
Abstract
Background
Representatives of the trematode family Fasciolidae are responsible for major socio-economic losses worldwide. Fascioloides magna is an important pathogenic liver fluke of wild and domestic ungulates. To date, only a limited number of studies concerning the molecular biology of F. magna exist. Therefore, the objective of the present study was to determine the complete mitochondrial (mt) genome sequence of F. magna, and assess the phylogenetic relationships of this fluke with other trematodes based on the mtDNA dataset.
Findings
The complete F. magna mt genome sequence is 14,047 bp. The gene content and arrangement of the F. magna mt genome is similar to those of Fasciola spp., except that trnE is located between trnG and the only non-coding region in F. magna mt genome. Phylogenetic relationships of F. magna with selected trematodes using Bayesian inference (BI) was reconstructed based on the concatenated amino acid sequences for 12 protein-coding genes, which confirmed that the genus Fascioloides is closely related to the genus Fasciola; the intergeneric differences of amino acid composition between the genera Fascioloides and Fasciola ranged 17.97–18.24 %.
Conclusions
The determination of F. magna mt genome sequence provides a valuable resource for further investigations of the phylogeny of the family Fasciolidae and other trematodes, and represents a useful platform for designing appropriate molecular markers.
Similar content being viewed by others


Background
Fascioloides magna (Bassi, 1875), the type- and only species of the genus Fascioloides Ward, 1917, was first described as Distomum magnum in 1875 [1]. Latter in 1917, Ward erected the genus Fascioloides for Fasciola magna (Bassi, 1875) [2]. Fascioloides magna, known as the large American liver fluke, giant liver fluke or deer fluke, is an important digenetic trematode of the family Fasciolidae [3, 4]. This species, which is of North America origin [5, 6] and invasive in European countries [7], has high potential to colonize new geographic territories (a variety of wild and domestic ungulates [3, 8–10]), and can establish expanding populations from a natural epidemic focus through translocated hosts [5, 6, 11]. Migration of F. magna immature flukes within the host body often leads to profound damage to the liver and other organ tissues [8, 12], causing economic losses worldwide [13].
The consequences of infection of various intermediate and definitive hosts by F. magna has been intensively studied [8, 12], but the relevant molecular research of this fluke has not received enough attention [4, 9]. To date, a sequence of nuclear ribosomal DNA (rDNA) of F. magna was obtained in 2008 [14], partial sequences of mitochondrial (mt) genes, such as cytochrome c oxidase subunit I (cox1) and nicotinamide dehydrogenase subunit I (nad1) were characterized [3]. According to these data, F. magna was divided into two mt haplotype groups [5, 14, 15], the first haplotype representing isolates from western North America and Italy, and the second haplotype representing isolates from eastern North America and some European countries such as Czech Republic, Poland and Croatia [3, 5]. Recently, the F. magna transcriptome was reported, which provides a useful platform for further fundamental studies of this fluke [16], but complete mt genome of F. magna is still unavailable.
Molecular tools, using genetic markers in mitochondrial DNA (mtDNA) sequences, have been proven reliable in identification and differentiation of trematode species [17–20]. In the present study we determined the mitochondrial genome sequence of F. magna (Czech isolate) using PCR-coupled sequencing technique combined with bioinformatic analysis, and for the first time assessed its phylogenetic relationship with selected trematodes based on the nucleotide- and inferred amino acid sequences of the protein-coding genes.
Methods
Sampling and DNA extraction
Three adult F. magna worms were isolated from livers of naturally infected red deer (Cervus elaphus), hunted at Kokořínsko area, Czech Republic. Worms were washed in 0.1 M phosphate-buffered saline (PBS), pH 7.2, fixed in 70 % (v/v) ethanol and preserved at -20 °C, until further use. Total genomic DNA was extracted from individual F. magna specimens using sodium dodecyl sulfate (SDS)/proteinase K treatment [21] and column-purification (Wizard® SV Genomic DNA Purification System, Promega, Madison, USA), according to the manufacturer’s protocol.
Acquisition of ITS rDNA and sample identification
The internal transcribed spacer (ITS) rDNA region of each of the three F. magna specimens, spanning partial 18S rDNA, the complete ITS-1, 5.8S rDNA, ITS-2, and partial 28S rDNA, was amplified using primers BD1 (forward; 5’-GTC GTA ACA AGG TTT CCG TA-3’ and BD2 (reverse; 5’-ATG CTT AAA TTC AGC GGG T-3’) [22] and sequenced using the same primers. These F. magna samples had ITS-1 and ITS-2 sequences identical to the corresponding sequences available on GenBank (EF051080).
Long-range PCR-based sequencing of mt genome
The primers were designed based on relatively conserved regions of mtDNA sequences from Fasciola hepatica and Fasciola gigantica. The entire mt genome from a single specimen of F. magna was amplified in 5 overlapping fragments, using the primers shown in Additional file 1: Table S1.
PCR reactions were conducted in a total volume of 50 μl, using 25 μl PrimeStar Max DNA polymerase premix (Takara, Dalian, China), 25 pmol of each primer (synthesized in Genewiz, Suzhou, China), 0.5 μl DNA templates, and H2O, in a thermocycler (Biometra, Göttingen, Germany). PCR cycling conditions started with an initial denaturation at 98 °C for 2 min, followed by 22 cycles of denaturation at 92 °C for 18 s, annealing at 52–65 °C for 12 s and extension at 60 °C for 1–5 min, followed by 92 °C denaturation for 2 min, plus 25 cycles of 92 °C for 18 s (denaturation), 50–67 °C for 12 s (annealing) and 66 °C for 3–6 min, with a final extension step for 10 min at 66 °C. A negative control (no DNA) was included in each amplification run. Amplicons (2.5 μl) were electrophoresed in a 2 % agarose gel, stained with Gold View I (Solarbio, Beijing, China) and photographed by GelDoc - It TS™ Imaging System (UVP, USA).
Assembly, annotation and bioinformatics analysis
Sequences were assembled manually and aligned against the entire mt genome sequences of Fa. hepatica (GenBank accession No. NC_002546) and Fa. gigantica (NC_024025) using MAFFT 7.122 to infer boundaries for each gene. Amino acid sequences of 12 protein-coding genes were translated using MEGA v.6.06 and NCBI translation Table 21 (Trematode Mitochondrial Code). The tRNA genes were affirmed using the programs tRNAscan-SE [23] and ARWEN (http://130.235.46.10/ARWEN/) or by comparison with those from the Fa. hepatica and Fa. gigantica mt genomes. The two rRNA genes were identified by comparison with those of Fa. hepatica and Fa. gigantica.
A comparative analysis of the nucleotide sequences of each protein-coding gene, the amino acid sequences, two ribosomal RNA genes, 22 tRNA genes as well as non-coding regions (NCRs) among F. magna, Fa. hepatica and Fa. gigantica was conducted.
Phylogenetic analysis
The concatenated amino acid sequences of F. magna mt genome, conceptually translated from individual genes of each mt genome, were aligned with those of published mt genomes from selected trematodes, including Opisthorchis felineus (GenBank acession No. EU_921260) and Clonorchis sinensis (FJ_381664) (Opisthorchiidae); Metagonimus yokogawai (KC_330755) and Haplorchis taichui (KF_214770) (Heterophyidae); Paragonimus westermani Japanese isolate (AF219379) and Paragonimus westermani Indian isolate (NC_027673) (Paragonimidae); Fa. hepatica, Fasciola sp. (KF_543343) and Fa. gigantica (Fasciolidae); Hypoderaeum sp. (KM111525) (Echinostomatidae); Paramphistomum leydeni (KP341657) and Fischoederius elongatus (KM397348) (Paramphistomatidae); Diplostomum spathaceum (KR269763) and Diplostomum pseudospathaceum (KR269764) (Diplostomidae); Ogmocotyle sikae (KR006934) (Notocotylidae); Eurytrema pancreaticum (KP241855) (Dicrocoeliidae); Schistosoma turkestanicum (HQ_283100) and Schistosoma japonicum (HM_120842) (Schistosomatidae). The sequence for the monogenean Gyrodactylus derjavinoides (NC_010976) (Gyrodactylidae), was included as the outgroup.
All inferred amino acid sequences were aligned using MAFFT 7.122. Poorly aligned sites and divergent regions of the alignment were eliminated using Gblocks Server v. 0.91b (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using default settings, selecting the option of less strict conservation of flanking positions. The alignment was then converted into nexus format using Clustal X1.83 and subjected to phylogenetic analysis using Bayesian inference (BI). A mixed model was used in BI analysis using MrBayes 3.1.1 [24], because the most suitable amino acid evolution model JTT + G + F, selected by ProTest 3.4 based on the Akaike information criterion (AIC) [25], was not available in the current MrBayes version. Four independent Markov chain were run for 10,000,000 metropolis-coupled MCMC generations, sampling trees every 1,000 generations. The first 2,500 trees (25 %) were discarded as ‘burn-in’, and the remaining trees were used for calculating Bayesian posterior probabilities. The analysis was regarded as completed when the potential scale reduction factor was close to 1, and the average standard deviation of split frequencies was below 0.01. Phylograms were prepared using FigTree v. 1.42 [26].
Findings
Genome content and organization
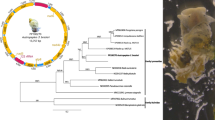
The complete mt genome sequence of F. magna (GenBank accession no. KR006934) is 14,047 bp in length (Fig. 1) and contains 36 genes that are transcribed in the same direction, including 12 protein-coding genes (nad1-6, nad4L, cox1-3, atp6 and cytb), 22 tRNA genes and two rRNA genes (rrnL and rrnS), lacking the atp8 gene (Table 1), consistent with those of selected trematode species available on GenBank [17–19, 27, 28]. There is only one NCR in F. magna mt genome, whereas the mt genomes of Fasciola flukes have two non-coding regions [17, 27].

Organization of the mitochondrial genome of Fascioloides magna. The scales are approximate. All genes are transcribed in the clockwise direction, using standard nomenclature. “NCR” refers to the only non-coding region in F. magna data. The A + T content is shown for each gene or region of the mt genome and represented by colour
The arrangement of genes in the F. magna mt genome is similar to that of Fasciola spp. [17], except that only one non-coding region (NCR) in F. magna mt genome is located between trnE (13,355–13,422) and cox3 (1–645) (Table 1). The gene order of F. magna mt genes is similar to that in species of the Paramphistomatidae, Notocotylidae, Echinostomatidae, Heterophyidae and Opisthorchiidae, but is distinct from some flukes of the Schistosomatidae (S. mansoni, S. spindale and S. haematobium) [29].
The nucleotide composition of F. magna mt genome is obviously biased towards A and T. The value of total A + T content for F. magna mtDNA is 61.42 %, within the range recognized in other trematode mt genomes (54.38 % in Paragonimus westermani Indian isolates [30], 72.71 % in Schistosoma spindale [29]). The content of C is low (10.3 %) and that of T is high (44.0 %). The A + T content for each gene or region of F. magna mt genome ranged from 48.48 % (trnL2) to 68.18 % (trnG) (nad3, 64.43 %; cox2, 59.7 %). All 12 protein-coding genes of F. magna mtDNA possess a lower A + T percentage than those of Fa. hepatica and Fa. gigantica [17, 27], except for nad5 (Additional file 2: Table S2).
Annotation of F. magna mt genome
In the mt genome of F. magna, the protein-coding genes had ATG or GTG as start codons and TAG or TAA as stop codons (Table 1). Half of the protein-coding genes of F. magna were initiated with GTG (nad4L, nad4, nad1, cox1, nad6 and nad5). Incomplete codons were not detected in the mt genome of F. magna.
The 22 tRNA genes of F. magna mt genome ranged from 57 to 69 bp in length. The structure of all tRNA sequences is similar to those of Fa. hepatica and Fa. gigantica [17, 27]. The large ribosomal RNA gene (rrnL) and the adjacent small ribosomal RNA gene (rrnS) are located between trnT and cox2, and separated by trnC (9,456–9,518) (Table 1). The length of the rrnL and rrnS RNA genes is 984 bp and 765 bp, respectively. The only NCR of F. magna mt genome is of 520 bp in length, and is located between trnE and cox3. It contains two complete direct repeats: six copies of a 23 nt - repeat A (AGA TAG GAT AGG CAT CTG GTA TA) and five copies of a 37 nt - repeat B (GGT GCC CCC GGT GAA GGG GGA AAA GGA AGG TTG TAA G). There are five AB repeats, with one A at the end (located at positions 13,620–13,642).
Comparative analysis among mt genomes of F. magna, Fa. hepatica and Fa. gigantica
The difference between complete mt genomes of F. magna and Fa. hepatica was 22.66 % (3,290 nt), which is close to that between F. magna and Fa. gigantica (22.65 %, 3,297 nt) (Table 2). Considering the 12 protein-coding genes, different nucleotides were present at 18.80 % of positions (1,897 nt) between F. magna and Fa. hepatica, and at 18.62 % of positions (1,879 nt) between F. magna and Fa. gigantica. At the inferred amino acid level, there were 605 substitutions (17.97 %) of amino acids between F. magna and Fa. hepatica, and 614 substitutions (18.24 %) between F. magna and Fa. gigantica (Table 2).
At the nucleotide level, sequence differences in protein-coding genes ranged from 13.1 to 24.2 % (between F. magna and Fa. hepatica) and from 12.8 to 26.2 % (between F. magna and Fa. gigantica), with cox1, nad1, nad4L and cytb being the most conserved genes, and nad6, nad5 and nad2 being the least conserved genes among those three species. At the amino acid level, sequence differences ranged from 9.2 to 25.4 % between F. magna and Fa. hepatica, and from 8.4 to 27.8 % between F. magna and Fa. gigantica: cox1, cytb, nad4L and nad1 were the most conserved protein-coding genes, while nad6, nad2 and nad5 were the least conserved.
Comparisons between the mt genomes of F. magna and Fasciola spp., at both nucleotide and amino acid levels, indicate that the most conserved and the least conserved gene in the Fasciolidae are cox1 and nad6, respectively. Besides, the nad5 is highly variable, and genes of nad4L and cytb are rather conserved. These characteristics are in accordance with flukes of the families Paramphistomatidae and Notocotylidae [18, 28].
Nucleotide differences were also found in ribosomal RNA genes: between F. magna and Fa. hepatica (rrnL, 18.3 %; rrnS, 22.2 %) and between F. magna and Fa. gigantica (rrnL, 16.6 %; rrnS, 21.4 %) as well as in tRNA genes (16.3 % between F. magna and Fa. hepatica and 16.0 % between F. magna and Fa. gigantica). Meaningful sequence comparisons of NCRs in mt genomes of the three fasciolid trematodes is not possible, because there is only one NCR present in F. magna mt genome, while in both Fa. hepatica and Fa. gigantica there are two NCRs.
Phylogenetic analysis
In the phylogenic tree inferred from the concatenated amino acid sequence dataset of all 12 mt proteins (Fig. 2) F. magna clustered with three other Fasciola species with strong support (Bpp = 1). The closest family to the Fasciolidae is Echinostomatidae, represented by Hypoderaeum sp. The taxonomic relationships of the selected trematodes are in concordance with results of previous studies [17–19, 28]. Each node received the maximum possible nodal support (Bpp = 1).

Phylogenetic relationships of Fascioloides magna and other trematodes. Tree inferred from the concatenated amino acid sequence dataset for 12 protein-coding genes from 19 trematodes was performed by Bayesian inference (BI). Gyrodactylus derjavinoides (NC_010976) was chosen as the outgroup
In several recent phylogenetic studies, the F. magna was characterized only based on partial 28S rDNA [31] and combined ITS1, ITS2 and nad1 sequences [32]. The relationship between the genera Fasciola and Fasciolopsis was considered as being very close and the genetic relationship between F. magna and Fasciola jacksoni (or Fascioloides jacksoni) is disputable [31–33]. Further studies are warranted to determine the mt genome of Fa. jacksoni and solve this controversy in the family Fasciolidae.
Conclusions
The present study determined the complete mt genome sequence of the pathogenic liver fluke F. magna and revealed its close relationship with the species of Fasciola. The complete mt genome data of F. magna provides a resource for further investigations of the phylogeny, epidemiology, biology and population genetics of the family Fasciolidae and other trematodes.
Abbreviations
mt, mitochondrial; mtDNA, mitochondrial DNA; rDNA, ribosomal DNA; BI, Bayesian inference; PBS, phosphate-buffered saline; SDS, sodium dodecyl sulphate; ITS, internal transcribed spacer; NCR, non-coding region
References
Bassi R. Sulla cachessia ittero-vermicosa, o marciaia dei Cervi, causata dal Distomum magnum. Medic Vet. 1875;4:497–515.
Ward HB. On the structure and classification of North American parasitic worms. J Parasitol. 1917;4:1–12.
Králová Hromadová I, Bazsalovicsová E, Štefka J, Špakulová M, Vávrová S, Szemes T, et al. Multiple origins of European populations of the giant liver fluke Fascioloides magna (Trematoda: Fasciolidae), a liver parasite of ruminants. Int J Parasitol. 2011;41:373–83.
Pybus M. Survey of hepatic and pulmonary helminths of wild cervids in Alberta Canada. J Wildlife Dis. 1990;26:453–9.
Bazsalovicsová E, Králová Hromadová I, Minárik G, Bokorová S, Pybus M. Genetic interrelationships of North American populations of giant liver fluke Fascioloides magna. Parasit Vectors. 2015;8:288.
Kašný M, Beran L, Siegelová V, Siegel T, Leontovyč R, Beránková K, et al. Geographical distribution of the giant liver fluke (Fascioloides magna) in the Czech Republic and potential risk of its further spread. Vet Med. 2012;57:101–9.
Ursprung J, Joachim A, Prosl H. Incidence and control of the American giant liver fluke, Fascioloides magna, in a population of wild ungulates in the Danubian wetlands east of Vienna. Berl Munch Tierarztl. 2005;119:316–23.
Wobeser BK, Schumann F. Fascioloides magna infection causing fatal pulmonary hemorrhage in a steer. Can Vet J. 2014;55:1093.
Foreyt W, Todd A. Development of the large American liver fluke, Fascioloides magna, in white-tailed deer, cattle, and sheep. J Parasitol. 1976;62:26–32.
Malcicka M. Life history and biology of Fascioloides magna (Trematoda) and its native and exotic hosts. Ecol Evol. 2015;5:1381–97.
Karamon J, Larska M, Jasik A, Sell B. First report of the giant liver fluke (Fascioloides magna) infection in farmed fallow deer (Dama dama) in Poland - pathomorphological changes and molecular identification. B Vet I Pulawy. 2015;59:339–44.
Pybus MJ. Liver flukes. In: Samuel WM, Pybus MJ, Kocan AA, editors. Parasitic diseases of wild mammals. Ames, Iowa: Iowa State Press; 2001. p. 121–49.
Semyenova SK, Morozova EV, Chrisanfova GG, Gorokhov VV, Arkhipov IA, Moskvin AS, et al. Genetic differentiation in eastern European and western Asian populations of the liver fluke, Fasciola hepatica, as revealed by mitochondrial nad1 and cox1 genes. J Parasitol. 2006;92(3):525–30.
Králová Hromadová I, Špakulová M, Horáčková E, Turčeková L, Novobilský A, Beck R, et al. Sequence analysis of ribosomal and mitochondrial genes of the giant liver fluke Fascioloides magna (Trematoda: Fasciolidae): intraspecific variation and differentiation from Fasciola hepatica. J Parasitol. 2008;94(1):58–67.
Bazsalovicsová E, Králová Hromadová I, Radvánszky J, Beck R. The origin of the giant liver fluke, Fascioloides magna (Trematoda: Fasciolidae) from Croatia determined by high-resolution melting screening of mitochondrial cox1 haplotypes. Parasitol Res. 2013;112:2661–6.
Cantacessi C, Mulvenna J, Young ND, Kašný M, Horák P, Aziz A, et al. A deep exploration of the transcriptome and “excretory/secretory” proteome of adult Fascioloides magna. Mol Cell Proteomics. 2012;11:1340–53.
Liu GH, Gasser RB, Young ND, Song HQ, Ai L, Zhu XQ. Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasit Vectors. 2014;7:150.
Ma J, He JJ, Liu GH, Zhou DH, Liu JZ, Liu Y, et al. Mitochondrial and nuclear ribosomal DNA dataset supports that Paramphistomum leydeni (Trematoda: Digenea) is a distinct rumen fluke species. Parasit Vectors. 2015;8:201.
Brabec J, Kostadinova A, Scholz T, Littlewood DTJ. Complete mitochondrial genomes and nuclear ribosomal RNA operons of two species of Diplostomum (Platyhelminthes: Trematoda): a molecular resource for taxonomy and molecular epidemiology of important fish pathogens. Parasit Vectors. 2015;8:336.
Yang X, Gasser RB, Koehler AV, Wang LX, Zhu KX, Chen L, et al. Mitochondrial genome of Hypoderaeum conoideum - comparison with selected trematodes. Parasit Vectors. 2015;8:97.
Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, et al. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2007;1:3121–8.
Morgan J, Blair D. Nuclear rDNA ITS sequence variation in the trematode genus Echinostoma: an aid to establishing relationships within the 37-collar-spine group. Parasitology. 1995;111:609–15.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Castoe TA, Parkinson CL. Bayesian mixed models and the phylogeny of pitvipers (Viperidae: Serpentes). Mol Phylogenet Evol. 2006;39:91–110.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Chen YY, Chen S, Kang J, Fang H, Dao H, Guo WZ, et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PLoS One. 2014;9:e107578.
Le TH, Blair D, McManus DP. Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda). Parasitology. 2001;123:609–21.
Ma J, He JJ, Liu GH, Blair D, Liu LZ, Liu Y, et al. Mitochondrial genome of Ogmocotyle sikae and implications for phylogenetic studies of the Notocotylidae trematodes. Infect Genet Evol. 2015;37:208–14.
Littlewood DTJ, Lockyer AE, Webster BL, Johnston DA, Le TH. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol. 2006;39:452–67.
Biswal DK, Chatterjee A, Bhattacharya A, Tandon V. The mitochondrial genome of Paragonimus westermani (Kerbert, 1878), the Indian isolate of the lung fluke representative of the family Paragonimidae (Trematoda). Peer J. 2014;2:e484.
Tkach VV, Kudlai O, Kostadinova A. Molecular phylogeny and systematics of the Echinostomatoidea Looss, 1899 (Platyhelminthes: Digenea). Int J Parasitol. 2015;46:15.
Heneberg P. Phylogenetic data suggest the reclassification of Fasciola jacksoni (Digenea: Fasciolidae) as Fascioloides jacksoni comb. nov. Parasitol Res. 2013;112:1679–89.
Lotfy WM, Brant SV, DeJong RJ, Le TH, Demiaszkiewicz A, Rajapakse RJ, et al. Evolutionary origins, diversification, and biogeography of liver flukes (Digenea, Fasciolidae). Am J Trop Med Hyg. 2008;79:248–55.
Acknowledgements
The authors thank Ms Miao-Miao Sun for technical assistance.
Funding
Project support was provided in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840) and the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037). This research was also supported by grants of the Grant Agency of the Charles University in Prague (Grant No. 502313), Masaryk University institutional support (Grant No. MUNI/A/1325/2015), and Charles University institutional/departmental support (Grant No. PRVOUK P41, UNCE 204017 and SVV 260202/2015).
Availability of data and material
The datasets supporting the conclusions of this article are included within the article and its additional files. The complete mt genome sequence of F. magna is deposited in the GenBank database under the accession number KR006934.
Authors’ contributions
XQZ and MK designed this study and critically revised the manuscript. JM and JJH performed the experiments, analysed data and drafted the manuscript. GHL and RL participated in sample collection and manuscript revision. All the authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Charles University, Czech Republic.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Sequences of primers used to amplify fragments of Fascioloides magna mitochondrial genome. (DOCX 13 kb)
Additional file 2: Table S2.
Comparison of A + T content of mitochondrial genomes of Fascioloides magna (Fm), Fasciola hepatica (Fh) and Fasciola gigantica (Fg). (DOCX 20 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ma, J., He, JJ., Liu, GH. et al. Complete mitochondrial genome of the giant liver fluke Fascioloides magna (Digenea: Fasciolidae) and its comparison with selected trematodes. Parasites Vectors 9, 429 (2016). https://doi.org/10.1186/s13071-016-1699-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1699-7