selected from the GenBank. Sequences originating from this study were limited to representative sequences in the case of 16S rRNA and COI, representative sequences and unique clones for ITS2, all sequences with 0–5 ambiguities and representative clones for TROSPA, and all sequences with a maximum of two ambiguous nucleotides for calreticulin (Additional file 2: Table S2).
Phylogenetic analyses were conducted by the ClustalW alignments built in Geneious R11.1.5 [38]. After manual editing of poorly aligned regions (especially the 16S rRNA gene), phylogenies were calculated by the maximum likelihood method using IQ-TREE multicore version 2.1.3 [39]. The best-fit evolution models were chosen based on the Bayesian information criterion (BIC) computed by ModelFinder [40]. Branch support was assessed by the ultrafast bootstrap (UFBoot) approximation [41] and by the SH-like approximate likelihood ratio test (SH-aLRT) [42]. Trees were visualized and edited in FigTree v1.4.4 and Inkscape 1.1.1.
Results
In total 374 adult ticks preselected by morphology as the I. ricinus/inopinatus complex were screened by multiplex PCR (Table 2) and three gel patterns were observed (Additional file 1: Figure S1). A single band of the size corresponding to the I. ricinus allele was observed in 321 ticks (317 from CZ and 4 from ALG) and a single band corresponding to the I. inopinatus allele was seen in 43 ticks from ALG and none from CZ. In 10 ticks from CZ, two bands were detected, each corresponding to one of the two species (Table 2, Additional file 1: Figure S1).
To validate the newly designed assay, the entire intron region of the TROSPA gene was amplified and sequenced from randomly selected representatives of both species from all localities and all 10 ambiguous samples. We were able to consistently amplify and sequence 670 bp out of the expected length of 824 bp, resulting in high-quality chromatograms from 112 I. ricinus and 19 I. inopinatus (based on the multiplex PCR) and all 10 ambiguous samples. Chromatograms commonly revealed double peaks; in fact, only 13 samples had no double peaks. In 117 samples, 1 to 15 clear double peaks in otherwise flawless chromatograms were observed, and in 10 samples (all assigned as ambiguous by multiplex PCR) 25 to 32 double peaks were detected in both strands (Fig. 1).

Schematic representation of the partial TROSPA gene sequences depicting the nucleotide positions with the double peaks (small yellow bars) (KF041821 is used as a reference sequence). The chromatograms depict the forward and reverse strands of sequencing for the uncloned PCR product. 441 cl.1 and 441 cl.5 are sequences after cloning resolving the double peaks of hybrid ticks
By cloning and sequencing of PCR products of the TROSPA gene from two ambiguous ticks and their alignment to sequences without double peaks from our study as well as with sequences from GenBank, we identified 23 positions consistently different between I. ricinus and I. inopinatus alleles (Fig. 1, Additional file 2: Table S1). In all 10 samples yielding bands corresponding to both I. ricinus and I. inopinatus in the multiplex PCR, the positions of double peaks corresponded to 23 single-nucleotide polymorphisms (SNPs) differentiating the two species. All other positions with double peaks showed no regular pattern and were detected in random positions.
To support the above-described analyses and to put the TROSPA species differentiation power in a larger context, we amplified and sequenced the same fragment from other Ixodes spp. (minimum of three individuals per species). In the phylogenetic analyses, I. ricinus and I. inopinatus sequences from this study together with sequences from the GenBank database form two well-supported sister clades (Fig. 2). The cloned sequences of the ambiguous samples based on the multiplex PCR, the two variants of alleles representing the I. ricinus and I. inopinatus species, fell within the respective clades. All other Ixodes spp. form well-supported and distinguished monophyletic clades (Fig. 2, Additional file 1: Figure S2).

Phylogenetic tree of ticks based on the TROSPA gene samples from this study are indicated in bold font. CZ Czech Republic, ALG Algeria. 441 M CZ cl.1/cl.5 and 331F CZ cl.3/13 are sequences after cloning of hybrid ticks showing a clear split to I. ricinus and I. inopinatus branches
Fragments of two mitochondrial genes, 16S rDNA and COI, the most commonly used molecular markers for ixodid ticks, were amplified and sequenced. The fragment of 16S rDNA was amplified and sequenced from 222 ticks from CZ and 43 ticks from ALG. Thirty-six unique haplotypes (26 CZ and 10 ALG) with sequence similarity 96.02–99.73% were identified. From these unique haplotypes, seven (three CZ and four ALG) had the “AG” haplotype assigned previously to I. inopinatus, 28 (23 CZ and five ALG) had the “CT” haplotype referring to I. ricinus [13, 16], and one new AT haplotype (1 ALG) was detected. In the phylogenetic analyses, all representative sequences from this study form a single, highly supported clade together with the I. ricinus and I. inopinatus sequences retrieved from the GenBank database. However, no structure based on the species or geography was detected within the clade(s) (Fig. 3). Other Ixodes spp. form well-distinguished and supported clades (with the exception of I. affinis and I. pararicinus forming a single clade, Additional file 1: Figure S3).

Phylogenetic tree of ticks based on the 16S rRNA gene showing the lack of power to distinguish I. ricinus and I. inopinatus. Samples from this study are indicated in bold font. CZ Czech Republic, ALG Algeria
Amplification and sequencing of the COI gene were done on 285 ticks (245 CZ and 40 ALG). In the phylogenetic analysis of the COI gene, all representative sequences from this study form a single clade together with the I. ricinus and I. inopinatus sequences retrieved from the GenBank database (Fig. 4). Although the bootstrap support of the clade is low, its resolution from the most closely related I. laguri is clear. All I. inopinatus sequences (as assigned based on the TROSPA analyses or by their name in GenBank) form a separate, highly supported subclade, although with very short branch length. Other Ixodes spp. form well-distinguished and supported clades, some with the intraspecific subclade structure (e.g., I. affinis and I. persulcatus) (Additional file 1: Figure S4).

Phylogenetic tree of ticks based on the COI gene depicting a very close relationship between I. ricinus and I. inopinatus. Samples from this study are indicated in bold font. CZ Czech Republic, ALG Algeria
In addition to the TROSPA gene, two other nuclear markers were also amplified and sequenced from the subset of our tick samples. After direct sequencing, the ITS2 region yielded high-quality chromatograms only from 24 ticks (13 CZ and 11 ALG). In other samples, a sudden loss of the sequencing signal followed by an apparently mixed product chromatogram was observed. PCR products from 15 samples (eight CZ including two ticks assigned as ambiguous by multiplex PCR, and seven ALG) were cloned and 71 individual clones were sequenced (4–7 clones per sample). Among these, 37 unique clones (18 CZ and 19 ALG) were observed. Phylogenetic analyses of the available sequences representing various Ixodes spp. showed a pattern similar to that of the COI gene. All I. ricinus and I. inopinatus sequences (directly sequenced and cloned in this study as well as from GenBank) form a single, highly supported clade. All sequences of I. inopinatus (as assigned by the TROSPA gene) assembled into the unsupported subclade (Fig. 5, Additional file 1: Figure S5). Clones originating from a single individual always fell into a single subclade (I. ricinus/inopinatus). All clones from the two ambiguous samples (80F and 42F) clustered within the I. ricinus subclade.

Phylogenetic tree of ticks based on the ITS2 gene Samples from this study are indicated in bold font. CZ Czech Republic, ALG Algeria. Matching colors indicate sequences after cloning to resolve sequence ambiguities
A part of the calreticulin gene was amplified and sequenced from 34 ticks (19 CZ and 15 ALG). Sequences with 0–2 double peaks (ambiguous bases were assigned) from 26 ticks (15 CZ and 11 ALG) were used for the phylogenetic analyses. The fragment was not suitable for distinguishing the Ixodes species since no clades were formed in the phylogeny (Additional file 1: Figure S6).
Discussion
Accurate identification of ticks at the species level is critical from several perspectives including distribution mapping, life cycle, and host range/preference, and most importantly for vector capacity for pathogens. Typically, methods based on the morphology and sequencing of 16S rDNA have been used for tick identification and description of new species [26, 28, 29], and other genes have rarely been used [27, 30, 33]. However, morphological identification relies on acarological expertise and specimen quality, and is rather time-consuming on large datasets. Furthermore, very low morphological variability makes it impossible to use for the identification of closely related taxa [9]. Sequencing of 16S rDNA is considered a gold standard for the identification of ticks and many other organisms, including bacteria. However, I. ricinus could not be differentiated from I. inopinatus by the commonly used 16S rDNA fragment [13, 16] due to the high haplotype diversity of this gene [8, 10, 11, 16, 22,23,24,25]. The COI gene is another common marker used for species delineation within the animal kingdom (e.g., the BOLD [Barcode of Life Data System] database); however, similarly to 16S rDNA, its analyses do not have the power to detect potential hybridization between closely related species. Mitochondrial markers are typically inherited uniparentally and therefore do not reflect the genetic history of an organism [43, 44].
Nuclear genes can reveal potential hybridization [35, 36], but these often have several copies resulting in mixed chromatograms in Sanger sequencing and the consequent need for cloning. So far, the TROSPA gene was the only marker that consistently distinguished tick populations from North Africa and Europe [5]. Internal transcribed spacers 1 and 2 (ITS1 and ITS2) are useful for subtyping due to the high intraspecific diversity [5]; however, for many tick species, sequences for these loci are not available in GenBank [45]. Calreticulin was found to be completely inappropriate for distinguishing tick species, which is consistent with Babkin et al. [46].
Clearly, the TROSPA and ITS2 genes seem to be good candidates for differentiation of the North African lineage of ticks referred to as I. inopinatus from the European population of I. ricinus. However, relatively small differences in these two markers between I. ricinus and I. inopinatus (in comparison to differences among other Ixodes species) opened a question of the natural gene flow between tick populations in North Africa and Europe. Our TROSPA data indicate natural hybridization followed by gene introgression and that hybrids of I. ricinus and I. inopinatus survive and may backcross the European parental population potentially resulting in unidirectional introgression [47]. However, this needs to be investigated further with larger sets of ticks, especially from North Africa.
Distribution patterns of arthropods, amphibians, reptiles, and mammals demonstrate biogeographical affinities between Europe and North Africa at the species level [48]. The distribution of primarily Palaearctic species across the Mediterranean has attracted considerable attention, showing North Africa as a refugium and differentiation center for Western Palaearctic thermophilic species. However, this applies to non-flying organisms only. Ixodes ricinus is a tick species commonly reported on birds (especially nymphs and larvae) [45], including migratory species [21, 49, 50]. We hypothesize, that I. inopinatus is adapted to climatic conditions in North Africa, and possibly the southernmost areas of Europe. African ticks are likely regularly carried by migratory birds between North Africa and Europe, as documented in the case of Hyalomma spp. [51, 52] as well as I. ricinus and I. inopinatus [53].
Since we did not find any signs of hybridization in North Africa, we hypothesize, that I. ricinus ticks from higher latitudes and their hybrids with the African population do not survive well in North African climate and that only North African ticks carried to Europe successfully hybridize and backcross with I. ricinus in Europe. However, it is important to point out that our data set from Algeria is much smaller than that from the Czech Republic, and follow-up studies are needed. To address this, we are currently conducting a surveillance study of ticks across Italy and additional sampling in Algeria (in preparation). The surveillance of ticks on migratory birds in the Czech Republic is also underway.
Similarly to the study from Germany [54], our data put into question studies reporting I. inopinatus from Central Europe based on morphology and/or sequencing of 16S rDNA, and we suggest that these should be re-examined. Even when TROSPA and other nuclear genes were used, double peaks and signs of hybridization and introgression have not been reported previously. In conclusion, we offer a fast and reliable multiplex PCR method for the identification of I. ricinus and I. inopinatus. Morphological similarity to I. ricinus and phylogenetic analyses both suggest African I. inopinatus to be “a species in statu nascendi” evolving from I. ricinus. Additional studies on the genetic diversity and the full genome sequencing of Ixodes ricinus/inopinatus in North Africa and regions of likely sympatry with I. ricinus in Europe (Spain, Portugal, Italy) are needed. Questions including the potential differences in vector competence between I. ricinus and I. inopinatus and their hybrids remain to be answered.
Availability of data and materials
The representative nucleotide sequences generated in the present study and used in the phylogenies have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/) under the accession numbers OQ981335-63 for 16S rRNA, OQ981450-76 for the COI gene, OQ991204-18 for ITS2, OQ999531-56 for the calreticulin gene, and OQ999557-628 for the TROSPA gene. The datasets used and/or analyzed during the current study are available from the corresponding author upon a reasonable request.
References
Estrada-Peña A, de la Fuente J. Host distribution does not limit the range of the tick Ixodes ricinus but impacts the circulation of transmitted pathogens. Front Cell Infect Microbiol. 2017;7:1–10. https://doi.org/10.3389/fcimb.2017.00405.
Al-Khafaji AM, Clegg SR, Pinder AC, Luu L, Hansford KM, Seelig F, et al. Multi-locus sequence typing of Ixodes ricinus and its symbiont Candidatus Midichloria mitochondrii across Europe reveals evidence of local co-cladogenesis in Scotland. Ticks Tick Borne Dis. 2019;10:52–62. https://doi.org/10.1016/j.ttbdis.2018.08.016.
Røed KH, Kvie KS, Hasle G, Gilbert L, Leinaas HP. Phylogenetic lineages and postglacial dispersal dynamics characterize the genetic structure of the tick, Ixodes ricinus, in Northwest Europe. PLoS ONE. 2016;11:e0167450. https://doi.org/10.1371/journal.pone.0167450.
Dinnis RE, Seelig F, Bormane A, Donaghy M, Vollmer SA, Feil EJ, et al. Multilocus sequence typing using mitochondrial genes (mtMLST) reveals geographic population structure of Ixodes ricinus ticks. Ticks Tick Borne Dis. 2014;5:152–60. https://doi.org/10.1016/j.ttbdis.2013.10.001.
Noureddine R, Chauvin A, Plantard O. Lack of genetic structure among Eurasian populations of the tick Ixodes ricinus contrasts with marked divergence from north-African populations. Int J Parasitol. 2011;41:183–92. https://doi.org/10.1016/j.ijpara.2010.08.010.
Poli P, Lenoir J, Plantard O, Ehrmann S, Røed KH, Leinaas HP, et al. Strong genetic structure among populations of the tick Ixodes ricinus across its range. Ticks Tick Borne Dis. 2020;11:101509. https://doi.org/10.1016/j.ttbdis.2020.101509.
Estrada-Peña A, Nava S, Petney T. Description of all the stages of Ixodes inopinatus n sp (Acari: Ixodidae). Ticks Tick Borne Dis. 2014;5:734–43. https://doi.org/10.1016/j.ttbdis.2014.05.003.
Fares W, Dachraoui K, Cherni S, Barhoumi W, Ben ST, Younsi H, et al. Tick-borne encephalitis virus in Ixodes ricinus (Acari: Ixodidae) ticks Tunisia. Ticks Tick Borne Dis. 2021;12:101606. https://doi.org/10.1016/j.ttbdis.2020.101606.
Chitimia-Dobler L, Bestehorn M, Bröker M, Borde J, Molcanyi T, Andersen NS, et al. Morphological anomalies in Ixodes ricinus and Ixodes inopinatus collected from tick-borne encephalitis natural foci in Central Europe. Exp Appl Acarol. 2017;72:379–97. https://doi.org/10.1007/s10493-017-0163-5.
Kahl O, Kämmer D, Bulling I, Komorek M, von Eiff C, Malerczyk C. Ticks on the turf: investigating the presence of ixodid ticks on and around football fields in Germany. Exp Appl Acarol. 2021;84:585–91. https://doi.org/10.1007/s10493-021-00628-0.
Król N, Obiegala A, Imholt C, Arz C, Schmidt E, Jeske K, et al. Diversity of Borrelia burgdorferi sensu lato in ticks and small mammals from different habitats. Parasit Vectors. 2022;15:195. https://doi.org/10.1186/s13071-022-05326-3.
Mechouk N, Mihalca AD, Deak G, Bouslama Z. Synopsis of the ticks of Algeria with new hosts and localities records. Parasit Vectors. 2022;15:302. https://doi.org/10.1186/s13071-022-05424-2.
del Cerro A, Oleaga A, Somoano A, Barandika JF, García-Pérez AL, Espí A. Molecular identification of tick-borne pathogens (Rickettsia spp., Anaplasma phagocytophilum, Borrelia burgdorferi sensu lato, Coxiella burnetii and piroplasms) in questing and feeding hard ticks from North-Western Spain. Ticks Tick Borne Dis. 2022;13:101961. https://doi.org/10.1016/j.ttbdis.2022.101961.
Hauck D, Springer A, Pachnicke S, Schunack B, Fingerle V, Strube C. Ixodes inopinatus in northern Germany: occurrence and potential vector role for Borrelia spp., Rickettsia spp., and Anaplasma phagocytophilum in comparison with Ixodes ricinus. Parasitol Res. 2019;118:3205–16. https://doi.org/10.1007/s00436-019-06506-4.
Hauck D, Springer A, Chitimia-Dobler L, Strube C. Two-year monitoring of tick abundance and influencing factors in an urban area (city of Hanover, Germany). Ticks Tick Borne Dis. 2020. https://doi.org/10.1016/j.ttbdis.2020.101464.
Hornok S, Daccord J, Takács N, Kontschán J, Tuska-Szalay B, Sándor AD, et al. Investigation on haplotypes of ixodid ticks and retrospective finding of Borrelia miyamotoi in bank vole (Myodes glareolus) in Switzerland. Ticks Tick Borne Dis. 2022;13:101865. https://doi.org/10.1016/j.ttbdis.2021.101865.
Chitimia-Dobler L, Rieß R, Kahl O, Wölfel S, Dobler G, Nava S, et al. Ixodes inopinatus—occurring also outside the Mediterranean region. Ticks Tick Borne Dis. 2018;9:196–200. https://doi.org/10.1016/j.ttbdis.2017.09.004.
Knoll S, Springer A, Hauck D, Schunack B, Pachnicke S, Strube C. Regional, seasonal, biennial and landscape-associated distribution of Anaplasma phagocytophilum and Rickettsia spp. infections in Ixodes ticks in northern Germany and implications for risk assessment at larger spatial scales. Ticks Tick Borne Dis. 2021;12:101657. https://doi.org/10.1016/j.ttbdis.2021.101657.
Hekimoğlu O. Phylogenetic placement of Turkish populations of Ixodes ricinus and Ixodes inopinatus. Exp Appl Acarol. 2022;88:179–89. https://doi.org/10.1007/s10493-022-00750-7.
Norte AC, Boyer PH, Castillo-Ramirez S, Chvostáč M, Brahami MO, Rollins RE, et al. The population structure of Borrelia lusitaniae Is reflected by a population division of its Ixodes vector. Microorganisms. 2021;9:933. https://doi.org/10.3390/microorganisms9050933.
Toma L, Mancuso E, d’Alessio SG, Menegon M, Spina F, Pascucci I, et al. Tick species from Africa by migratory birds: a 3-year study in Italy. Exp Appl Acarol. 2021;83:147–64. https://doi.org/10.1007/s10493-020-00573-4.
Elati K, Khbou MK, Kahl O, Mwacharo JM, El Shamaa K, Rekik M, et al. Preliminary study on the seasonal questing of Ixodes ricinus group ticks in Ain Draham forest (north-western Tunisia) with analyses of their phylogenetic diversity. Vet Parasitol Reg Stud Rep. 2022;36:100786. https://doi.org/10.1016/j.vprsr.2022.100786.
Glass A, Springer A, Strube C. A 15-year monitoring of Rickettsiales (Anaplasma phagocytophilum and Rickettsia spp.) in questing ticks in the city of Hanover. Germany Ticks Tick Borne Dis. 2022;13:101975. https://doi.org/10.1016/j.ttbdis.2022.101975.
Reynolds C, Kontschán J, Takács N, Solymosi N, Sándor AD, Keve G, et al. Shift in the seasonality of ixodid ticks after a warm winter in an urban habitat with notes on morphotypes of Ixodes ricinus and data in support of cryptic species within Ixodes frontalis. Exp Appl Acarol. 2022;88:127–38. https://doi.org/10.1007/s10493-022-00756-1.
Vogelgesang JR, Walter M, Kahl O, Rubel F, Brugger K. Long-term monitoring of the seasonal density of questing ixodid ticks in Vienna (Austria): setup and first results. Exp Appl Acarol. 2020;81:409–20. https://doi.org/10.1007/s10493-020-00511-4.
D’Amico G, Estrada-Peña A, Kalmár Z, Fuh T, Petrželková K, Mihalca AD. Redescription of the adult stages of Ixodes (Afrixodes) rasus Neumann 1899, with notes on its phylogenetic position within the genus Ixodes. Ticks Tick Borne Dis. 2018;9:654–9. https://doi.org/10.1016/j.ttbdis.2018.02.006.
Backus LH, Foley JE, Hobbs GB, Bai Y, Beati L. A new species of tick, Ixodes (Ixodes) mojavensis (Acari: Ixodidae), from the Amargosa Valley of California. Ticks Tick Borne Dis. 2022;13:102020. https://doi.org/10.1016/j.ttbdis.2022.102020.
Onofrio VC, Guglielmone AA, Barros-Battesti DM, Gianizella SL, Marcili A, Quadros RM, et al. Description of a new species of Ixodes (Acari: Ixodidae) and first report of Ixodes lasallei and Ixodes bocatorensis in Brazil. Ticks Tick Borne Dis. 2020;11:101423. https://doi.org/10.1016/j.ttbdis.2020.101423.
Orkun Ö, Vatansever Z. Rediscovery and first genetic description of some poorly known tick species: Haemaphysalis kopetdaghica Kerbabaev, 1962 and Dermacentor raskemensis Pomerantzev, 1946. Ticks Tick Borne Dis. 2021;12:101726. https://doi.org/10.1016/j.ttbdis.2021.101726.
Rar V, Yakimenko V, Tikunov A, Vinarskaya N, Tancev A, Babkin I, et al. Genetic and morphological characterization of Ixodes apronophorus from Western Siberia Russia. Ticks Tick Borne Dis. 2020;11:101284. https://doi.org/10.1016/j.ttbdis.2019.101284.
Saracho-Bottero MN, Venzal JM, Tarragona EL, Thompson CS, Mangold AJ, Beati L, et al. The Ixodes ricinus complex (Acari: Ixodidae) in the Southern Cone of America: Ixodes pararicinus, Ixodes aragaoi, and Ixodes sp. cf. I. affinis. Parasitol Res. 2020;119:43–54. https://doi.org/10.1007/s00436-019-06470-z.
Fukunaga M, Yabuki M, Hamase A, Oliver JH, Nakao M. Molecular phylogenetic analysis of ixodid ticks based on the ribosomal DNA spacer, internal transcribed spacer 2, sequences. J Parasitol. 2000;86:38–43. https://doi.org/10.1645/0022-3395(2000)086[0038:MPAOIT]2.0.CO;2.
Kovalev SY, Fedorova SZh, Mukhacheva TA. Molecular features of Ixodes kazakstani: first results. Ticks Tick Borne Dis. 2018;9:759–61. https://doi.org/10.1016/j.ttbdis.2018.02.019.
Numan M, Islam N, Adnan M, Zaman Safi S, Chitimia-Dobler L, Labruna MB, et al. First genetic report of Ixodes kashmiricus and associated Rickettsia sp. Parasit Vectors. 2022;15:378. https://doi.org/10.1186/s13071-022-05509-y.
Kovalev SY, Mikhaylishcheva MS, Mukhacheva TA. Natural hybridization of the ticks Ixodes persulcatus and Ixodes pavlovskyi in their sympatric populations in Western Siberia. Infect Genet Evol. 2015;32:388–95. https://doi.org/10.1016/j.meegid.2015.04.003.
Kovalev SY, Golovljova IV, Mukhacheva TA. Natural hybridization between Ixodes ricinus and Ixodes persulcatus ticks evidenced by molecular genetics methods. Ticks Tick Borne Dis. 2016;7:113–8. https://doi.org/10.1016/j.ttbdis.2015.09.005.
Latrofa MS, Giannelli A, Persichetti MF, Pennisi MG, Solano-Gallego L, Brianti E, et al. Ixodes ventalloi: morphological and molecular support for species integrity. Parasitol Res. 2017;116:251–8. https://doi.org/10.1007/s00436-016-5286-9.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. https://doi.org/10.1093/molbev/msu300.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. https://doi.org/10.1038/nmeth.4285.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95. https://doi.org/10.1093/molbev/mst024.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. https://doi.org/10.1080/10635150390235520.
Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–44. https://doi.org/10.1046/j.1365-294X.2003.02063.x.
Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21:3907–30. https://doi.org/10.1111/j.1365-294X.2012.05664.x.
Gray J, Kahl O, Zintl A. What do we still need to know about Ixodes ricinus? Ticks Tick Borne Dis. 2021;12:101682. https://doi.org/10.1016/j.ttbdis.2021.101682.
Babkin IV, Tikunov AY, Gologljova I, Tikunova NV. High level of identity of calreticulin gene sequences of Ixodes persulcatus and Ixodes pavlovskyi ticks. J Parasitol. 2018;104:337–41. https://doi.org/10.1645/17-186.
Rhymer JM, Simberloff D. Extinction by hybridization and introgression. Annu Rev Ecol Syst. 1996;1:83–109.
Husemann M, Schmitt T, Zachos FE, Ulrich W, Habel JC. Palaearctic biogeography revisited: evidence for the existence of a North African refugium for Western Palaearctic biota. J Biogeogr. 2014;41:81–94. https://doi.org/10.1111/jbi.12180.
Hasle G. Transport of ixodid ticks and tick-borne pathogens by migratory birds. Front Cell Infect Microbiol. 2013;4:1–6. https://doi.org/10.3389/fcimb.2013.00048.
Sándor AD, Mărcuţan DI, D’Amico G, Gherman CM, Dumitrache MO, Mihalca AD. Do the ticks of birds at an important migratory hotspot reflect the seasonal dynamics of Ixodes ricinus at the migration initiation site? a case study in the Danube Delta. PLoS ONE. 2014;9:e89378. https://doi.org/10.1371/journal.pone.0089378.
Estrada-Peña A, D’Amico G, Fernández-Ruiz N. Modelling the potential spread of Hyalomma marginatum ticks in Europe by migratory birds. Int J Parasitol. 2021;51:1–11. https://doi.org/10.1016/j.ijpara.2020.08.004.
Lesiczka PM, Daněk O, Modrý D, Hrazdilová K, Votýpka J, Zurek L. A new report of adult Hyalomma marginatum and Hyalomma rufipes in the Czech Republic. Ticks Tick Borne Dis. 2022. https://doi.org/10.1016/j.ttbdis.2021.101894.
Toma L, Mancuso E, Alessio SG, Menegom M, Spina F, Pascucci I, et al. Tick species from Africa by migratory birds: a 3-year study in Italy. Exp Appl Acarol. 2021;83:147–64. https://doi.org/10.1007/s10493-020-00573-4.
Rollins RE, Margos G, Brachmann A, Krebs S, Mouchet A, Dingemanse NJ, et al. German Ixodes inopinatus samples may not actually represent this tick species. Int J Parasitol. 2023. https://doi.org/10.1101/2023.02.14.528458.
Jizhou Lv, Shaoqiang Wu, Yongning Zhang, Tianyi Zhang, Chunyan Feng, Guangle Jia & Xiangmei Lin Development of a DNA barcoding system for the Ixodida (Acari: Ixodida), Mitochondrial DNA, 2014;25:(2)142–149, https://doi.org/10.3109/19401736.2013.792052
Funding
This project was supported by the Czech Science Foundation GACR 21-11661S to LZ, DM, and PA. KH was supported by the project National Institute of Virology and Bacteriology (Programme EXCELES, ID Project No. LX22NPO5103) Funded by the European Union—Next Generation EU.
Author information
Authors and Affiliations
Contributions
KH analyzed the data and wrote the manuscript. OD, AH, and EN performed the lab. work and analyzed the data. BC analyzed the data. JV and PA secured the funding. ADM arranged the collection of ticks in Algeria. MN collected ticks in Algeria. DM designed the project, secured the funding, and wrote the manuscript. LZ designed and supervised the project, secured the funding, and wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Multiplex PCR. Figure S2. TROSPA gene phylogeny. Figure S3. 16S rRNA phylogeny. Figure S4. COI gene phylogeny. Figure S5. ITS2 phylogeny. Figure S6. Calreticulin gene phylogeny.
Additional file 2: Table S1.
List of 23 SNPs in the TROSPA intron. Table S2. Sequences used for phylogenetic analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hrazdilova, K., Danek, O., Hrbatova, A. et al. Genetic analysis challenges the presence of Ixodes inopinatus in Central Europe: development of a multiplex PCR to distinguish I. inopinatus from I. ricinus. Parasites Vectors 16, 354 (2023). https://doi.org/10.1186/s13071-023-05971-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-023-05971-2