
Abstract
Background
Triatomine bugs are vectors of the protozoan parasite Trypanosoma cruzi, which causes Chagas disease. Rhodnius pallescens is a major vector of Chagas disease in Panama. Understanding the microbial ecology of disease vectors is important in the development of vector management strategies that target vector survival and fitness. In this study we examined the whole-body microbial composition of R. pallescens from three locations in Panama.
Methods
We collected 89 R. pallescens specimens using Noireau traps in Attalea butyracea palms. We then extracted total DNA from whole-bodies of specimens and amplified bacterial microbiota using 16S rRNA metabarcoding PCR. The 16S libraries were sequenced on an Illumina MiSeq and analyzed using QIIME2 software.
Results
We found Proteobacteria, Actinobacteria, Bacteroidetes and Firmicutes to be the most abundant bacterial phyla across all samples. Geographical location showed the largest difference in microbial composition with northern Veraguas Province having the most diversity and Panama Oeste Province localities being most similar to each other. Wolbachia was detected in high abundance (48–72%) at Panama Oeste area localities with a complete absence of detection in Veraguas Province. No significant differences in microbial composition were detected between triatomine age class, primary blood meal source, or T. cruzi infection status.
Conclusions
We found biogeographical regions differ in microbial composition among R. pallescens populations in Panama. While overall the microbiota has bacterial taxa consistent with previous studies in triatomine microbial ecology, locality differences are an important observation for future studies. Geographical heterogeneity in microbiomes of vectors is an important consideration for future developments that leverage microbiomes for disease control.
Similar content being viewed by others


Background
Insect microbiota are composed of a wide variety of microbial species [1, 2] that serve as commensals, pathogens, or have mutualistic benefits that impact the reproduction, nutrition, and immune systems of the insect host [1,2,3,4]. The symbiotic relationship between an insect disease vector and its microbiota can have an important influence on the competence and transmission potential of human diseases [4, 5], increasing or decreasing pathogen transmission from vector to host [6], including in blood-feeding species [3, 7]. Insect microbiota research can lead to improved methods of vector control [8,9,10,11,12,13], with substantial research in some mosquito species, but limited research in many other vector species, often conflicting results, leaves many questions unanswered [6]. Vector life stage, distribution, species, methods/sampling strategies and environment (e.g. habitat type or geographical region) may influence vector microbiota [6, 14,15,16,17,18,19,20,21,22,23,24,25].
In this study, we evaluated patterns of whole-body microbiota of a Chagas disease vector. Chagas disease, caused by the kinetoplastid protozoan parasite Trypanosoma cruzi, is transmitted between a wide range of potential mammalian hosts and humans by hematophagous (blood-feeding) triatomine insect vectors. Despite widespread control programmes, Chagas disease remains a significant health threat to millions of inhabitants in Latin America, particularly those that live in poverty [26]. The idea of using bacterial symbionts of triatomine bugs to control Chagas disease has long been proposed [27]. Recent studies describe microbial community composition within triatomines [23, 28,29,30,31,32,33], including R. pallescens from Colombia [22] and Panama [34], and a sister species, R. prolixus [35]. Studies thus far describe triatomine microbiota as having low complexity in terms of diversity and species-specific patterns [23, 29, 30], yet the microbiota for many taxa remain to be studied.
Infection of triatomines with trypanosomes has been associated with reduction in gut microbial diversity [28, 35], and blood meal identity may influence composition of the predominant bacterial taxa [30, 31]. However, other important comparisons among triatomines are lacking, such as differences between location and habitat type. Microbial composition variation between different geographical locations has been observed in ticks [15, 18, 20, 24, 25] and with mixed observations in mosquitoes [16, 36]. Habitat has also been shown to be a main driver of microbial species composition in mosquitoes [37, 38].
There are more than 150 species of triatomines, with different distributions, habitat requirements and life histories that can impact microbiota, or microbiota that can affect vectorial capacity. This complexity requires extensive research into different triatomine microbiomes. Currently, we still lack basic microbial community composition descriptions for many triatomine species and these large gaps in our knowledge make informed research for vector biocontrol difficult. Therefore, advancing research in triatomine microbiota is crucial for gaining a better understanding of T. cruzi infection, triatomine vector capacity, and developmental biology.
Rhodnius pallescens is the main vector Trypanosoma cruzi and T. rangeli in Panama [39] where they are widely distributed throughout Panama and into the neighboring countries of Costa Rica and Colombia. Rhodnius pallescens is commonly found and associated with Attalea butyracea palms [40, 41] that are common and widely distributed across many different habitats in Panama [40, 41]. The sylvatic behavior has complicated control efforts [42]. New endemic regions are still being described [43,44,45] and a darker chromatic variation of R. pallescens associated with distinct genetic groups of T. cruzi and T. rangeli has also been found recently in Santa Fe District, Panama [44].
Here, we describe the whole-body bacterial microbiota of wild-caught R. pallescens from three separate geographical locations in Panama. We used the entire triatomine body to encompass all potential microbial taxa relevant to R. pallescens that could affect their fitness and survival as a benchmark for future studies of localized anatomy microbiota. We hypothesize that both habitat type and geographical location will be associated with differences in whole-body microbiota composition similar to previous findings in other vectors [15, 16, 18, 20, 24, 25, 37, 38]. We further hypothesize that complex environments, such as forest patches, will be associated with a more diverse microbiota composition in insects in more homogenous environments (i.e. cattle pastures), as previous studies in insects show that environmental diversity leads to increased microbial diversity [46,47,48]. We used Illumina 16S rRNA amplicon sequencing to characterize and evaluate the bacterial microbiota of R. pallescens between different locations and habitats, comparing infection status, age class, and primary blood-meal source to evaluate a range of variables that may be associated with whole-body bacterial community composition.
Methods
Sample collection and DNA extraction
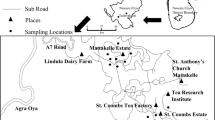
All R. pallescens evaluated specimens (n = 89) were collected in Panama, Central America using Noireau traps [49] in Attalea butyracea palms (the main habitat of this species) and placed directly in 95% molecular grade ethanol before use. We sampled from a total of 8 palms, in three habitats (pasture, peridomestic and peridomestic-forest), from three geographic locations in lowland moist tropical forest (Las Pavas and Trinidad de las Minas) and moist tropical forest (Santa Fe and Veraguas) (Fig. 1). We consider peridomestic to be home yards or areas within 100 m of a dwelling and peridomestic-forest to be patches of regenerated forest within a peridomestic landscape matrix. Samples from Las Pavas, La Chorrera District (9.104167° N, 79.885833° W) (n = 27 from two habitats) and Trinidad de las Minas, Capira District (8.775556° N, 79.995833° W) (n = 32, from one habitat) were from a previous study examining blood meals [50]. We further collected 30 samples from four sites comprising three habitats located in Santa Fe District, Veraguas (8.509232° N, 81.077800° W) from 8–11 July 2017. The Santa Fe region has recently been described as a new endemic focus for Chagas disease in Panama, where a dark morph of R. pallescens predominates [43, 44]. All specimens were nymphs, primarily N3 and below (92%, 95% CI: 84.4–96.39%), with the exception of one male from Trinidad de las Minas (Additional file 1: Table S1). DNA was extracted from whole specimens following Kieran et al. [50]. Briefly, samples were macerated and digested overnight in digest buffer with proteinase K and extracted with phenol-chloroform-isoamyl alcohol. Extractions were reconstituted in TLE buffer (10 mM Tris, pH 8; 0.1 mM EDTA), and impurities were removed with Sera-Mag SpeedBeads™ (Thermo Fisher Scientific, Waltham, MA, USA; [51]) with a final reconstitution in 30 µl TLE.

Map of Panama showing the locations of the three collection sites
DNA amplification and sequencing
We amplified bacterial 16S rRNA DNA using the S-D-Bact-0341-b-S-17 (5′-CCT ACG GGN GGC WGC AG-3′) forward and S-D-Bact-0785-a-A-21 (5′-GAC TAC HVG GGT ATC TAA TCC-3′) reverse primer pair [52] to which we added modifications following previous studies [50, 53, 54]. We added Illumina TruSeq sequences to the 5′-end of the forward (Read 1) and reverse (Read 2) primer creating fusion primers. We synthesized 8 forward and 12 reverse fusion primers, each with a unique variable length (5–8 bp) index sequence between the 16S and TruSeq sequences. We then performed two rounds of PCR. For the first-round we performed replicate PCRs using 12.5 μl reactions of KAPA HiFi HotStart Kits (Kapa Biosystems, Wilmington, MA, USA) consisting of 2.5 μl of 5× buffer, 0.375 μl of 10 mM dNTPs, 0.25 μl hot start Taq, 5.4 μl molecular grade water, 1 μl of 5 μM forward primer, 1 μl of 5 μM reverse primer, and 2 μl of DNA. The ranges of DNA sample concentrations were from 10 ng/μl to 60 ng/μl. Each DNA sample had a unique primer-index combination with the following thermocycler conditions: 98 °C for 3 min, followed by 30 cycles at 95 °C for 30 s, 63 °C for 1 min, 72 °C for 1 min and a final extension at 72 °C for 5 min. Amplification success was verified on a 1.5% agarose gel.
Amplicons were pooled in equal concentrations and cleaned using a 1:1 ratio of SPRI-beads and reconstituted in 25 μl TLE. The second-round PCR primers consisted of Illumina TruSeqHT compatible 8 nt indexed primers [55]. We used 25 μl reaction of KAPA HiFi HotStart Kits using 5 μl of 5× Buffer, 0.75 μl of 10 mM dNTPs, 0.5 μl HotStart, 3.75 μl molecular grade water, 2.5 μl of 5 μM forward primer, 2.5 μl 5 μM reverse primer, and 10 μl of 16S amplicon pool. We performed two replicate PCRs with the following thermocycler conditions: 98 °C for 2 min, followed by 10 cycles at 98 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s and a final extension at 72 °C for 5 min. Library product was cleaned with Sera-Mag SpeedBeads™ (1:1 ratio) and pooled with other uniquely indexed samples prior to sequencing.
For blood-meal source data, we used previous data from Kieran et al. [50] and for newly collected samples, we amplified 12S rRNA gene following Kieran et al. [50]. All libraries were sent to the Georgia Genomics and Bioinformatics Core (http://dna.uga.edu) for sequencing on an Illumina MiSeq using a v3 PE300 kit (Illumina, San Diego, CA, USA). We also screened for the presence of T. cruzi and T. rangeli amplifying telomeric kinetoplastid DNA with Tc189 and Tr primers [56]. Samples were also verified for T. cruzi using 121/122 primers targeting the kinetoplastid minicircle [57]. Amplification success was verified on a 1.5% agarose gel.
Data processing and analysis
We demultiplexed the amplicon indices using Mr. Demuxy 1.2.0 (https://pypi.org/project/Mr_Demuxy/) and resulting fastq files were imported into Geneious 10.0.1 [58]; (https://www.geneious.com) where we trimmed primers, paired and merged the reads using FLASH [59]. Subsequent data were exported as fastq files for importation into Qiime2 [60]. The quality of the sequences was checked and filtered using QIIME2 v. 2018.8 plugin DADA2 [61] and chimeric sequences were removed. The remaining forward sequences were truncated to a final length of 292 bp and the reverse sequences were truncated to a final length of 240 bp. Amplicon sequence variants (ASV) were analyzed using the q2-diversity Qiime2 plugin to calculate multiple alpha diversity metrics, including Shannonʼs index H’, Simpsonʼs index Ds, Chao1, Faith’s phylogenetic diversity, and observed-ASV’s.
The Qiime2 plugin q2-phylogeny was used to complete a multiple sequence alignment to reconstruct rooted and unrooted phylogenetic trees from the filtered alignment based on maximum-likelihood approximation with FastTree 2 [62]. Alpha and beta diversity metrics were assigned using the q2-diversity Qiime2 plugin. An alpha rarefaction was used to evaluate sampling depth, and the data was rarefied at 1000 sequences per sample, removing two samples and retaining 87 samples for final analyses.
The Qiime2 plugin q2-feature-classifier was used to align the sequences against the Greengenes 13.8 database [63]. OTUs were identified from phyla down to the genus level, we removed archaea, chloroplasts, mitochondria, not available (NAs), and uncharacterized taxa at the kingdom level.
Alpha diversity (species diversity) was calculated using Shannonʼs (species richness), Simpson’s (evenness or relative abundance), and Chao1 (estimate of diversity from abundance) diversity metrics. To compare alpha diversities from individuals across location, habitat type, and infection status, a one‐way analysis of variance (ANOVA) and post‐hoc Tukeyʼs honest significant difference (HSD) tests for multiple comparisons were performed to evaluate differences in taxonomic abundance and alpha diversities. A P-value less than 0.05 was considered statistically significant.
Beta diversity (compositional variation) was calculated for the whole-body microbiota comparison between triatomines across location, habitat type, and infection status using Bray-Curtis dissimilarity. Bray-Curtis is based on shared OTU counts between individuals. Finally, we used non-metric multidimensional scaling (nMDS) [64] to visualize differences between the microbial communities, and a permutational MANOVA for hypothesis testing [65]. All diversity analyses and visualizations were conducted using qiime2 artifact outputs in R (v. 3.5.1) and with the packages phyloseq [66], vegan [67], dplyr [68], ggplot2 [69] and metacoder [70]. To further assess our findings of a location effect we removed older age classes (N4, N5, Adult) and repeated the analysis (n = 80).
Results
16S rRNA sequences and classification of entire microbiota community
We obtained a total of 4,995,733 16S rRNA V3–V4 region sequences from 101 samples, including the negative controls of molecular grade water and positive controls of E. coli. After quality filtering and the exclusion of 14 samples due to low read numbers, the number of sequences obtained per sample ranged from 1000 to 34,792 reads, with a mean frequency of 9811.63. The total number of OTUs within the 87 final samples was 4033, with the top 4 phyla consisting of Proteobacteria (60.67%), Actinobacteria (16.93%), Bacteroidetes (9.55%) and Firmicutes (4.11%) out of the total phyla present in the dataset. Rhodnius pallescens, in Las Pavas and Trinidad de las Minas respectively, is primarily composed of Proteobacteria (71.63% and 76.11%, respectively), Actinobacteria (6.13% and 15.88%, respectively), and Bacteroidetes (13.98% and 2.66%, respectively) (Fig. 2, Additional file 2: Table S2, Additional file 3: Figure S1). This contrasts with specimens from northern Veraguas with the most abundant phylum shifting from Proteobacteria (26.43%) to Actinobacteria (27.56%) and introducing more Firmicutes (11.48%). At the family-level (Fig. 3, Additional file 3: Figure S1), the top 3 taxa overall were Anaplasmataceae (45.76%), Pseudonocardiaceae (6.04%), Moraxellaceae (2.77%), although these relative proportions differ by location (Additional file 4: Table S3, Additional file 3: Figure S1). Most notably, as seen in Fig. 3, Anaplasmataceae was the dominant family throughout samples from Las Pavas (48.30%) and Trinidad de las Minas (72.51%) (Fig. 4, Additional file 5: Table S4, Additional file 3: Figure S1) but is not present within samples collected in northern Veraguas. The high abundance of Anaplasmataceae was due to a single genus, Wolbachia spp., comprising greater than 70% and 42% of the composition in more than half the specimens from Trinidad de las Minas and Las Pavas, respectively (Additional file 6: Table S5). No differences in microbial composition were detected between triatomine age class (Shannon, F(5, 85) = 1.07, P > 0.09) or primary blood-meal source (Shannon, F(9, 77) = 1.07, P > 0.38).

Taxonomic composition at the phylum level by location

Top 20 taxonomic composition per location at the family level

Top 20 taxonomic composition per location at the genus level
Infection rates
Trypanosma cruzi and T. rangeli were detected in sampled vectors at all locations (Additional file 1: Table S1). Rates of positive T. cruzi infection were 7.41% (2/27, 95% CI: 0.96–24.47%) at Las Pavas (n palms = 2), 75% (24/32, 95% CI: 57.67–86.97%) at Trinidad de las Minas (n palms = 2), and 46.67% (14/30, 95% CI: 30.23–63.46%) at Santa Fe District, Veraguas (n palms = 4). When examining T. cruzi infection by habitat we find rates of 12.5% (1/8, 95% CI: 0.11–49.2%) in Las Pavas pasture compared to 50% (8/16, 95% CI: 28–72%) in Veraguas pasture. In peridomestic areas, rates varied from 5.26% (1/19, 95% CI: < 0.01–26.48%) in Las Pavas and 42.85% (3/7, 95% CI: 15.75–75.02%) in Veraguas. Peridomestic-forest habitat in Veraguas had an infection rate of 42.85% (3/7, 95% CI: 15.75–75.02%).
For T. rangeli, positive detection rates were 66.67% (18/27, 95% CI: 47.71–81.47%) in Las Pavas, 78.13% (25/32, 95% CI: 60.96–89.27%) in Trinidad de las Minas, and 30% (9/30, 95% CI: 16.52–48.02) in Veraguas. There were low rates of T. rangeli infection in both Las Pavas (0%) and Veraguas (6.25%, 1/16, 95% CI: < 0.01–30.31%) pastures, while peridomestic areas were high (94.74%, 18/19, 95% CI: 73.52– > 99.99%) to moderate (28.57%, 2/7, 95% CI: 7.56–64.76%) at each respective location. Veraguas peridomestic-forest infection rate was 85.7% (6/7, 95% CI: 46.65–99.47).
There was an overall rate of coinfections of both trypanosomes of 31.46% (28/89, 95% CI: 22.72–41.73%). Coinfections were less abundant at the lower infection sites of Las Pavas (3.7%, 95% CI: < 0.01–19.8%) and Veraguas (13.33%, 95% CI: 4.7–30.3%) compared to Trinidad de las Minas which had a higher coinfection rate (71.88%, 95% CI: 54.46–84.6%). At the habitat level, coinfection rates varied from non-existent (0%) in pastures, to low (5.26%, 1/19, 95% CI: < 0.01–26.48%) and moderate (28.57%, 2/7, 95% CI: 7.56–64.76) in peridomestic areas for Las Pavas and Veraguas, respectively. Coinfection rate for peridomestic-forest was the same as peridomestic areas in Veraguas.
Location, habitat type, and infection status on microbial composition
Alpha diversity
Alpha richness between locations (Additional file 7: Figure S2) was significantly different using three different diversity metrics (ANOVA, F(2, 85) = 12.16, P < 0.001; Shannon’s index H’ ANOVA, F(2, 85) = 11.7, P < 0.001; Chao1 ANOVA, F(2, 85) = 12.04, P < 0.001) even when older aged individuals were removed (ANOVA, F(2, 77) = 10.89, P < 0.001; Shannon’s index H’ ANOVA, F(2, 77) = 11.38, P < 0.001; Chao1 ANOVA, F(2, 77) = 10.78, P < 0.001). Individuals from Veraguas had significantly greater alpha richness when compared to individuals from Trinidad de las Minas (Chao1, Shannon, TukeyHSD, P < 0.0001; Simpson, Tukey HSD P = 0.008) and Las Pavas (TukeyHSD, P < 0.0004, Chao1, TukeyHSD, P = 0.0004; Shannon, TukeyHSD, P = 0.0096). These results remained the same for all metrics when older age classes were removed (P < 0.001 for Trinidad de las Minas; P < 0.0093 for Las Pavas). Alpha richness across habitat type (Additional file 8: Figure S3) showed significance for one metric (Shannon, TukeyHSD, F(2, 85) = 4.72, P = 0.011; older classes removed F(2, 77) = 2.24, P = 0.006) between peridomestic and peridomestic-forest types (P = 0.009; older classes removed P = 0.004). This trend holds when examining habitat across the Veraguas site (P = 0.001). Trypanosoma cruzi infection status (Additional file 9: Figure S4), however, was not significantly different (Simpson, F(2, 78) = 3.54, P > 0.063).
Beta diversity
All PERMANOVA results are reported using Bray-Curtis dissimilarity indices. The community differences (beta diversity) of triatomine microbiota showed significant differences at pasture sites across Trinidad de las Minas and Veraguas (PERMANOVA, F(1, 20) = 5.61, P = 0.001; Additional file 10: Figure S5) and at peridomestic sites across all three locations (PERMANOVA, F(2, 61) = 12.09, P = 0.001; Additional file 11: Figure S6). Community differences across the three habitat types also showed some significant differences within Veraguas (PERMANOVA, F(2, 25) = 1.41, P = 0.001; Additional file 12: Figure S7) and Las Pavas (PERMANOVA, F(1, 25) = 3.96, P = 0.001; Additional file 13: Figure S8). There was no observed difference between pasture habitat composition between sites (PERMANOVA, F(1, 22) = 1.88, P = 0.818). When examining T. cruzi infection status, the only significant differences in microbial composition between T. cruzi-positive and negative samples were observed at Las Pavas (PERMANOVA, F(2, 24) = 1.31, P = 0.029; Additional file 14: Figure S9); however, positive infection was extremely low (2/27). Microbial composition and T. cruzi infection status was also significant between peridomestic habitats among all locations (PERMANOVA, F(1, 56) = 2.21, P = 0.01, Additional file 15: Figure S10). Sites at Trinidad de las Minas (PERMANOVA, F(2, 29) = 1.59, P = 0.65; Additional file 16: Figure S11) and Veraguas (PERMANOVA, F(2, 25) = 1.29, P = 0.76; Additional file 17: Figure S12) did not show significant compositional difference between infected and non-infected samples. Results with the removal of older age classes were not substantially different. The lack of effect of older nymphal stages is further supported by multivariate PCA in Additional file 18: Figure S13.
Discussion
Here, we characterized the bacterial microbiota of 87 wild individuals of the Chagas disease vector Rhodnius pallescens from three populations in Panama. We explored comparisons in composition between location, microhabitat, nymphal stage, T. cruzi infection, and blood-meal status. Overall, the microbiota of R. pallescens exhibited relatively low complexity in its bacterial composition which is consistent with other triatomine studies [23, 29, 30]. Proteobacteria has also been found to be the most abundant phylum in other vector species [1, 71,72,73,74,75] including the triatomines R. neglectus, R. prolixus, Triatoma vitticeps, T. infestans, T. brasiliensis, T. pseudomaculata, Dipetalogaster maximus and Panstrongylus megistus [29, 30, 73], while a predominance of Actinobacteria has been found previously in R. pallescens [22], both consistent with our findings.
Common bacterial genera found in other triatomines include Burkholderia, Dietzia, Gordonia, Williamsia [22, 30, 73], Actinomycetospora, Arsenophonus, Corynebacterium, Rhodococcus, Staphylococcus [23, 30, 31], and Enterococcus, Enterobacteriaceae, Bacillus [23, 33]. Of these only Actinomycetospora was one of the top 20 genera found across all studied sites (Additional file 5: Table S4). Enterobacteriaceae and Bacillus were found at all sites, but at much lower abundance (0.34–6.03% and 0.21–0.76%, respectively) than found by Waltmann et al. (36.7% and 2%, respectively) [33]; however, the present study differs in that we analyzed wild and not laboratory-reared samples. Dietzia and Gordonia were each in the top 20 taxa for Las Pavas and Trinidad de las Minas (Additional file 5: Table S4). Arsenophonus was detected with a very low abundance in only a single specimen from northern Veraguas Province. All other taxa were found across all sites, but at lower abundance (Additional file 19: Table S6) than in other studies.
High levels of Proteobacteria observed in specimens from Panama Oeste Province localities are due to the very high levels of Wolbachia sp. Specimens from northern Veraguas Province, interestingly, did not have any Wolbachia present. It has been estimated that Wolbachia infects 52% of all aquatic insect species [76] and can infect a high proportion of the number of individuals in a species [77]. However, while many arthropod species may be infected with Wolbachia, a majority of the individuals within a species may not be. In a comparative study, Sazama et al. [76] found that less than half of the individuals were infected in most (69%) Wolbachia-infected species. Wolbachia has been found previously in Rhodnius sp. [29, 34] and is common in hematophagous insects [78], but has not been found in other triatomines [29, 31, 73]. In triatomines [29] and sandflies [74, 79, 80], the role of Wolbachia remains unknown. In mosquitoes, Wolbachia can affect reproduction and insecticide resistance among others [7], thus creating opportunities for vector biocontrol. However, without further knowledge of the role of Wolbachia in triatomines, identifying microbes that can serve as effective control agents for triatomines requires further research. To this end, we need more characterized microbiomes under multiple environmental conditions, and functional analysis of microbial taxa.
In our study, geographical location was associated with differences between microbial communities of R. pallescens. This observation is most evident between the two most disparate geographical locations (northern Veraguas vs Panama Oeste localities). This observation may be the result of quite different environments between these locations. Veraguas Province is located in the highlands of the western isthmus of Panama where the climate is cooler (mean temperature of 21 °C) and more humid with mountainous topography. In the two Panama Oeste area locations (Las Pavas and Trinidad de las Minas) the topography is flatter with warmer temperatures (mean 27 °C). Of particular interest is that the evaluated specimens from Santa Fe District in northern Veraguas correspond with a darker chromatic variation of R. pallescens infected by specific genetic groups of T. rangeli and T. cruzi [44]. Although the genetic characteristics of this population have not been studied, the reported phenotypic differences and the differences found in their microbial composition could be explained by the presence of this dark chromatic variant in this geographical region. However, it is not known if this dark variant represents a separate geographical population, a new subspecies, or a new distinct species of R. pallescens.
Geographical differences in microbiota have been observed in ticks [15, 18, 20, 24, 25], but not in mosquitoes where species-specific microbiota is thought to be stable [36]. One study in triatomines did not observe any difference in the microbiota between three distant locations in the southern USA for Triatoma protracta [23], which is a trend that has been confirmed [81] and opposed [82] in other hemipterans. As in other insect species, taxonomic clade-wide stability of microbiota composition with regard to one environmental variable or another does not appear to be consistent. Generalizations about geographical variation in insect microbiomes will have to remain at the species level for now. However, this is still very much an open question in triatomine microbiota research.
Contrary to our expectations, there was no significant difference in bacterial community composition between T. cruzi-infected and uninfected individuals. This contrasts with previous studies that have found significant differences between microbiomes of T. cruzi-positive and negative individuals [23, 30]. However, our small sample size, limited number of palms sampled (n = 8) with skewed infection ratios, and a skewed abundance of younger stage nymphs (N1–N3), may confound true observable differences. Furthermore, detectable levels of differences may be localized to a portion of the triatomine gut where T. cruzi develops; this deserves further study and experimental controls. A similar situation may occur during infection with T. rangeli, which can colonize not only the insect intestine but also the hemocoel and salivary glands [83]. We also did not find any significant difference in the microbiota as a result of the dominant blood-meal source found, as previously observed [31]. This could be due to the complexity of variables that influence the microbiota. While blood-meal source potentially has an effect on the bacterial composition in the gut, these samples often have mixed blood-meal sources with differing abundance (Additional file 1: Table S1; [50]), making discrete differences difficult to observe. Habitat associated microbial composition was found to be different between peridomestic and peridomestic-forest; however, this observation represents a few peridomestic-forest samples from northern Veraguas only and likely is an artifact of location difference. Similarly, microbial composition between age classes showed no differences, but because this dataset is highly skewed toward a couple of nymphal classes, distinctions are impossible to detect. This study examined the whole-body microbiota, which may obscure anatomically localized differences observed in other studies and, on the whole, result in a more comprehensive measure of microbial composition where local environment has a bigger impact.
Conclusions
In conclusion, we examined the whole-body microbiota of Rhodnius pallescens, which can serve as a benchmark for future comparative studies examining the microbiota of specific organs or anatomical regions. Interestingly, the largest difference in R. pallescens microbial community composition was between geographical locations. While we did not find any definitive differences in relation to other variables (e.g. habitat type, age class, blood-meal source, infection status) these remain important aspects of vector biology that require further study. The effects of geographical environmental diversity can be minimized through the use of more comparative studies using laboratory-reared insects and controlled studies to tease apart more complex variables such as blood-meal sources and infection status.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. Demultiplexed reads are available from NCBI Sequence Read Archive under BioProject ID PRJNA543522 and Submission ID SUB5631094. Metadata and NCBI accession numbers are in Additional file 1: Table S1.
References
Engel P, Moran NA. The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735.
Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90.
Oliver KM, Martinez AJ. How resident microbes modulate ecologically-important traits of insects. Curr Opin Insect Sci. 2014;4:1–7.
Weiss B, Aksoy S. Microbiome influences on insect host vector competence. Trends Parasitol. 2011;27:514–22.
Cirimotich CM, Ramirez JL, Dimopoulos G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe. 2011;10:307–10.
Finney CA, Kamhawi S, Wasmuth JD. Does the arthropod microbiota impact the establishment of vector-borne diseases in mammalian hosts? PLoS Pathog. 2015;11:e1004646.
Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors. 2013;6:146.
Crotti E, Balloi A, Hamdi C, Sansonno L, Marzorati M, Gonella E, et al. Microbial symbionts: a resource for the management of insect-related problems. Microb Biotechnol. 2012;5:307–17.
Saldana MA, Hegde S, Hughes GL. Microbial control of arthropod-borne disease. Mem Inst Oswaldo Cruz. 2017;112:81–93.
Iturbe-Ormaetxe I, Walker T, SL ON. Wolbachia and the biological control of mosquito-borne disease. EMBO Rep. 2011;12:508–18.
Bourtzis K, Dobson SL, Xi Z, Rasgon JL, Calvitti M, Moreira LA, et al. Harnessing mosquito-Wolbachia symbiosis for vector and disease control. Acta Trop. 2014;132(Suppl):S150–63.
Shaw WR, Catteruccia F. Vector biology meets disease control: using basic research to fight vector-borne diseases. Nat Microbiol. 2019;4:20–34.
Caragata EP, Dutra HL, Moreira LA. Inhibition of Zika virus by Wolbachia in Aedes aegypti. Microb Cell. 2016;3:293–5.
Akorli J, Gendrin M, Pels NA, Yeboah-Manu D, Christophides GK, Wilson MD. Seasonality and locality affect the diversity of Anopheles gambiae and Anopheles coluzzii midgut microbiota from Ghana. PloS One. 2016;11:e0157529.
Carpi G, Cagnacci F, Wittekindt NE, Zhao F, Qi J, Tomsho LP, et al. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PloS One. 2011;6:e25604.
Coon KL, Brown MR, Strand MR. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol Ecol. 2016;25:5806–26.
Coon KL, Vogel KJ, Brown MR, Strand MR. Mosquitoes rely on their gut microbiota for development. Mol Ecol. 2014;23:2727–39.
Fryxell RTT, DeBruyn JM. The microbiome of Ehrlichia-infected and uninfected lone star ticks (Amblyomma americanum). PloS One. 2016;11:e0146651.
Gimonneau G, Tchioffo MT, Abate L, Boissiere A, Awono-Ambene PH, Nsango SE, et al. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Inf Genet Evol. 2014;28:715–24.
Gurfield N, Grewal S, Cua LS, Torres PJ, Kelley ST. Endosymbiont interference and microbial diversity of the Pacific coast tick, Dermacentor occidentalis, in San Diego County, California. PeerJ. 2017;5:e3202.
Kwan JY, Griggs R, Chicana B, Miller C, Swei A. Vertical vs. horizontal transmission of the microbiome in a key disease vector, Ixodes pacificus. Mol Ecol. 2017;26:6578–89.
Montoya-Porras LM, Omar TC, Alzate JF, Moreno-Herrera CX, Cadavid-Restrepo GE. 16S rRNA gene amplicon sequencing reveals dominance of Actinobacteria in Rhodnius pallescens compared to Triatoma maculata midgut microbiota in natural populations of vector insects from Colombia. Acta Trop. 2018;178:327–32.
Rodriguez-Ruano SM, Skochova V, Rego ROM, Schmidt JO, Roachell W, Hypsa V, et al. Microbiomes of North American Triatominae: the grounds for Chagas disease epidemiology. Front Microbiol. 2018;9:1167.
Van Treuren W, Ponnusamy L, Brinkerhoff RJ, Gonzalez A, Parobek CM, Juliano JJ, et al. Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl Environ Microbiol. 2015;81:6200–9.
Williams-Newkirk AJ, Rowe LA, Mixson-Hayden TR, Dasch GA. Characterization of the bacterial communities of life stages of free living lone star ticks (Amblyomma americanum). PloS One. 2014;9:e102130.
Gurtler RE, Kitron U, Cecere MC, Segura EL, Cohen JE. Sustainable vector control and management of Chagas disease in the Gran Chaco, Argentina. Proc Natl Acad Sci USA. 2007;104:16194–9.
Beard CB, Cordon-Rosales C, Durvasula RV. Bacterial symbionts of the triatominae and their potential use in control of Chagas disease transmission. Annu Rev Entomol. 2002;47:123–41.
Castro DP, Moraes CS, Gonzalez MS, Ratcliffe NA, Azambuja P, Garcia ES. Trypanosoma cruzi immune response modulation decreases microbiota in Rhodnius prolixus gut and is crucial for parasite survival and development. PloS One. 2012;7:e36591.
da Mota FF, Marinho LP, Moreira CJ, Lima MM, Mello CB, Garcia ES, et al. Cultivation-independent methods reveal differences among bacterial gut microbiota in triatomine vectors of Chagas disease. PLoS Negl Trop Dis. 2012;6:e1631.
Diaz S, Villavicencio B, Correia N, Costa J, Haag KL. Triatomine bugs, their microbiota and Trypanosoma cruzi: asymmetric responses of bacteria to an infected blood meal. Parasit Vectors. 2016;9:636.
Dumonteil E, Ramirez-Sierra MJ, Perez-Carrillo S, Teh-Poot C, Herrera C, Gourbiere S, et al. Detailed ecological associations of triatomines revealed by metabarcoding and next-generation sequencing: implications for triatomine behavior and Trypanosoma cruzi transmission cycles. Sci Rep. 2018;8:4140.
Orantes LC, Monroy C, Dorn PL, Stevens L, Rizzo DM, Morrissey L, et al. Uncovering vector, parasite, blood meal and microbiome patterns from mixed-DNA specimens of the Chagas disease vector Triatoma dimidiata. PLoS Negl Trop Dis. 2018;12:e0006730.
Waltmann A, Willcox AC, Balasubramanian S, Borrini Mayori K, Mendoza Guerrero S, Salazar Sanchez RS, et al. Hindgut microbiota in laboratory-reared and wild Triatoma infestans. PLoS Negl Trop Dis. 2019;13:e0007383.
Espino CI, Gomez T, Gonzalez G, do Santos MF, Solano J, Sousa O, et al. Detection of Wolbachia bacteria in multiple organs and feces of the triatomine insect Rhodnius pallescens (Hemiptera, Reduviidae). Appl Environ Microbiol. 2009;75:547–50.
Vieira CS, Mattos DP, Waniek PJ, Santangelo JM, Figueiredo MB, Gumiel M, et al. Rhodnius prolixus interaction with Trypanosoma rangeli: modulation of the immune system and microbiota population. Parasit Vectors. 2015;8:135.
Novakova E, Woodhams DC, Rodriguez-Ruano SM, Brucker RM, Leff JW, Maharaj A, et al. Mosquito microbiome dynamics, a background for prevalence and seasonality of West Nile virus. Front Microbiol. 2017;8:526.
Dada N, Jumas-Bilak E, Manguin S, Seidu R, Stenstrom TA, Overgaard HJ. Comparative assessment of the bacterial communities associated with Aedes aegypti larvae and water from domestic water storage containers. Parasit Vectors. 2014;7:391.
Dickson LB, Jiolle D, Minard G, Moltini-Conclois I, Volant S, Ghozlane A, et al. Carryover effects of larval exposure to different environmental bacteria drive adult trait variation in a mosquito vector. Sci Adv. 2017;3:e1700585.
Rodriguez IG, Loaiza JR. American trypanosomiasis, or Chagas disease, in Panama: a chronological synopsis of ecological and epidemiological research. Parasit Vectors. 2017;10:459.
Christensen HA, Whitlaw JT Jr, Chaniotis BN, de Vasquez AM. Sylvatic hosts of Rhodnius pallescens (Hemiptera: Reduviidae) nymphs in the Panama Canal Zone. J Med Entomol. 1980;17:182.
Whitlaw JT, Chaniotis BN. Palm trees and Chagas’ disease in Panama. Am J Trop Med Hyg. 1978;27:873–81.
Vasquez AM, Samudio FE, Saldana A, Paz HM, Calzada J. Eco-epidemiological aspects of Trypanosoma cruzi, Trypanosoma rangeli and their vector (Rhodnius pallescens) in Panama. Rev Inst Med Trop Sao Paulo. 2004;46:217–22.
Saldana A, Pineda V, Martinez I, Santamaria G, Santamaria AM, Miranda A, et al. A new endemic focus of Chagas disease in the northern region of Veraguas Province, Western Half Panama, Central America. PloS One. 2012;7:e34657.
Saldana A, Santamaria AM, Pineda V, Vasquez V, Gottdenker NL, Calzada JE. A darker chromatic variation of Rhodnius pallescens infected by specific genetic groups of Trypanosoma rangeli and Trypanosoma cruzi from Panama. Parasit Vectors. 2018;11:423.
Calzada JE, Pineda V, Garisto JD, Samudio F, Santamaria AM, Saldana A. Human trypanosomiasis in the eastern region of the Panama Province: new endemic areas for Chagas disease. Am J Trop Med Hyg. 2010;82:580–2.
Bosmans L, Pozo MI, Verreth C, Crauwels S, Wilberts L, Sobhy IS, et al. Habitat-specific variation in gut microbial communities and pathogen prevalence in bumblebee queens (Bombus terrestris). PloS One. 2018;13:e0204612.
Tiede J, Scherber C, Mutschler J, McMahon KD, Gratton C. Gut microbiomes of mobile predators vary with landscape context and species identity. Ecol Evol. 2017;7:8545–57.
Zytynska SE, Meyer ST, Sturm S, Ullmann W, Mehrparvar M, Weisser WW. Secondary bacterial symbiont community in aphids responds to plant diversity. Oecologia. 2016;180:735–47.
Noireau F, Abad-Franch F, Valente SA, Dias-Lima A, Lopes CM, Cunha V, et al. Trapping Triatominae in silvatic habitats. Mem Inst Oswaldo Cruz. 2002;97:61–3.
Kieran TJ, Gottdenker NL, Varian CP, Saldana A, Means N, Owens D, et al. Blood meal source characterization using Illumina sequencing in the Chagas disease vector Rhodnius pallescens (Hemiptera: Reduviidae) in Panama. J Med Entomol. 2017;54:1786–9.
Faircloth BC, Glenn TC. Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PloS One. 2012;7:e42543.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Glenn TC, Pierson TW, Bayona-Vásquez NJ, Kieran TJ, Hoffberg SL, Thomas JC, et al. Adapterama II: Universal amplicon sequencing on Illumina platforms (TaggiMatrix). PeerJ. 2019;7:e7786.
Wang J, Tang L, Glenn TC, Wang JS. Aflatoxin B1 induced compositional changes in gut microbial communities of male F344 rats. Toxicol Sci. 2016;150:54–63.
Glenn TC, Nilsen RA, Kieran TJ, Sanders JG, Bayona-Vásquez NJ, Finger JW, et al. Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ. 2019;7:e7755.
Chiurillo MA, Crisante G, Rojas A, Peralta A, Dias M, Guevara P, et al. Detection of Trypanosoma cruzi and Trypanosoma rangeli infection by duplex PCR assay based on telomeric sequences. Clin Diagn Lab Immunol. 2003;10:775–9.
Wincker P, Britto C, Pereria JB, Cardoso MA, Oelemann W, Morel CM. Use of a simplified polymerase chain reaction procedure to detect Trypanosoma cruzi in blood samples from chronic chagasic patients in a rural endemic area. Am J Trop Med Hyg. 1994;51:771–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, et al. Qiime2: Reproducible, interactive, scalable, and extensible microbiome data science. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PloS One. 2010;5:e9490.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
Kruskal JB. Multidimensional scaling by optimizing goodness-of-fit to a nonmetric hypothesis. Psychometrika. 1964;29:1–28.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One. 2013;8:e61217.
Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30.
Wickham H, François R, Henry L, Müller K. dplyr: A grammar of data manipulation. R package version 0.8.0. 2018.
Wickham H, Chang W, Henry L, Pedersen TL, Takahashi K, Wilke C, et al. ggplot2: Create elegant data visualisations using the grammar of graphics. R package version 3.2.1. 2018.
Foster ZS, Sharpton TJ, Grunwald NJ. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput Biol. 2017;13:e1005404.
Azambuja P, Garcia ES, Ratcliffe NA. Gut microbiota and parasite transmission by insect vectors. Trends Parasitol. 2005;21:568–72.
Boissiere A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012;8:e1002742.
Gumiel M, da Mota FF, Rizzo Vde S, Sarquis O, de Castro DP, Lima MM, et al. Characterization of the microbiota in the guts of Triatoma brasiliensis and Triatoma pseudomaculata infected by Trypanosoma cruzi in natural conditions using culture independent methods. Parasit Vectors. 2015;8:245.
Vivero RJ, Jaramillo NG, Cadavid-Restrepo G, Soto SI, Herrera CX. Structural differences in gut bacteria communities in developmental stages of natural populations of Lutzomyia evansi from Colombiaʼs Caribbean coast. Parasit Vectors. 2016;9:496.
Yun JH, Roh SW, Whon TW, Jung MJ, Kim MS, Park DS, et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol. 2014;80:5254–64.
Sazama EJ, Bosch MJ, Shouldis CS, Ouellette SP, Wesner JS. Incidence of Wolbachia in aquatic insects. Ecol Evol. 2017;7:1165–9.
Jiggins FM, Hurst GDD, Schulenburg JHVD, Majerus MEN. Two male-killing Wolbachia strains coexist within a population of the butterfly Acraea encedon. Heredity. 2001;86:161–6.
Jimenez-Cortes JG, Garcia-Contreras R, Bucio-Torres MI, Cabrera-Bravo M, Cordoba-Aguilar A, Benelli G, et al. Bacterial symbionts in human blood-feeding arthropods: patterns, general mechanisms and effects of global ecological changes. Acta Trop. 2018;186:69–101.
Fraihi W, Fares W, Perrin P, Dorkeld F, Sereno D, Barhoumi W, et al. An intergrated overview of the midgut bacterial flora composition of Phlebotomus pernicoisus, a vector of zoonotic visceral leishmaniasis in the Western Mediterranean Basin. PLoS Negl Trop Dis. 2017;11:e0005484.
Monteiro CC, Villegas LEM, Campolina TB, Pires ACMA, Miranda JC, Pimenta PFP, et al. Bacterial diversity of the American sand fly Lutzomyia intermedia using high-throughput metagenomic sequencing. Parasit Vectors. 2016;9:480.
Sudakaran S, Salem H, Kost C, Kaltenpoth M. Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae). Mol Ecol. 2012;21:6134–51.
Welch EW, Macias J, Bextine B. Geographic patterns in the bacterial microbiome of the glassy-winged sharpshooter, Homalodisca vitripennis (Hemiptera: Cicadellidae). Symbiosis. 2015;66:1–12.
Azambuja P, Garcia ES. Trypanosoma rangeli interactions within the vector Rhodnius prolixus - a mini review. Mem Inst Oswaldo Cruz. 2005;100:567–71.
Acknowledgements
We thank Franklyn Samudio and Jose Montenegro for assistance in the field. The authors also thank the Ministerio de Ambiente in Panama (MiAMBIENTE), the Smithsonian Tropical Research Institute (STRI), and the University of Georgia Graduate School.
Funding
This study was funded by the University of Georgia (UGA) Office of the Vice President for Research; Secretaria Nacional de Ciencia Tecnología e Inovación (SENACYT), grant COL11-043 awarded to ICGES; UGA Graduate School Innovative and Interdisciplinary Research Grant awarded to TJK; National Science Foundation Grant No. DGE-1545433 awarded to the UGA Interdisciplinary Disease Ecology Across Scales program; UGA Odum School of Ecology.
Author information
Authors and Affiliations
Contributions
TJK, JCT, TCG and NLG designed the study. AS, JEC, TCG and NLG provided project supervision and coordination. TJK, KMHA, CPV, AS, JEC and NLG collected samples. TJK, KMHA and CPV performed laboratory work. TJK and KMHA conducted data analysis. TJK drafted the manuscript. All authors edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Smithsonian Tropical Research Institute IACUC approval numbers 2013-0601-2016-02, and 2016-0801-2019-A1. MINAM (Ministerio de Ambiente) permit numbers SE/A-93-13 and SE/AP-27-16. AUP 2013 05-001-43-Y3-A0 and A2018 02-024-Y1-A0.
Consent for publication
Not applicable.
Competing interests
JEC and AS are members of the SNI (Sistema Nacional de Investigación from SENACYT of Panama). TJK, KMHA, JCT, CPV, AS, JEC, TCG and NLG declare no further competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Metadata for all samples used in this study including NCBI accession numbers.
Additional file 2: Table S2.
Phylum level read counts and proportions for each of the three locations.
Additional file 3: Figure S1.
Heat-trees showing the abundance of reads for each taxonomic group (branches) across the three collection localities.
Additional file 4: Table S3.
Top 20 Family level read counts and proportions for each of the three locations.
Additional file 5: Table S4.
Top 20 Genus level read counts and proportions for each of the three locations.
Additional file 6: Table S5.
Total number reads and the number and proportion of Wolbachia reads for each sample across all locations.
Additional file 7: Figure S2.
Alpha richness plot across locations.
Additional file 8: Figure S3.
Alpha richness plot across habitat type.
Additional file 9: Figure S4.
Alpha richness plot across infection status.
Additional file 10: Figure S5.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across pastures in Veraguas.
Additional file 11: Figure S6.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across location at peridomestic sites with (A) and without Veraguas (B) shown.
Additional file 12: Figure S7.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across palm type at Veraguas sites.
Additional file 13: Figure S8.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across palm type at Las Pavas sites.
Additional file 14: Figure S9.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across Infection status at Las Pavas sites.
Additional file 15: Figure S10.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across infection status at peridomestic sites.
Additional file 16: Figure S11.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across infection status at Trinidad de las Minas sites.
Additional file 17: Figure S12.
Non-metric multidimensional scaling plot (based on Bray-Curtis distances) of OTU frequency for the microbial communities of triatomines across infection status at Veraguas sites.
Additional file 18: Figure S13.
Multivariate Principal Coordinate Analyses results showing location by age class (a) and age class by location (b).
Additional file 19: Table S6.
Total read counts and proportions for all observed bacterial taxa down to the genus level for each of the three sampling locations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kieran, T.J., Arnold, K.M.H., Thomas, J.C. et al. Regional biogeography of microbiota composition in the Chagas disease vector Rhodnius pallescens. Parasites Vectors 12, 504 (2019). https://doi.org/10.1186/s13071-019-3761-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3761-8