
Abstract
Background
Blastocystis sp. is an anaerobic protozoan that parasitizes many animal hosts and the human gastrointestinal tract, and its pathogenicity is controversial. Captive wildlife may be potential reservoirs for human infection with Blastocystis sp. The present study was performed to investigate the prevalence and subtype distribution of Blastocystis sp. in zoo animals in Sichuan Province, southwestern China.
Methods
A total of 420 fresh fecal samples were collected from 54 captive wildlife species in four zoos in southwestern China between June 2017 and September 2019. The prevalence and subtype (ST) genetic characteristics of Blastocystis sp. were determined by PCR amplification of the barcode region of the SSU rRNA gene and phylogenetic analysis.
Results
Overall, 15.7% (66/420) of the animal samples and 20.7% (14/54) of the species tested were shown to be infected with Blastocystis sp. The highest prevalence of Blastocystis sp. was found in Panzhihua Zoo (24.3%), which was significantly higher than that in Chengdu Zoo (6.9%), and Xichang Zoo (2.9%) (P < 0.05). There are also significant differences in the prevalence of Blastocystis sp. among different species (P < 0.05), and the highest of Blastocystis sp. prevalence was observed in white-cheeked gibbon, black great squirrel, and red giant flying squirrel (100%). Subtype analysis of Blastocystis sp. revealed nine subtypes, including six zoonotic STs (ST1-5, and ST8) and three animal-specific STs (ST10, ST14, and ST17), with ST17 as the predominant subtype (26/66) in Blastocystis sp.-positive isolates.
Conclusions
To our knowledge, this is the first report on the prevalence and subtype distribution of Blastocystis sp. among captive wildlife in zoos in southwestern China. This study highlights that these animals may serve as reservoirs for human Blastocystis sp. infections.
Graphical Abstract

Similar content being viewed by others


Background
Blastocystis sp., belonging to the phylum stramenopiles, is a common anaerobic eukaryotic protist that inhabits the gastrointestinal tract of a wide range of hosts, including humans. It is estimated that this parasite has colonized 1 to 2 billion people worldwide based on epidemiological surveys [1]. Blastocystis sp. is mainly transmitted through the fecal-oral route, food, and water [2,3,4]. There is supporting evidence that some human infections may be caused by zoonotic transmission of Blastocystis sp. [5, 6].
Based on polymorphisms of small subunit (SSU) gene of Blastocystis sp., 22 subtypes (STs) consisting of ST1 to ST17, ST21, and ST23 to ST26 have been identified in humans and domestic and wild animals worldwide [7]. ST1-9 and -12 are able to infect humans, with ST1-4 being the most common, accounting for more than 90% of human Blastocystis sp. infections [8]. Interestingly, the prevalence of different STs among countries and among regions within the same country seems to vary greatly [9]. Remarkable genetic diversity was observed among various STs [10], and different STs exhibit diverse biological features, such as drug resistance, pathogenicity, and effects on microbiota [11,12,13].
Although Blastocystis sp. has been reported > 100 years, the clinical significance of this common parasite remains controversial [14]. Accumulating evidence shows Blastocystis sp. long-term colonization in asymptomatic carriers, accompanied with a healthy gut microbiota [15, 16], suggesting that it should be regarded as a member of the normal intestinal microbiota. Blastocystis sp. has been found in patients with irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD), [17, 18], but not the presence of the protist that causes them. In vitro experiments using cell lines have also determined the potential pathogenicity of some specific STs of Blastocystis sp., such as disrupting the epithelial barrier [19] by increasing the epithelial permeability [20, 21]. Moreover, experimental infections with Blastocystis sp. in mouse models have shown that it can decrease the abundance of beneficial bacteria Bifidobacterium and Lactobacillus [12].
Blastocystis sp. has been reported in a substantial number of animal hosts, including livestock, companion animals, and captive wildlife, with greatly varying prevalence [22,23,24]. In recent years, several important intestinal zoonotic pathogens (e.g., Cryptosporidium, Giardia, and Microsporidia) have been reported in captive wildlife in China [25, 26], highlighting wildlife may be potential reservoirs for human to contract these infectious agents. However, less information is currently available regarding the prevalence and subtype distribution of Blastocystis sp. in zoo animals in China [27]. The purpose of the present study was thus to determine the genetic characteristics and subtype distribution of Blastocystis sp. in various zoo animals in southwestern China to better assess its zoonotic potential.
Methods
Sample collection
A total of 420 fresh fecal samples were collected from wildlife in Chengdu Zoo (n = 144), Ya’an Zoo (n = 204), Xichang Zoo (n = 35), and Panzhihua Zoo (n = 37) between June 2017 and September 2019 in Sichuan Province, southwestern China (Fig. 1). The collected samples include a large variety of mammalian groups and several avian species. The animals were either housed individually or in monospecific groups of 5–10 individuals sharing the same enclosures. For those animals housed individually, only one sample was collected per animal. In group housing, between two and five samples were collected, each of which was considered as individual sample. All the fresh fecal samples were collected by zookeepers before the cleaning of animal cages in the morning and were strictly controlled to minimize potential contamination between animal species. Feces samples from some avian species were collected carefully directly on the ground or in their nests. All fecal samples were collected in sterilized plastic containers using disposable sterile gloves and preserved at 4 ℃ until DNA extraction.

Geographical distribution of the sampled cities (filled circle) in Sichuan Province, Southwestern China
DNA extraction
Fecal specimens were sieved and washed three times with distilled water by centrifugation at 3000g for 5 min. Genomic DNA was extracted using a QIAamp DNA
Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA quality was verified by NanoDrop (Thermo Fisher Scientific, Carlsbad, CA, USA) measurements. DNA was eluted in 50 μl of nuclease-free water and stored at − 20 °C until use.
PCR amplification
All extracted DNA was screened for the presence of Blastocystis sp. by a polymerase chain reaction (PCR) amplification of the barcode region of the SSU rRNA gene, using primers RD5 (5′-ATCTGGTTGATCCTGCCAGT-3′) and BhRDr (5′-GAGCTTTTTAACTGCAACAACG-3′) [28]. The PCR mixture (25 μl) contained 12.5 μl Taq PCR Master Mix (Sangon Biotech Co., Ltd., Shanghai, China), 1 μl each primer (0.4 μM), 2 μl genomic DNA, 1.5 mM MgCl2, and nuclease-free water up to desired volume. The PCR was started at 94 °C for 4 min followed by 30 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s, with an extension at 72 °C for 5 min. Positive and negative controls were included in all the PCR tests. PCR products were subjected to 1.5% agarose gel (AddGene, Watertown, MA, USA) electrophoresis and visualized by staining with SYBR Safe DNA Gel Stain (Thermo Fisher Scientific).
Sequencing and phylogenetic analysis
PCR products with the expected size (∼ 600 bp) were excised from the agarose gel and purified using a QIAquick Gel Extraction Kit (Qiagen) according to the manufacturer’s instructions. Purified products were directly sequenced on an ABI PRISMTM 3730 DNA Analyzer (Applied Biosystems, USA), using a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster, CA, USA).
Nucleotide sequences obtained in the present study were subjected to BLAST searches (http://www.ncbi.nlm.nih.gov/blast/), and the reference sequences were downloaded from the GenBank database. Blastocystis sp. subtypes were identified by BLAST searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi), and the alleles were identified at the Blastocystis sp. database (http://pubmlst.org/blastocystis). The sequences obtained in this study and reference sequences were aligned using the MUSCLE algorithm of MEGA-X [29]. The ambiguous positions were removed from the alignment, and then the alignment was trimmed using MEGA 6 (http://www.megasoftware.net/) before phylogenetic analysis. ST11 is not available in the barcode region, and ST21 and ST23-26 need further data for determination, so we did not include these subtypes in the phylogenetic analysis [7]. The phylogenetic tree was constructed with the neighbor-joining (NJ) method using the Mega 6 software. Evolutionary distances were calculated using the Kimura two-parameter model. Bootstrap analysis (with 1000 replicates) was performed to define the robustness of the findings. Proteromonas lacertae was used as outgroup for the phylogenetic analysis.
Statistical analysis
The difference in Blastocystis sp. prevalence between different zoos and the order of animals was analyzed with the chi-square (χ2) test, using SPSS 20.0 (IBM, Chicago, IL, USA). The difference was considered statistically significant when P < 0.05. Ninety-five percent confidence intervals (95% CIs) and odds ratios (ORs) were also calculated to explore the strengths of association between Blastocystis sp. positivity and each factor.
Results
Prevalence of Blastocystis sp. in captive wild animals
It is worth noting that the prevalences herein are the minimum since we used barcoding primers rather than qPCR. In the present study, 66 of 420 (15.7%) fecal samples collected from four animal zoos in China were determined to be Blastocystis sp. positive by PCR amplification of the barcode region of the SSU rRNA gene. Specifically, 10 of 144 (6.9%) animals sampled from Chengdu Zoo, 46 of 204 (22.5%) samples from Ya’an Zoo, 1 of 35 (2.9%) samples from Xichang Zoo, and 9 of 37 (24.3%) samples collected from Panzhihua Zoo were Blastocystis sp. positive (Table 1). The difference in Blastocystis sp. prevalence was significant in four zoos (P < 0.05). The prevalence of Blastocystis sp. in nonhuman primates (NHPs) was 30.5%, which is higher than that in Rodentia (18.2%), Artiodactyla (12.8%), birds (8.8%), and Carnivora (5.9%). Similarly, the difference of Blastocystis sp. prevalence among different order animals was significant (P < 0.05).
In general, of the 54 species tested in this study, 14 (20.7%) were positive for Blastocystis sp. (Table 2). Specifically, of the 29 species tested at the Chengdu Zoo, 6 (20.7%) were positive for Blastocystis sp. At the Ya’an Zoo, the prevalence of the parasite was 33.3% (5/15) among the species screened; 7.7% (1/13) and 33.3% (3/9) species at Xichang Zoo and Panzhihua Zoo in the present study were shown to be infected with Blastocystis sp. respectively.
Interestingly, the prevalence of Blastocystis sp. varies greatly among different species (Table 3). The highest Blastocystis sp. prevalence was observed in white-cheeked gibbon (100%, 4/4). By comparison, sika deer and green peafowl showed lower Blastocystis sp. prevalence, accounting for 9.1% and 8.3% respectively.
Subtype distributions of Blastocystis sp. in captive wild animals
Among the 66 Blastocystis sp.-positive samples, 9 subtypes were identified, including 6 zoonotic STs (ST1-5, and ST8) and 3 animal-specific STs (ST10, ST14, and ST17). ST17 (allele 160) (39.4%, 26/66) was the dominant subtype in zoo animals examined in the present study (Table 4), followed by ST1 (allele 1) (22.7%, 15/66), ST5 (allele 118) (10.6%, 7/66), ST2 (allele 15) (9.1%, 6/66), and ST8 (allele 21) (6.1%, 4/66). ST3 (allele 34), ST4 (allele 42), ST10 (allele 152), and ST14 (allele 157) were only found in two fecal samples respectively (Table 4). Notably, ST1 has the widest host range in zoo animals, detected in rhesus macaque, ring-tailed lemur, leopard, and sika deer (Table 4). Meanwhile, ST17 was identified in four species of animals, including squirrel monkey, Asiatic black bear, and chinchilla (Table 4).
Genetic characteristics of Blastocystis sp. subtypes
The identity analysis of the SSU rRNA gene revealed that 15 sequences of ST1 isolates identified in NHPs, leopard, and sika deer were identical to those from Philippine long-tailed macaque in the Philippines (KY929113). Similarly, six ST2 sequences from NHPs (ring-tailed lemur, and white-cheeked gibbon) showed 100% identity with GenBank sequences MN585877 (from human in Brazil) and MF581567 (from human in Angola). ST3 and ST4 sequences had the largest identity (99.62% and 98.97%) related to that from human in China (MT645668), and human in Czech Republic (MT042813), with two and six nucleotide substitutions respectively. Meanwhile, one leopard and six ostrich-derived ST5 sequences had 98.95% identity with that from an ostrich in China (MK930351), with six single-nucleotide polymorphisms (SNPs). In terms of four pigeon-derived ST8 isolates, the sequences had 99.65% identity with that from human in Brazil (MN585867), with two nucleotide substitutions being observed. The sequences of ST10 and ST14 from alpaca were identical to the GenBank sequences MN316600 and MN316667, both from goat in Iran respectively. For 26 ST17 isolates, two representative sequences were obtained from NHPs, Asiatic black bear, and chinchillas. The sequence (MN227379) of ST17 isolates showed 100% identity to the sequence of ST17 isolated from a gundi in Libya (KC148208). The sequence (MN227380) exhibited 99.15% identity to the sequence of ST17 isolated from a chinchilla in China (MN124518), with five SNPs.
Phylogenetic analysis of Blastocystis sp.
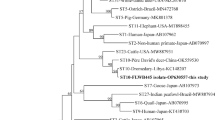
A total of 10 representative sequences were obtained from 66 Blastocystis sp. isolates in the present study. The sequences obtained in this study showed high identity with the reference sequences of Blastocystis sp. in GenBank. Newly acquired sequences belong to ST1, ST2, ST3, ST4, ST5, ST8, ST10, ST14, and ST17. ST1 and ST2 along with sequences originating from humans and Macaca clustered together. ST3 and ST8 grouped together with sequences mainly from humans. ST4 clustered together with sequences from rats and humans. ST14 along with sequences isolated from goat and cattle grouped together. ST5 along with sequences isolated from ostrich, cattle, and human clustered together. ST10 formed a clade with sequences from dog, cattle, and goat. Similarly, ST17 formed a separate branch with sequences from gundi and chinchilla (Fig. 2).

Phylogenetic relationships among nucleotide sequences of barcode regions of small subunit ribosomal RNA (SSU rRNA) of Blastocystis sp. The neighbor-joining method was used to construct the trees by the Kimura-2-parameter model. The numbers on the branches are percent bootstrapping values from 1000 replicates, with values > 50% shown in the tree. Each sequence is identified by its subtypes, hosts, accession number, and country. Blastocystis subtypes identified in the present study are indicated in bold type
Discussion
Blastocystis sp. is a common intestinal protozoan parasite with uncertain pathogenicity. It is believed that zoonotic STs often spread between animals and humans, as some STs of animal origin are a large potential reservoir for human infections [30,31,32]. Blastocystis sp. has been identified in many animal hosts, such as NHPs, pigs, cattle, sheep, goats, dogs, birds, and various captive wildlife [23, 24, 33]. The prevalence of Blastocystis sp. in zoo animals examined in this study was 15.7% (66/420), which was lower than that in captive wild animals in Qinling Mountains, China (40.2%, 200/497) [34], in zoo animals in the UK (35.5%, 82/231) [35], in zoo animals in Japan (39.0%, 46/118) [36], in various captive animals in France (32.2%, 99/307) [37], and in zoo animals in Western Australia (42%, 32/76) [5], while the Blastocystis sp. prevalence in the present study was higher than that in zoo animals in three cities in China (6.0%, 27/450) [27]. However, it is difficult to explain the discrepancy of Blastocystis sp. prevalence between different countries or within the same country because many factors contribute to the effect the prevalence, such as the size of examined samples, animal species, or management methods.
The prevalence of Blastocystis sp. among the four zoos was significantly different in the present study. The prevalence of Blastocystis sp. in Panzhihua Zoo was the highest (24.3%), which was significantly higher than that in Chengdu Zoo (6.9%) and Xichang Zoo (2.9%) (P < 0.05). The difference in the prevalence of Blastocystis sp. may be related to the zoo’s sanitary conditions and management methods. The sanitary conditions are relatively poor, and there is no good deworming and immunization program in the Panzhihua Zoo. In addition, the host and number of samples collected in each zoo are different, which may also be one of the reasons for the difference in Blastocystis sp. prevalences.
In the present study, 9 Blastocystis STs, including ST1-5, ST8, ST10, ST14, and ST17, were identified in 66 Blastocystis sp.-positive samples from captive wildlife. Of them, four subtypes (ST1-3, and ST17) were identified in NHPs (rhesus macaque, ring-tailed lemur, squirrel monkey, and white-cheeked gibbon). Generally, ST1-3 has been frequently determined in NHPs, while ST17 was identified to a lesser extent [34, 37]. Interestingly, a more recent study showed zoo animals and staff were infected with ST1-3 and showed high consistency with known sequences from NHPs and humans, highlighting that these STs have zoonotic potential [38]. In China, ST1, ST2, ST3, ST4, ST5, ST9, ST10, and ST13 were identified in NHPs [27, 31, 39, 40], with ST1 and ST2 being the most common. In this study, Blastocystis sp. was first discovered in squirrel monkey, and subtype analysis showed all isolates from squirrel monkey were ST17.
To date, several zoonotic STs have been identified in Carnivora, such as ST1-5, ST7-8, and ST10 [32, 33, 38]. In the current study, sequences obtained from Carnivora belonged to ST1, ST5, and ST17. These isolates were infecting the leopard (ST1 and ST5) and Asiatic black bear (ST17). As previously reported, the animals belonging to the order of Carnivora such as South American coati, Arctic fox, and dogs were determined to be infected with ST1 [38, 41]. Hussein et al. reported that ST1 inoculated into Wistar rats could cause moderate and severe degrees of pathological changes, suggesting the potential pathogenicity of this subtype [42]. ST5 was the most predominant subtype in pigs [43], but it was also identified in a various animals, such as NHPs, cattle, sheep, rodents, and birds [23]. However, ST5 was rarely found in carnivores, and only a few wild carnivores, such as Northern tiger cat, and meerkat were determined to have ST5 infection [27, 38]. In addition, ST5 infection has also been reported in stray and domestic dogs in India [44] and the Philippines [45]. ST17 has been identified in gundi in Libya [46] and in cattle in the USA [47]. While there is no study on ST17 infections in Carnivora, this is the first report of ST17 infection in Asiatic black bear in China, demonstrating a wider host range of this subtype.
Previous studies reported many animals in the order of Artiodactyla harboring the Blastocystis sp., such as pigs, cattle, sheep, goats, camels, and deer [23, 33]. The majority of STs (ST1-7, ST10, ST13-15, and ST17) have been identified in Artiodactyla to date [46, 48]. Among them, ST10 was the most common subtype in cattle in the US [49], Denmark [50], and China [51, 52]. In the present study, ST1 was found in sika deer. Several studies have reported on Blastocystis sp. infections in deer, with different subtype distributions. For example, sika deer, fallow deer, and white-lipped deer were reported to be infected with ST10 in China [34]. ST4 and ST10 were also reported in red deer, and muntjac deer were found with ST14 infection in the UK [53]. Strikingly, a rare ST (ST13) was also determined in a mouse deer in the UK [46] and in Java mouse-deer in France [37]. The distribution of STs in alpaca in the present study was consistent with a previous study in the Qinglin Mountains in China, in which all isolates identified belonged to ST10 and ST14 [34]. Similarly, ST10 infection in alpaca was also reported in French zoos [37]. Overall, these data suggest that deer and alpaca may serve as natural hosts of Blastocystis sp.
In this study, ST4 and ST17 were identified in rodents, corroborating previous data on pet rodents in Sichuan Province [54]. ST4 was originally isolated from a healthy Wistar rat in Singapore [55], and rodents were proposed to be a main reservoir of ST4. Recent studies confirmed that other STs, such as ST1-3, ST5, ST7, ST8, ST10, and ST17, can also be found in rodents [46, 53]. ST4 infection has been the reported for the first time in red giant flying squirrel and black great squirrel, expanding its host range. The observation of ST17 in shrew-faced squirrels in the United Arab Emirates suggests rodents may be the natural host of this subtype [56]. In the present study, ST17 was also observed in chinchilla in China for the first time to our knowledge, indicating a novel host for this subtype.
Regarding the non-mammalian groups, birds have already been considered potential reservoirs of Blastocystis sp. transmission to humans [57]. It is believed that birds usually harbor ST6 and ST7, which are considered ‘avian STs’ because of their predominance in this host group. Nevertheless, of the 11 avian isolates characterized in the present study, none were identified as belonging to the “avian” ST6 or ST7. Six of them belonged to ST5, four to ST8, and one to ST3; these results are similar to those of a previous study in birds in French zoos where ST5 was the dominant subtype [37]. It should be noted that our previous study reported that ST8 was the predominant subtype in captive birds in Sichuan Province [58], suggesting that this subtype may circulate among birds in the investigated areas. However, the transmission characteristics of these zoonotic subtypes warrant further examination in future studies.
Conclusions
The present study described the prevalence, subtype distribution and genetic characterizations of Blastocystis sp. for the first time in zoo animals in southwestern China. The data demonstrated that Blastocystis sp. could be maintained and transmitted between wildlife. Moreover, the zoonotic subtypes were identified in captive wildlife suggesting that these animals may serve as natural reservoirs for human Blastocystis sp. infections. The present results could provide fundamental information for the evaluation of potential zoonotic transmission between wildlife and humans.
Availability of data and materials
The nucleotide sequences generated in the present study have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/) under accession numbers MN227371, MN227378, MN227374, MN227376, MN227362, MN227363, MW078483, MW078491, MN227379, and MN227380. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- SSU rRNA:
-
Small subunit ribosomal RNA
- STs:
-
Subtypes
- IBS:
-
Irritable bowel syndrome
- IBD:
-
Inflammatory bowel disease
- PCR:
-
Polymerase chain reaction
- ORs:
-
Odds ratios
- NHPs:
-
Nonhuman primates
- SNPs:
-
Single nucleotide polymorphisms
References
Scanlan PD, Stensvold CR. Blastocystis: getting to grips with our guileful guest. Trends Parasitol. 2013;29:523–9.
Tan KSW. New insights on classification, identification, and clinical relevance of Blastocystis spp. Clin Microbiol Rev. 2008;21:639.
Li IL, Tan TC, Karmacharya BM, Govind SK. Blastocystis sp.: waterborne zoonotic organism, a possibility? Parasit Vectors. 2012;5:130–130.
Leelayoova S, Siripattanapipong S, Thathaisong U, Naaglor T, Taamasri P, Piyaraj P, et al. Drinking water: a possible source of Blastocystis spp. subtype 1 infection in schoolchildren of a rural community in central Thailand. Am J Trop Med Hyg. 2008;79:401–6.
Parkar U, Traub RJ, Vitali S, Elliot A, Levecke B, Robertson I, et al. Molecular characterization of Blastocystis isolates from zoo animals and their animal-keepers. Vet Parasitol. 2010;169:8–17.
Wang W, Owen H, Traub RJ, Cuttell L, Inpankaew T, Bielefeldtohmann H. Molecular epidemiology of Blastocystis in pigs and their in-contact humans in Southeast Queensland, Australia, and Cambodia. Vet Parasitol. 2014;203:264–9.
Stensvold CR, Clark CG. Pre-empting Pandora’s Box: Blastocystis subtypes revisited. Trends Parasitol. 2020;36:229–32.
Stensvold CR, Tan KSW, Clark CG. Blastocystis. Trends Parasitol. 2020;36:315–6.
Clark CG, van der Giezen M, Alfellani MA, Stensvold CR. Recent developments in Blastocystis research. Adv Parasitol. 2013;82:1–32.
Gentekaki E, Curtis BA, Stairs CW, Klimeš V, Eliáš M, Salas-Leiva DE, et al. Extreme genome diversity in the hyper-prevalent parasitic eukaryote Blastocystis. PLoS Biol. 2017;15:e2003769.
Ajjampur SS, Tan KS. Pathogenic mechanisms in Blastocystis spp.–interpreting results from in vitro and in vivo studies. Parasitol Int. 2016;65:772–9.
Yason JA, Liang YR, Png CW, Zhang Y, Tan KSW. Interactions between a pathogenic Blastocystis subtype and gut microbiota: in vitro and in vivo studies. Microbiome. 2019;7:30.
Rajamanikam A, Hooi HS, Kudva M, Samudi C, Kumar S. Resistance towards metronidazole in Blastocystis sp.: a pathogenic consequence. PLoS ONE. 2019;14:e0212542.
Andersen LO, Stensvold CR. Blastocystis in health and disease: Are we moving from a clinical to a public health perspective? J Clin Microbiol. 2016;54:524–8.
Scanlan PD, Stensvold CR, Rajilić-Stojanović M, Heilig HG, De Vos WM, O’Toole PW, et al. The microbial eukaryote Blastocystis is a prevalent and diverse member of the healthy human gut microbiota. FEMS Microbiol Ecol. 2014;90:326–30.
Audebert C, Even G, Cian A, Group BI, Loywick A, Merlin S, et al. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci Rep. 2016; 6: 25255.
Shirvani G, Fasihi-Harandi M, Raiesi O, Bazargan N, Zahedi MJ, Sharifi I, et al. Prevalence and molecular subtyping of Blastocystis from patients with irritable bowel syndrome, inflammatory bowel disease and chronic urticaria in Iran. Acta Parasitol. 2020;65:90–6.
Peña S, Carrasco G, Rojas P, Castillo D, Ozaki LS, Mercado R. Determination of subtypes of Blastocystis sp. in chilean patients with and without inflammatory bowel syndrome, A preliminary report. Parasite Epidemiol Control. 2020;8:e00125.
Wu Z, Mirza H, Tan KS. Intra-subtype variation in enteroadhesion accounts for differences in epithelial barrier disruption and is associated with metronidazole resistance in Blastocystis subtype-7. PLoS Negl Trop Dis. 2014;8:e2885.
Mirza H, Wu Z, Teo JD, Tan KS. Statin pleiotropy prevents rho kinase-mediated intestinal epithelial barrier compromise induced by Blastocystis cysteine proteases. Cell Microbiol. 2012;14:1474–84.
Puthia MK, Sio SW, Lu J, Tan KS. Blastocystis ratti induces contact-independent apoptosis, F-actin rearrangement, and barrier function disruption in IEC-6 cells. Infect Immun. 2006;74:4114–23.
Rauff-Adedotun AA, Mohd Zain SN, Farah Haziqah MT. Current status of Blastocystis sp. in animals from Southeast Asia: a review. Parasitol Res. 2020; 1–12.
Jiménez PA, Jaimes JE, Ramírez JD. A summary of Blastocystis subtypes in North and South America. Parasit Vectors. 2019;12:376.
Mohammadpour I, Bozorg-Ghalati F, Gazzonis AL, Manfredi MT, Motazedian MH, Mohammadpour N. First molecular subtyping and phylogeny of Blastocystis sp. isolated from domestic and synanthropic animals (dogs, cats and brown rats) in southern Iran. Parasit Vectors. 2020;13:365.
Li W, Deng L, Yu X, Zhong Z, Wang Q, Liu X, et al. Multilocus genotypes and broad host-range of Enterocytozoon bieneusi in captive wildlife at zoological gardens in China. Parasit Vectors. 2016;9:395.
Li J, Qi M, Chang Y, Wang R, Li T, Dong H, et al. Molecular characterization of Cryptosporidium spp., Giardia duodenalis, and Enterocytozoon bieneusi in captive wildlife at Zhengzhou Zoo. China J Eukaryot Microbiol. 2015;62:833–9.
Li XD, Zou Y, Pan J, Liang QL, Zeng Z, Meng YM, et al. Prevalence and subtypes of Blastocystis sp. infection in zoo animals in three cities in China. Parasitol Res. 2020;119:465–71.
Scicluna SM, Tawari B, Clark CG. DNA Barcoding of Blastocystis. Protist. 2006;157:77–85.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Sharifi Y, Abbasi F, Shahabi S, Zaraei A, Mikaeili F, Sarkari B. Comparative genotyping of Blastocystis infecting cattle and human in the south of Iran. Comp Immunol Microbiol Infect Dis. 2020;72:101529.
Zhu W, Wei Z, Li Q, Lin Y, Yang H, Li W. Prevalence and subtype diversity of Blastocystis in human and nonhuman primates in North China. Parasitol Res. 2020;119:2719–25.
Li J, Karim MR, Li D, Rahaman Sumon SMM, Siddiki S, Rume FI, et al. Molecular characterization of Blastocystis sp. in captive wildlife in Bangladesh National Zoo: non-human primates with high prevalence and zoonotic significance. Int J Parasitol Parasites Wildl. 2019;10:314–20.
Deng L, Chai Y, Zhou Z, Liu H, Zhong Z, Hu Y, et al. Epidemiology of Blastocystis sp. infection in China: a systematic review. Parasite. 2019;26:41.
Zhao GH, Hu XF, Liu TL, Hu RS, Yu ZQ, Yang WB, et al. Molecular characterization of Blastocystis sp. in captive wild animals in Qinling Mountains. Parasitol Res. 2017;116:2327–33.
Betts EL, Gentekaki E, Tsaousis AD. Exploring micro-eukaryotic diversity in the gut: Co-occurrence of Blastocystis subtypes and other protists in zoo animals. Front Microbiol. 2020;11:288.
Abe N, Nagoshi M, Takami K, Sawano Y, Yoshikawa H. A survey of Blastocystis sp. in livestock, pets, and zoo animals in Japan. Vet Parasitol. 2002;106:203–12.
Cian A, El Safadi D, Osman M, Moriniere R, Gantois N, Benamrouz-Vanneste S, et al. Molecular epidemiology of Blastocystis sp. in various animal groups from two French zoos and evaluation of potential zoonotic risk. PLoS ONE. 2017;12:e0169659.
Oliveira-Arbex AP, David ÉB, Tenório MDS, Cicchi PJP, Patti M, Coradi ST, et al. Diversity of Blastocystis subtypes in wild mammals from a zoo and two conservation units in southeastern Brazil. Infect Genet Evol. 2020;78:104053.
Ma L, Qiao H, Wang H, Li S, Zhai P, Huang J, et al. Molecular prevalence and subtypes of Blastocystis sp. in primates in northern China. Transbond Emerg Dis. 2020;26:46.
Li TC, Li Z, Zhang YL, Chen WJ, Dong XL, Yang JF, et al. Assessment of the subtypes and the zoonotic risk of Blastocystis sp. of experimental macaques in Yunnan province, southwestern China. Parasitol Res. 2020;119:741–8.
Wang J, Gong B, Liu X, Zhao W, Bu T, Zhang W, et al. Distribution and genetic diversity of Blastocystis subtypes in various mammal and bird species in northeastern China. Parasit Vectors. 2018;11:522.
Hussein EM, Hussein AM, Eida MM, Atwa MM. Pathophysiological variability of different genotypes of human Blastocystis hominis Egyptian isolates in experimentally infected rats. Parasitol Res. 2008;102:853–60.
Song JK, Hu RS, Fan XC, Wang SS, Zhang HJ, Zhao GH. Molecular characterization of Blastocystis from pigs in Shaanxi province of China. Acta Trop. 2017;173:130–5.
Wang W, Cuttell L, Bielefeldt-Ohmann H, Inpankaew T, Owen H, Traub RJ. Diversity of Blastocystis subtypes in dogs in different geographical settings. Parasit Vectors. 2013;6:215.
Belleza ML, Cadacio JL, Borja MP, Solon JA, Padilla MA, Tongol-Rivera PN, et al. Epidemiologic study of Blastocystis infection in an urban community in the Philippines. J Environ Publ Heal. 2015;2015:894297.
Alfellani MA, Taner-Mulla D, Jacob AS, Imeede CA, Yoshikawa H, Stensvold CR, et al. Genetic diversity of Blastocystis in livestock and zoo animals. Protist. 2013;164:497–509.
Maloney JG, Lombard JE, Urie NJ, Shivley CB, Santin M. Zoonotic and genetically diverse subtypes of Blastocystis in US pre-weaned dairy heifer calves. Parasitol Res. 2019;118:575–82.
Wawrzyniak I, Poirier P, Viscogliosi E, Dionigia M, Texier C, Delbac F, et al. Blastocystis, an unrecognized parasite: an overview of pathogenesis and diagnosis. Therap Adv Infect Dis. 2013;1:167.
Ronald F, Monica S, Dumitru M. Detection of concurrent infection of dairy cattle with Blastocystis, Cryptosporidium, Giardia, and Enterocytozoon by molecular and microscopic methods. Parasitol Res. 2012;111:1349–55.
Stensvold CR, Alfellani MA, Nørskov-Lauritsen S, Prip K, Victory EL, Maddox C, et al. Subtype distribution of Blastocystis isolates from synanthropic and zoo animals and identification of a new subtype. Int J Parasitol. 2009;39:473–9.
Zhu W, Tao W, Gong B, Yang H, Li Y, Song M, et al. First report of Blastocystis infections in cattle in China. Vet Parasitol. 2017;246:38–42.
Wang J, Gong B, Yang F, Zhang W, Zheng Y, Liu A. Subtype distribution and genetic characterizations of Blastocystis in pigs, cattle, sheep and goats in northeastern China’s Heilongjiang Province. Infect Genet Evol. 2018;57:171–6.
Betts EL, Gentekaki E, Thomasz A, Breakell V, Carpenter AI, Tsaousis AD. Genetic diversity of Blastocystis in non-primate animals. Parasitol. 2018;145:1228–34.
Chai Y, Deng L, Liu H, Yao J, Zhong Z, Fu H, et al. First subtyping of Blastocystis sp. from pet rodents in southwestern China. Int J Parasitol Parasites Wildl. 2020;11:143–8.
Chen XQ, Singh M, Ho LC, Moe KT, Tan SW, Yap EH. A survey of Blastocystis sp. in rodents. Lab Anim Sci. 1997;47:91–4.
AbuOdeh R, Ezzedine S, Madkour M, Stensvold CR, Samie A, Nasrallah G, et al. Molecular subtyping of Blastocystis from diverse animals in the United Arab Emirates. Protist. 2019;170:125679.
Noël C, Dufernez F, Gerbod D, Edgcomb VP, Delgado-Viscogliosi P, Ho LC, et al. Molecular phylogenies of Blastocystis isolates from different hosts: implications for genetic diversity, identification of species, and zoonosis. J Clin Microbiol. 2005;43:348–55.
Deng L, Yao JX, Liu HF, Zhou ZY, Chai YJ, Wang WY, et al. First report of Blastocystis in giant pandas, red pandas, and various bird species in Sichuan province, southwestern China. Int J Parasitol ParasitesWildl. 2019;9:298–304.
Acknowledgements
We thank Wuyou Wang for the commets on the manuscript and Tong Jie Xin for critical eiditing. Lei Deng was the recipient of a scholarship from the Chinese Scholarship Council (CSC).
Funding
This work was funded by the Chengdu Giant Panda Breeding Research Foundation (CPF2017-12) and Special Fund for Forestry Reform and Development of Wolong Special Administrative Region of Sichuan Province (510000-02-064387).
Author information
Authors and Affiliations
Contributions
This study was conceived and designed by GP. Experiments were performed by LD, JY, and SC. Fecal samples were collected by TH, XS, ZZ (Zhijun Zhong). and YC. Data were analyzed by ZZ (Ziyao Zhou), HF, and HL. All authors have read and approved the submitted version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study protocol was reviewed and approved by the Research Ethics Committee and the Animal Ethical Committee of Sichuan Agricultural University. Permission was obtained from zoo managers prior to collection of fecal specimens.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deng, L., Yao, J., Chen, S. et al. First identification and molecular subtyping of Blastocystis sp. in zoo animals in southwestern China. Parasites Vectors 14, 11 (2021). https://doi.org/10.1186/s13071-020-04515-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04515-2