
Abstract
Background
Fasciolopsis buski is a zoonotic intestinal fluke infecting humans and pigs, but it has been seriously neglected. It is yet to know whether there is any genetic diversity among F. buski from different geographical locations, particularly in sequences of nuclear ribosomal DNA (rDNA) and mitochondrial (mt) DNA. Therefore, we determined the sequences of partial 18S, the complete internal transcribed spacer (ITS) rDNA and the complete mt genome of F. buski from China, compared the rDNA and mtDNA sequences with those of isolates from India and Vietnam, and assessed the phylogenetic relationships of this fluke and related fasciolid trematodes based on the mtDNA dataset.
Results
The complete mt genome sequence of F. buski from China is 14,833 bp, with 36 genes, including 12 protein-coding genes (PCGs), 22 tRNA genes, and two rRNA genes (rrnL and rrnS). The AT content of F. buski from China is 65.12%. The gene content and arrangement of the F. buski mt genome is similar to that of Fascioloides magna. Genetic distances between isolates of F. buski from China and India were high (28.2% in mtDNA, 13.2% in ITS-1 and 9.8% in ITS-2) and distinctly higher than the interspecific differences between Fasciola hepatica and Fasciola gigantica. The rDNA and mtDNA datasets for F. buski from China (isolate from pigs) and Vietnam (isolates from humans) were identical. The intergeneric differences in amino acid and nucleotide sequences among the genera Fasciolopsis, Fascioloides and Fasciola ranged between 24.64–25.56% and 26.35–28.46%, respectively.
Conclusions
Our results indicate that F. buski from China and India may represent distinct taxa, while F. buski in Vietnam and China represent the same species. These findings might have implications for the implementation of appropriate control strategies in different regions. Further studies are needed to decode mtDNA and rDNA sequences of F. buski from various geographical isolates for the better understanding of the species complex of F. buski.
Similar content being viewed by others


Background
Fasciolopsis buski (Lankester, 1857) Looss, 1899, the iconic species of the genus Fasciolopsis, is the etiological agent of fasciolopsiasis of many mammals, such as humans, pigs, monkeys, dogs and rabbits [1]. Known as the giant intestinal fluke [2], F. buski is one of the largest digeneans that infect humans worldwide [3] and is largely confined to Asian countries [4–6], including China [7]. The infection with F. buski occurs through the consumption of raw or insufficiently cooked aquatic food contaminated with the metacercariae [8, 9]. Fasciolopsis buski is also found in co-infection with other soil-transmitted helminths [8] especially trematodes [10]. Fasciolopsiasis may present with moderate and heavy infection in the digestive tract [11], sometimes even followed by various complications such as acute kidney injury [12], decrease of the host immune system [13], and death [6, 13–15], leading to significant losses and public health issues in endemic areas [16, 17].
Despite the severe damage and morbidity [18] caused by F. buski, molecular research on this species has not attracted enough attention. Incomplete sequences of the first and the second internal transcribed spacer (ITS-1 and ITS-2) regions of nuclear ribosomal DNA (rDNA) of F. buski were determined and deposited [15, 19]; however, the ITS-2 sequences of isolates of F. buski from India and Vietnam differ by 6.7% [18], a divergence that is much larger than that between Fasciola hepatica and Fasciola gigantica (1.7%) [20]. Meanwhile, as the complete mitochondrial (mt) genome sequence of F. buski from India has been decoded [21], we have noticed that the sequence difference in the nicotinamide dehydrogenase subunit 1 (nad1) gene between the F. buski isolates from India and Vietnam is 17.7%, which is higher than that between Fascioloides magna and Fa. hepatica (15%) [22]. Still, no information exists to illustrate the relationships between isolates of F. buski from India, Vietnam and mainland China, and none of the publications have explained the sequence discrepancies among these F. buski isolates.
The accurate identification of trematode species is relevant in relation to better understand their biology, epidemiology and ecology, and has implications for the diagnosis of infections. Genetic markers in mtDNA and rDNA have been proven effective for identifying trematode species [23, 24] that cannot be differentiated by morphological analysis, including flukes in the family Fasciolidae [10]. Here, we determined the sequences of the complete mt genome and ITS-1, ITS-2 and partial 18S rDNA sequences of F. buski from China, and compared these with sequences derived from isolates from India and Vietnam to test the hypothesis that the isolate of F. buski sampled in China may represent a genetically distinct taxon from that sampled in India.
Methods
Sampling and DNA extraction
Four adult fluke specimens were obtained from intestine of naturally infected pigs from Jiangxi Province, China. These flukes were identified morphologically as F. buski according to the existing keys [25]. All worms were washed in 0.1 M phosphate-buffered saline (PBS), pH 7.2, fixed in 70% (v/v) ethanol and preserved at -20 °C. Total genomic DNA was extracted from individual F. buski specimens using sodium dodecyl sulfate (SDS)/proteinase K treatment [26] and column-purification (Wizard® SV Genomic DNA Purification System, Promega, Madison, USA), according to the manufacturer’s protocol.
Amplification of 18S and ITS rDNA
The partial 18S rDNA region of each of the four F. buski specimens was amplified and sequenced using primers FBJ.18SF1 (forward; 5′-ACG GGG AGG TAG TGA CGA AAA A-3′) and FBJ.18SR1 (reverse; 5′-CAC CAA CCA CCG AAT CAA GAA A-3′) which were designed based on corresponding sequences available in GenBank. The ITS rDNA region, spanning part of the 18S rRNA gene, the complete ITS-1, the 5.8S rRNA gene, ITS-2, and part of the 28S rRNA gene, was amplified and sequenced using primers BD1 (forward; 5′-GTC GTA ACA AGG TTT CCG TA-3′) and BD2 (reverse; 5′-ATG CTT AAA TTC AGC GGG T-3′) [27]. Sequence comparisons of ITS among fasciolid species were also conducted.
Long-range PCR-based sequencing of mt genome
The primers, designed based on relatively conserved regions of mtDNA sequences from Fas. magna and Fa. hepatica, were used to amplify the sequence of the entire mt genome of F. buski from a single specimen in 12 overlapping fragments (Additional file 1: Table S1).
PCR reactions were conducted in a total volume of 50 μl, using 25 μl PrimeStar Max DNA polymerase premix (Takara, Dalian, China), 25 pmol of each primer (synthesized in Genewiz, Suzhou, China), 0.7 μl DNA templates, and H2O, in a thermocycler (Biometra, Göttingen, Germany). PCR cycling conditions started with an initial denaturation at 98 °C for 2 min, followed by 18 cycles of denaturation at 92 °C for 15 s, annealing at 55–63 °C for 15 s and extension at 60 °C for 1–5 min, followed by 92 °C denaturation for 2 min, plus 22 cycles of 92 °C for 15 s (denaturation), 55–63 °C for 15 s (annealing) and 66 °C for 1–6 min, with a final extension step for 10 min at 68 °C. A negative control (no DNA) was included in each amplification run. Positive amplicons were sent to Genewiz Company (Beijing, China) for sequencing.
Assembly, annotation and bioinformatics analysis
Sequences were assembled manually and aligned against the entire mt genome sequences of Fas. magna (GenBank accession No. KU060148) and Fa. hepatica (NC002546) using MAFFT 7.122 to infer boundaries for each gene. Amino acid sequences of 12 protein-coding genes were translated using MEGA v.6.06 and NCBI translation Table 21 (Trematode Mitochondrial Code). The tRNA genes were affirmed using the programs tRNAscan-SE [28] or by comparison with those from the Fas. magna and Fa. hepatica mt genomes. The two rRNA genes were identified by comparison with those of Fas. magna and Fa. hepatica.
A comparative analysis of the nucleotide sequences of each protein-coding gene, the amino acid sequences, two ribosomal RNA genes, 22 tRNA genes as well as non-coding regions (NCRs) among F. buski, Fas. magna and Fa. hepatica was conducted.
Phylogenetic analysis
The concatenated amino acid sequences of F. buski mt genome from the Chinese isolate, conceptually translated from individual genes of each mt genome, were aligned with those of published mt genomes of fasciolid trematodes, including Fa. hepatica (NC002546), Fasciola sp. (KF_543343), Fa. gigantica (NC024025) and Fas. magna (KU060148). The inferred genetic relationships of these fasciolid flukes were reconstructed based on sequences of partial nad1 gene (488 bp), including the isolates of F. buski from India (SRR924085) [21], China (NC030528) and Vietnam (EF612501) [29] and those from other fasciolid flukes. The sequence of Paramphisitomum leydeni (KP341657) (Paramphistomatidae) served as the outgroup.
All nucleotide or inferred amino acid sequences of were aligned using MAFFT 7.122. Poorly aligned sites and divergent regions of the alignment were eliminated using Gblocks Server v. 0.91b (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using default settings, selecting the option of less strict conservation of flanking positions. The alignment was then converted into nexus format using Clustal X1.83 and subjected to phylogenetic analysis using Bayesian inference (BI). The mixed model was used in inferred amino acid matrix, while the 4by4 was used as a nucleotide substitution model in BI analysis using MrBayes 3.1.1 [30]. Four independent Markov chain were run for 10,000,000 metropolis-coupled MCMC generations, sampling trees every 1,000 generations. The first 2,500 trees were discarded as ‘burn-in’, and the remaining trees were used for calculating Bayesian posterior probabilities. The analysis was regarded as completed when the potential scale reduction factors were close to 1, and the average standard deviation of split frequencies was below 0.01. Phylograms were prepared using FigTree v. 1.42 [31].
Results and discussion
Comparison of 18S and ITS rDNA
The amplicons of 18S and ITS rDNA were approximately 900 bp and 2,130 bp in length, respectively. The ITS-1 and ITS-2 were 1,505 bp and 379 bp in length, respectively. The gene boundaries of ITS were inferred by comparing with those of Fas. magna (GenBank: EF051080). The sequences from four F. buski samples were identical.
The sequences of 18S amplicons of four F. buski specimens from China were all 99.9% similar to those from Vietnam (AY311386 and L06668) [18]. The ITS differences between isolates from Vietnam [EF612477 (ITS-1) and EF612489 (ITS-2)] and China were 0.95% in ITS-1 and 0% in ITS-2. These divergence levels support the suggestion that F. buski isolates from China and Vietnam represent the same species. For the ITS rDNA sequences, differences between isolates from China and India [DQ351843 (ITS-1) and DQ351841 (ITS-2)] were 3.31% (in ITS-1) and 6.75% (in ITS-2). The difference of ITS rDNA between isolates from Vietnam [EF612477 (ITS-1) and EF612489 (ITS-2)] and India were 2.36% in ITS-1 and 6.75% in ITS-2. These were higher than the interspecific differences between Fa. hepatica and Fa. gigantica (1.2% in ITS-1 and 1.7% in ITS-2) [20], but lower than intergeneric difference between Fas. magna and Fa. hepatica (7.04% in ITS-1, 13.8% in ITS-2) [22]. The results of these comparative sequence analyses suggest that F. buski isolates from China and India may represent distinct fluke species.
Mitochondrial genome content, organization and annotation
The complete mt genome sequence of F. buski from China (GenBank accession No. NC030528) is 14,833 bp in length (Fig. 1), and contains 36 genes, including 12 protein-coding genes (atp6, cytb, cox1–3, nad1–6 and nad4L), 22 tRNA genes and two rRNA genes (rrnL and rrnS) (Table 1). The gene content and organization of F. buski mt genome is consistent with other fasciolid flukes [22]. There is only one NCR in F. buski mt genome, which is located between trnE (13,458–13,519) and cox3 (1–645) (Table 1), which is consistent with Fas. magna mt genome [22], whereas two non-coding regions exist in the mt genomes of Fasciola spp. flukes [20, 32]. The arrangement of the genes in F. buski mt genome is similar to that in Fas. magna [22].

Organization of the mitochondrial genome of Fasciolopsis buski. The scales are approximate. All genes are transcribed in the clockwise direction, using standard nomenclature. “NCR” refers to the only non-coding region in F. buski data. The A + T content is shown for each gene or region of the mt genome and represented by colour
The value of total A + T content for F. buski mtDNA is 65.12%, which is slightly higher than that for Fas. magna (61.42%). The A + T content for each gene or region of F. buski mt genome ranged from 53.97% (trnI) to 74.60% (trnV). The nucleotide composition of F. buski mt genome is obviously biased towards A and T, with the low content of C (8.67%) and high content of T (46.71%) (Additional file 2: Table S2).
In the protein-coding genes of F. buski mt genome, ATG or GTG were used as start codons, and TAG or TAA were used as stop codons (Table 1). Incomplete codons were not detected in the mt genome of F. buski from China. The 22 tRNA genes of F. buski mt genome ranged from 57 to 70 bp in length, with their structures similar to those of Fas. magna and Fa. hepatica [20, 32]. The large ribosomal RNA gene (rrnL), 989 bp in length, is located between trnT and trnC. The small ribosomal RNA gene (rrnS) are located between trnC and cox2, which is 775 bp in length (Table 1).
The only NCR of F. buski mt genome (1,314 bp in length), is located between trnE and cox3, with the A + T content being 63.55%. It contains nine copies of a 101 nt complete direct repeats (TGA TTG TGT GTT GGA TAG GAT AGG TAT GTT GGG TAT TTG TTT TGG TGG ATT GGA TTG TGC CTA GGG GCT GAG TGT TAA TGA TAA TGG GAT GTG TAT TAT AT). Every two repeats are separated by 3 nt (GGG).
Comparative analysis of mt genomes among Fasciolopsis, Fascioloides and Fasciola
The genetic distance across the complete mt genome between F. buski from China and India is 28.2%, which is higher than the interspecific difference between Fa. gigantica and Fa. hepatica (12.2%) [20], between Paramphistomum cervi and P. leydeni (14.7%) [23], and between Paragonimus westermani isolates from Korea and India (20.74%) [33].
The sequence difference of cytochrome c oxidase subunit 1 (cox1) gene between isolates from India (GenBank: SRR924085; 1,542 bp in length) and China is 11.8%, which is higher than that between Fa. hepatica and Fa. gigantica (9.13%) [20]. These comparisons suggest that F. buski from China may represent a distinct species.
Although no complete mt DNA data of F. buski from Vietnam were reported, the sequence of nad1 from the Chinese isolate is 98.6% similar to that from Vietnam (EF612501) [29], indicating that the isolates of F. buski from China and Vietnam might be the same species. It is noteworthy that the Chinese isolate of F. buski came from pigs [7], while the Vietnamese isolate infected both humans [13, 18] and pigs [29], suggesting a possible cross-transmission between humans and pigs.
The sequence differences in the complete mt genomes between F. buski and Fas. magna (28.46% at nucleotide level, 24.64% at the inferred amino acid level) are very close to those between F. buski and Fasciola spp. (26.35–26.51% and 25.56–25.48%, respectively). Considering the 12 protein-coding genes, sequence differences range between 16.12 and 29.36% at the nucleotide level, and between 14.44 and 32.55% at the amino acid level. For fasciolid flukes, at both nucleotides and amino acid levels, the comparisons among the mt genomes of the species showed that cox1 and nad4L are the most conserved genes while nad4 is the least conserved. In addition, nad1 and cox2 are conserved, whereas nad5 and nad6 are very variable (Table 2), which is in accordance with the characteristics of trematodes of the family Paramphistomatidae [23]. Nucleotide differences between F. buski and other fasciolid species ranged between 18.48 and 20.72% in rrnL, between 23.95 and 25.80% in rrnS, and between 22.03 and 26.54% in tRNA genes. We also found that rrnL is more conserved than rrnS, as observed in parasites of the Paramphistomatidae [23] and the Dicrocoeliidae [34].
Phylogenetic analysis
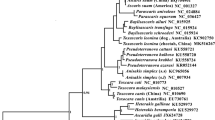
The phylogenetic tree was constructed based on the concatenated amino acid sequence dataset of all 12 mt protein-coding genes of six selected trematodes (Fig. 2a). Fasciolopsis buski clustered with flukes of the genera Fasciola and Fascioloides within the Fasciolidae. The taxonomic status of these trematodes is in concordance with those of previous studies [20–23], with each node receiving strong nodal support (Bpp = 1).

Phylogenetic relationships of Fasciolopsis buski and related fasciolid trematodes. Tree inferred from the concatenated amino acid sequence dataset for 12 protein-coding genes (a) and nucleotides for partial nad1 gene (b) from selected trematodes were constructed using Bayesian inference analysis (BI). Paramphistomum leydeni (KP341657) (Paramphistomatidae) served as the outgroup
The complete mitochondrial genome of F. buski from Vietnam is not decoded, only the nad1 gene sequence is available in GenBank. Thus, nad1 gene is the only mt gene with sequences available on GenBank for F. buski from China, India and Vietnam. Therefore, this gene was used as a molecular marker for assessing the phylogenetic relationships among species in the family of Fasciolidae. The evolutionary tree of six fasciolid species based on sequences of partial nad1 gene highlighted the close phylogenetic relationship between the isolates of F. buski from Vietnam and China, while the isolate from India appeared very distinct from these two isolates, although nodal values were marginally supported (Bpp > 0.94) (Fig. 2b).
Conclusions
The present study determined the sequences of the complete mt genome as well as the ITS and partial 18S rDNA sequences of the giant intestinal fluke F. buski from China. Comparative sequence analyses indicated that F. buski from China and India may represent distinct taxa, while F. buski from Vietnam and China represent the same species. Further studies are required to better understand the F. buski species complex by determining the mtDNA and rDNA sequences of F. buski from more geographical locations.
Abbreviations
- BI:
-
Bayesian inference
- cox1:
-
Cytochrome c oxidase subunit 1
- ITS:
-
Internal transcribed spacer
- mt:
-
Mitochondrial
- mtDNA:
-
Mitochondrial DNA
- nad1:
-
Nicotinamide dehydrogenase subunit 1
- NCR:
-
Non-coding region
- PBS:
-
Phosphate-buffered saline
- rDNA:
-
Ribosomal DNA
- SDS:
-
Sodium dodecyl sulphate
References
Malviya HC. The susceptibility of mammals to Fasciolopsis buski. J Helminthol. 1985;59:19–22.
Cao YH, Ma YM, Qiu F, Zhang XQ. Rare cause of appendicitis: Mechanical obstruction due to Fasciolopsis buski infestation. World J Gastroenterol. 2015;21:3146–9.
Basnuevo JG, Seuc CA. Fasciolopsis buski in Cuba; two cases treated with chloroquine, gentian violet, hexylresorcinol and tetrachloroethylene. Rev Kuba Med Trop Parasitol. 1950;6:91–6.
Gilman RH, Mondal G, Maksud M, Alam K, Rutherford E, Gilman JB, et al. Endemic focus of Fasciolopsis buski infection in Bangladesh. Am J Trop Med Hyg. 1982;31:796–802.
Jaroonvesama N, Charoenlarp K, Areekul S, Aswapokee N, Leelarasmee A. Prevalence of Fasciolopsis buski and other parasitic infections in residents of three villages in Sena District, Ayudhaya province, Thailand. J Med Assoc Thai. 1980;63:493–9.
Gupta A, Xess A, Sharma HP, Dayal VM, Prasad KM, Shahi SK. Fasciolopsis buski (giant intestinal fluke) - a case report. Indian J Pathol Microbiol. 1999;42:359–60.
Chen MX, Hu W, Li J, He JJ, Ai L, Chen JX. Identification and characterization of microRNAs in the zoonotic fluke Fasciolopsis buski. Parasitol Res. 2016;115:2433–8.
Bhattacharya S, Khurana S, Bhatti HS, Singhi S, Malla N. Polyparasitism: Fasciolopsis buski, Ascaris lumbricoides and discussion Hookworm coinfection in a child. Trop Gastroenterol. 2010;31:126–7.
Graczyk TK, Alam K, Gilman RH, Mondal G, Ali S. Development of Fasciolopsis buski (Trematoda: Fasciolidae) in Hippeutis umbilicalis and Segmentina trochoideus (Gastropoda: Pulmonata). Parasitol Res. 2000;86:324–6.
Mas-Coma S, Bargues MD, Valero MA. Fascioliasis and other plant-borne trematode zoonoses. Int J Parasitol. 2005;35:1255–78.
Singh UC, Kumar A, Srivastava A, Patel B, Shukla VK, Gupta SK. Small bowel stricture and perforation: an unusual presentation of Fasciolopsis buski. Trop Gastroenterol. 2011;32:320–2.
Karthikeyan G, Ramkumar V, Kumar SP, Ramkumar S, Selvamani S, Vetriveeran B, et al. Intestinal infestation with Fasciolopsis buski leading to acute kidney injury. J Assoc Physicians India. 2013;61:936–8.
Fiamma M, Longoni SS, Ngo TM, Le Phan MT, Santona A, Ton Nu PA, et al. Fasciolopsis buski infection in a Vietnamese pregnant woman with systemic lupus erythematosus. J Infect Dev Ctries. 2015;9:670–3.
Hadidjaja P, Dahri HM, Roesin R, Margono SS, Djalins J, Hanafiah M. First autochthonous case of Fasciolopsis buski infection in Indonesia. Am J Trop Med Hyg. 1982;31:1065.
Prasad PK, Goswami LM, Tandon V, Chatterjee A. PCR-based molecular characterization and insilico analysis of food-borne trematode parasites Paragonimus westermani, Fasciolopsis buski and Fasciola gigantica from Northeast India using ITS2 rDNA. Bioinformation. 2011;6:64–8.
Wiwanitkit V, Suwansaksri J, Chaiyakhun Y. High prevalence of Fasciolopsis buski in an endemic area of liver fluke infection in Thailand. MedGenMed. 2002;4:6.
Sen Sarma M, Yachha SK, Srivastava A, Poddar U. Endoscopic extraction of Fasciolopsis buski presenting as acute upper GI bleeding in a child. Gastrointest Endosc. 2015;82:743.
Le TH, Nguyen VD, Phan BU, Blair D, McManus DP. Case report: unusual presentation of Fasciolopsis buski in a Vietnamese child. Trans R Soc Trop Med Hyg. 2004;98:193–4.
Prasad PK, Tandon V, Chatterjee A, Bandyopadhyay S. PCR-based determination of internal transcribed spacer (ITS) regions of ribosomal DNA of giant intestinal fluke, Fasciolopsis buski (Lankester, 1857) Looss, 1899. Parasitol Res. 2007;101:1581–7.
Liu GH, Gasser RB, Young ND, Song HQ, Ai L, Zhu XQ. Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasit Vectors. 2014;7:150.
Biswal DK, Ghatani S, Shylla JA, Sahu R, Mullapudi N, Bhattacharya A, et al. An integrated pipeline for next generation sequencing and annotation of the complete mitochondrial genome of the giant intestinal fluke, Fasciolopsis buski (Lankester, 1857) Looss, 1899. PeerJ. 2013;1, e207.
Ma J, He JJ, Liu GH, Leontovyc R, Kasny M, Zhu XQ. Complete mitochondrial genome of the giant liver fluke Fascioloides magna (Digenea: Fasciolidae) and its comparison with selected trematodes. Parasit Vectors. 2016;9:429.
Ma J, He JJ, Liu GH, Zhou DH, Liu JZ, Liu Y, et al. Mitochondrial and nuclear ribosomal DNA dataset supports that Paramphistomum leydeni (Trematoda: Digenea) is a distinct rumen fluke species. Parasit Vectors. 2015;8:201.
Ma J, He JJ, Liu GH, Blair D, Liu LZ, Liu Y, et al. Mitochondrial genome of Ogmocotyle sikae and implications for phylogenetic studies of the Notocotylidae trematodes. Infect Genet Evol. 2015;37:208–14.
Yamaguti S. Systema Helminthum, vol. I. New York: The Digenetic Trematodes of Vertebrates. Interscience, Inc.; 1958.
Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, et al. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2007;1:3121–8.
Morgan J, Blair D. Nuclear rDNA ITS sequence variation in the trematode genus Echinostoma: an aid to establishing relationships within the 37-collar-spine group. Parasitology. 1995;111:609–15.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Lotfy WM, Brant SV, DeJong RJ, Le TH, Demiaszkiewicz A, Rajapakse RJ, et al. Evolutionary origins, diversification, and biogeography of liver flukes (Digenea, Fasciolidae). Am J Trop Med Hyg. 2008;79:248–55.
Castoe TA, Parkinson CL. Bayesian mixed models and the phylogeny of pitvipers (Viperidae: Serpentes). Mol Phylogenet Evol. 2006;39:91–110.
Chen YY, Chen S, Kang J, Fang H, Dao H, Guo WZ, et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PLoS One. 2014;9, e107578.
Le TH, Blair D, McManus DP. Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda). Parasitology. 2001;123:609–21.
Biswal DK, Chatterjee A, Bhattacharya A, Tandon V. The mitochondrial genome of Paragonimus westermani (Kerbert, 1878), the Indian isolate of the lung fluke representative of the family Paragonimidae (Trematoda). PeerJ. 2014;2, e484.
Liu GH, Yan HB, Otranto D, Wang XY, Zhao GH, Jia WZ, et al. Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol Phylogenet Evol. 2014;79:325–31.
Acknowledgements
The authors thank Ms Rong Li for technical assistance.
Funding
Project support was provided by the Fundamental Research Funds of Chinese Academy of Agricultural Sciences (Grant No. Y2016JC05) and the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. The complete mt genome sequence of F. buski is deposited in the GenBank database under the accession number KX169163.
Authors’ contributions
XQZ, MXC and JM designed this study and critically revised the manuscript. JM, MMS and JJH performed the experiments, analysed data and drafted the manuscript. GHL and LA participated in sample collection and manuscript revision. All the authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Sequences of primers used to amplify fragments of Fasciolopsis buski mitochondrial genome. (DOCX 17 kb)
Additional file 2: Table S2.
Nucleotide composition and skews of the Fasciolopsis buski mitochondrial protein-coding genes. (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ma, J., Sun, MM., He, JJ. et al. Fasciolopsis buski (Digenea: Fasciolidae) from China and India may represent distinct taxa based on mitochondrial and nuclear ribosomal DNA sequences. Parasites Vectors 10, 101 (2017). https://doi.org/10.1186/s13071-017-2039-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2039-2