
Abstract
The escalating threat posed by the Monkeypox virus (MPXV) to global health necessitates the urgent discovery of effective antiviral agents, as there are currently no specific drugs available for its treatment, and existing inhibitors are hindered by toxicity and poor pharmacokinetic profiles. This study aimed to identify potent MPXV inhibitors by screening a diverse library of small molecule compounds derived from marine fungi, focusing on the viral protein VP39, a key methyltransferase involved in viral replication. An extensive virtual screening process identified four promising compounds—CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569—alongside a control molecule. Rigorous evaluations, including re-docking, molecular dynamics (MD) simulations, and hydrogen bond analysis, were conducted to assess their inhibitory potential against MPXV VP39. CMNPD15724 and CMNPD30883, in particular, demonstrated a superior binding affinity and stable interactions within the target protein's active site throughout the MD simulations, suggesting a capacity to overcome the limitations associated with sinefungin. The stability of these VP39-compound complexes, corroborated by MD simulations, provided crucial insights into the dynamic behavior of these interactions. Furthermore, Principal Component Analysis (PCA) based free energy landscape assessments offered a detailed understanding of the dynamic conformational changes and energetic profiles underlying these compounds' functional disruption of VP39. These findings establish CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 as promising MPXV inhibitors and highlight marine fungi as a valuable source of novel antiviral agents. These compounds represent potential candidates for further experimental validation, advancing the development of safer and more effective therapeutic options to combat this emerging viral infection.
Similar content being viewed by others
Introduction
Monkeypox virus (MPXV) is a zoonotic viral infection that has garnered attention as it emerges as a global health threat. Bearing significant homology to the Variola virus, the causative agent of smallpox, MPXV has been newly implicated in outbreaks revealing its pathogenic potential [1]. At the heart of these pathogenic processes is the viral methyltransferase VP39, a replicase enzyme essential for mRNA capping, a process required to cap viral mRNAs to stabilize them [2], and conceal them from the immune system [3, 4]. Understanding the molecular mechanics of VP39 in the epidemiology and pathogenesis of Monkeypox is vital to the development of the next generation of antiviral therapeutics.
The MPXV is an Orthopoxvirus belonging to the Poxviridae family [5], which possesses a complicated double-stranded DNA genome [6]. This genome encodes a variety of proteins, including the methyltransferase VP39 [7], which is essential to the viral life cycle. The role of VP39 in the mRNA capping process is crucial for preventing viral mRNA degradation and ensuring efficient translation [4]. This distinct process distinguishes the virus from its host and opens the door to focused therapeutic interventions. Understanding the genetic architecture of the virus, as well as the specific role of VP39 in the virus's replication machinery, is critical for the development of effective antiviral medications. Because of its specific involvement in the MPXV, VP39 is an appealing target for antiviral medication research. Inhibiting VP39 may impair the virus's capacity to multiply and elude detection by the host immune system. This method holds the potential for creating specialized antiviral medicines with few off-target impacts on host cells. The development of VP39 inhibitors necessitates a thorough understanding of the enzyme’s structure and role in the viral replication cycle. Targeting VP39 may not only impede viral replication but also increase the virus’s visibility to the immune system, giving a dual mode of action against Monkeypox. Recent Monkeypox outbreaks, characterized by close contact and respiratory droplets [8], have underlined the need for better therapies. The virus's global expansion, with instances recorded on various continents, emphasizes the importance of developing targeted antiviral therapies. Because of its crucial role in the virus's life cycle, the enzyme VP39 emerges as a key target in this attempt. Inhibiting VP39 effectively could result in a considerable reduction in viral replication, assisting in the control and management of Monkeypox epidemics. Understanding the epidemiology of Monkeypox, including transmission mechanisms and the function of VP39 in pathogenicity, is critical for developing targeted and effective antiviral medicines. With their unique ability to create a diverse variety of bioactive chemicals, marine fungi represent an untapped resource for antiviral medication discovery [9]. Adapted to the harsh maritime environment, these organisms have evolved to generate secondary metabolites with a wide range of chemical structures and biological functions [9]. Recent studies have begun to look into the possibility of these chemicals as sources of novel antiviral medicines [10]. The chemical diversity of marine fungi-derived chemicals represents a promising pathway for the discovery of new treatments, notably in the fight against developing viral infections.
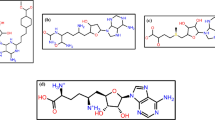
Silhan and his team conducted a study on designing sinefungin analogs (Fig. 1) [2]. This study demonstrated that the inhibitor TO1119 features a tiny iodine atom at the 7-deaza position. This compound causes minimal disruption to the water network within the binding site, resulting in an IC50 of 128 nM. Despite its relatively small substituent, TO1119 effectively binds within the water cavity adjacent to the sinefungin binding groove, illustrating the importance of subtle modifications for maintaining inhibitory activity. In contrast, inhibitors such as TO427, TO494, TO500, TO504, and TO507 possess large aromatic or heterocyclic substituents at the 7-deaza position. These modifications lead to a significant rearrangement of the water molecules in the binding site. These inhibitors demonstrate markedly improved efficacy compared to sinefungin, with IC50 values ranging from 0.08 to 0.17 μM. Notably, TO500 includes a benzimidazole heterocycle capable of forming a hydrogen bond, which contributes to its effective inhibition profile. These structural changes enhance the binding affinity and inhibitory potency, making them promising candidates for antiviral drug development [2].

Structures of previously reported sinefungin (SFG) analogs with potential inhibitory effects against Mpox methyltransferase VP39
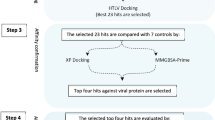
We used a computational drug discovery strategy to identify potential inhibitors of Monkeypox methyltransferase VP39 from a library of marine fungus compounds in this study [11]. Virtual screening with Lipinski's rule of five, re-docking, molecular dynamics simulations, free binding energy calculations, and PCA-based free energy landscape analysis were all part of our process. This thorough approach enabled us to uncover interesting compounds that could block VP39, opening up new avenues for the development of effective Monkeypox antiviral treatments. Not only do our findings advance antiviral research, but they also highlight the potential of marine fungi as a rich source of novel medicinal compounds.
Materials and methods
Data collection, virtual screening and re-docking
This study focused on computational techniques to discover possible inhibitors of Monkeypox methyltransferase VP39. We utilized a Marine Fungi Compounds library, focusing on compounds that would demonstrate a putative inhibitory effect against VP39 [12]. The process involved virtual screening with Lipinski’s rule of five filters, followed by re-docking of the selected compounds. The initial step in our methodology was the collection of target protein and ligand data. The 3D structure of Monkeypox methyltransferase VP39 was retrieved from the Protein Data Bank (PDB) with the PDB ID 8B07 [2]. The PDB is a widely recognized archive for the 3D structures of major biological molecules [13]. The Marine Fungi Compounds library was downloaded from the Comprehensive Marine Natural Products Database (CMNPD) [14]. This database provides a large number of compounds, each with unique structures and characteristics, making them good candidates for virtual screening. The VP39 protein was prepared for docking using the Dock Prep function in the Chimera program [15]. This preparation involves many key steps such as the insertion of hydrogen atoms to ensure the right ionization and tautomeric states of amino acids, the removal of water molecules to simulate an anhydrous environment, and the assignment of Kollman charges to the protein. These procedures were necessary for accurately the docking of ligands with the protein. The MtiOpenScreen webserver was deployed for virtual screening [16]. This tool allows us to apply Lipinski's rule of five as a filter. Lipinski's rule is vital for assuring the drug-likeness of the compounds by analyzing factors such as molecular weight, lipophilicity, and hydrogen bond donors and acceptors [17]. We conducted a thorough screening of the Marine Fungi Compounds library against the VP39 protein structure. The screening procedure was automated, allowing us to efficiently examine 330 compounds obtained after applying Lipinski's rule (Supplementary Table S1). For this screening, the center coordinates (X = − 3.5, Y = 18.0 and Z = − 33.38) and grid size (X = 20, Y = 20 and Y = 20). The upshot of this screening was the selection of four compounds that displayed the highest docking scores, indicating a substantial potential for interaction with the VP39 protein. Following the virtual screening, a re-docking process was done to further validate the results. For this, we used the AutoDock Vina [18] plugin within the Chimera interface [15]. Similar methodologies were used in recent research [19, 20]. This plugin is noted for its quickness and accuracy in docking simulations. The selected four compounds, together with a reference molecule for comparison, were re-docked against the VP39 protein. This phase was crucial to confirm the initial binding affinities and to acquire a more complete understanding of the interaction between the chemicals and the protein. We verified that the re-docking circumstances were consistent with the first screening to retain the validity of our comparisons. The re-docking method not only corroborated our previous findings but also gave further insights into the binding mechanisms and orientations of the compounds within the VP39 active region. This knowledge is essential for understanding how these substances could affect the action of the VP39 enzyme.
Molecular dynamic simulation
We conducted 200 ns Molecular Dynamics (MD) simulations and trajectory analysis to study the interactions between Monkeypox methyltransferase VP39 with the top four scoring putative inhibitors identified as virtual screening and redocking [21]. Our methodology includes the utilization of the CHARMM—36 force field [22, 23] within the GROMACS version 2023.3 [24, 25] simulation software, focused on four selected chemical complexes and one contol molecule. The MD simulations begin with the creation of the VP39-inhibitor complexes. Each complex containing a ligand was placed in the center of a cubic simulation box. The complexes were solvated with TIP3P water [26], guaranteeing a buffer distance of at least 1 nm from the protein to the border of the box. To simulate physiological ionic strength, sodium and chloride ions were introduced, neutralizing the system and obtaining a 0.15 M concentration. Before the dynamics, an energy minimization phase was required to remove steric conflicts and geometric inconsistencies. This was achieved using the steepest descent minimization method [27], continuing until the maximal force was less than 1000 kJ/mol/nm. This procedure guaranteed that the systems were appropriately relaxed for the upcoming MD simulations after that we performed equilibration in two phases—NVT followed by NPT. In the NVT phase, each system was equilibrated at 310 K using a V-rescale thermostat. This was followed by the NPT phase, where the pressure was maintained at 1 bar using the Parrinello-Rahman barostat [28]. Each phase lasted 100 ps, allowing the systems to establish a stable temperature and pressure. The production MD simulations were done using the CHARMM force field [22]. We ran simulations for a time scale sufficient to capture the relevant dynamic characteristics of the complexes, generally lasting 200 ns. During this interval, the temperature and pressure were controlled as per the equilibration phases, and periodic boundary conditions were applied in all directions to resemble an endless system. The obtained trajectories were evaluated for several metrics including RMSD, RMSF, hydrogen bonding patterns, and binding interactions. RMSD gave insights into the general stability of the complexes, while RMSF revealed the flexibility of individual residues in the protein. Additionally, analysed the quantity and length of hydrogen bonds between VP39 and the inhibitors to assess the strength and specificity of the interactions. PCA was utilized to deconstruct the MD trajectories into primary motions. This study helps in detecting the significant conformational changes during the simulation. The free energy landscapes were built based on these primary components, offering a visual representation of the conformational states explored by the protein–ligand complexes. These landscapes allowed us to pinpoint stable conformations and transitional states, affording vital insights into the dynamic behavior of the complexes.
Principal component analysis based on free energy landscape and structural analysis
The PCA was conducted for MD simulation trajectories of the VP39 complexes. This selection attempted to capture the most relevant dynamic interactions between the protein and the inhibitors. We focused on the backbone atoms of the VP39 protein to underline the key motions controlling ligand binding. Utilizing the gmx sham module within GROMACS [24, 25], we retrieved the principal components (PCs) from the trajectory data. This procedure required reducing the multidimensional dataset to determine the fundamental axes of motion. PCA showed substantial conformational changes and patterns within the protein–ligand interactions, providing insight into the dynamic features of binding. For generating the scattered plot based on the PC1 and PC2 obtained the ‘gmx anaproj’ command was utilized [29]. The PCs served as coordinates for creating the free energy landscapes. Using Python's Matplotlib tool, we plotted these PCs against each other to depict the energy states traversed by the complexes during the simulations [30, 31]. The landscapes displayed the distribution of structural states, with valleys suggesting stable low-energy conformations. The free energy landscapes were studied to determine the stability and flexibility of the VP39 complexes. Key observations included identifying stable conformational basins and transition states, which are indicative of the binding affinities and specificities of the inhibitors. This approach allowed us to find energetically favorable binding modes and to forecast the probable efficacy of the inhibitors. The extracted structures were superimposed and visualized in UCSF Chimera [32].
Results
Virtual screening
Virtual screening is a computational-based drug discovery approach utilized for the identification of possible therapeutic compounds from the enormous database of chemical compounds. Virtual screening aims to prioritize chemical compounds that are expected to exhibit biological activity against a target molecule, such as a protein linked to a disease, this will assist in lowering the quantity of chemicals that require experimental testing, saving time and money in the process. Virtual screening was utilized in this study to identify the four natural chemicals derived from the Marine Fungi Compounds library following the virtual screening, the top compounds were chosen based on binding energy. For the screening purpose, a total of 4683 compounds library was selected, based on the binding score obtained in the virtual screening, the top four compounds named CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 with binding scores of − 8.60 kcal/mol, − 7.63 kcal/mol, − 7.53 kcal/mol, and − 7.52 kcal/mol respectively were chosen for further study along with one control (sinefungin).
Redocking and interaction analysis
Redocking is a technique used in the computational-based drug discovery strategy to evaluate the precision of the molecular docking protocol and investigate the significant interaction between the ligands and binding site residues of the protein of interest. During the re-docking procedure, nine poses for each complex were obtained. The one with the highest negative binding energy was chosen to analyze the selected complex's stability and binding affinity. Additionally, sinefungin as a control molecule was used for the comparative study. During the redocking individual compounds' obtained good scores such as compounds CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 with binding scores of − 9.4 kcal/mol, − 8.9 kcal/mol, − 9.2 kcal/mol, and − 9.4 kcal/mol respectively and control sinefungin scored -8.9 kcal/mol in the redocking (Center gride was used X = − 3.5, Y = 18.0 and Z = − 33.38), as illustrated in Table 1. The list of methyltransferase VP39 amino acid residues involved in various types of molecular interactions with the selected compounds is given in Table 2 and plotted in Fig. 2. The CMNPD15724-protein complex has a strong contact, as evidenced by the formation of eight hydrogen bonds between the chemical and the protein residues Asp138, Ser141, Val116, and Arg97. Arg140 and Asp95 also form two hydrogen bonds with Asp95 and Arg140, a pi-pi stacking interaction with Phe115, and two halogen bonds with Ile94 and Val116 and eight hydrophobic interactions with Leu42, Val139, Ala70, Ile67, Leu159, Val116, Ile94, Phe115 (Fig. 2a). These numerous interactions point to CMNPD15724 having a robust and well-defined binding mode. Through docking interactions, compound CMNPD28811 displays significant binding affinity, including one hydrogen bond with Asp138 and two pi-pi stacking interactions with Phe115 and seven hydrophobic interactions with Leu159, Val116, Phe115, Ile94, Val139, Ile67 and Ala70 (Fig. 2c). These interactions imply that CMNPD28811 adopts a unique orientation relative to the protein, which contributes to its high binding potential. The CMNPD30883-protein complex interacts in a variety of ways, including two hydrogen bonds with Gly72 and Gln39, a salt bridge with Arg97 and ten hydrophobic interactions with Val116, Phe115, Leu159, Tyr66, Ile67, Ala70, Pro71, Leu42, Ile75, Val139 (Fig. 2e). This broad set of contacts suggests that CMNPD30883 operates in a flexible binding mechanism that allows it to adapt to different protein conformational states. Gly68, Asp138, Arg140, Asp95, Arg97, and Val116 form seven hydrogen bonds in the CMNPD18569 complex Val116 formed a double bond (Fig. 2g). Furthermore, Phe115 interacts via one pi-pi stacking and seven hydrophobic bonds with Leu159, Val116, Phe115, Val139, Ala70, Ile67 and Ile94. These interactions imply that CMNPD18569 has a highly structured binding mechanism, which may contribute to its specificity and selectivity. The control complex forms eight hydrogen bonds with Asp95, Arg140, Asp138, Try66, Gln39, and Gly72 residues, as well as a double hydrogen bond with Val116, one Pi-Pi stacking with Phe115 and eleven hydrophobic interaction Ile94, Phe115, Val116, Leu159, Ile75, Leu42, Pro71, Ala70, Ile67, Tyr66 and Val139. This differential binding pattern indicates that the control compound has a different conformation and orientation than the CMNPD compounds. Overall, the examination of these interactions reveals the varied binding mechanisms and affinities of CMNPD compounds, providing important insights into their possible therapeutic applications.

The representation of molecular interactions in three dimensions and two dimensions for the ligands CMNPD15724 (a, b), CMNPD28811 (c, d), CMNPD30883 (e, f), CMNPD18569 (g, h), and (i, j) sinefungin (control). These structures were generated using free academic maestro [33]
Molecular dynamic simulation
Bioinformatics extensively utilizes molecular simulation to examine the structural behaviours and interplays of biomolecules like proteins and their ligands, focusing on atomic and molecular detail [34, 35]. The four most promising results were selected for molecular dynamics (MD) simulation, aiming to investigate the complex's stability and adaptability. To analyze the protein–ligand complex's interactions, the Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) of both the protein and ligand were calculated throughout 200 ns. The first and last pose of the MD simulation is shown in Figure S1.
RMSD and RMSF analysis
Root Mean Square Deviation (RMSD) is a key metric in molecular dynamics (MD) simulations that measures the average deviation of a protein–ligand complex from its initial position over time. This metric provides valuable insights into the stability of the complex, as lower RMSD values generally indicate greater stability and less deviation from the starting structure. In this study, RMSD values for the selected marine fungal compounds (CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569) were analysed over a 200 ns simulation period to assess their stability in complex with the VP39 protein as shown in Fig. 3. Compound CMNPD15724 exhibited an average RMSD of approximately 0.225 nm over the simulation period. The RMSD remained below 0.25 nm for the first 100 ns, indicating stable interactions between the compound and the protein. After 100 ns, there were minor fluctuations, with RMSD values consistently below 0.2 nm. These minor fluctuations suggest that while the complex is largely stable, it undergoes slight conformational adjustments to maintain optimal interactions within the binding site. Overall, the low RMSD indicates a stable binding configuration throughout the simulation. While the CMNPD28811 average RMSD for this compound was about 0.18 nm. During the initial 100 ns, the complex showed slight variations in RMSD, likely due to structural adjustments of the protein as it accommodates the ligand. However, these variations were minimal, and the complex quickly reached a stable equilibrium that was maintained for the rest of the 200 ns period. The low and stable RMSD values reflect a robust binding conformation, suggesting that CMNPD28811 forms a strong and stable interaction with the VP39 protein. On the other hand compound CMNPD30883 demonstrated the highest stability among the tested compounds, with an average RMSD of around 0.15 nm. Although there were major fluctuations in the RMSD values up to 125 ns, the complex stabilized significantly afterwards, with RMSD values settling at approximately 0.1 nm. The initial fluctuations indicate that the compound and the protein undergo substantial conformational changes to achieve a stable binding mode. Once this stable configuration is reached, the strong and stable interactions are reflected in the low RMSD values, making CMNPD30883 the most stable complex in this study. Compound CMNPD18569 displayed more significant fluctuations with an average RMSD of approximately 0.23 nm throughout the simulation. The higher degree of fluctuation suggests that the binding mode of CMNPD18569 is more flexible, potentially allowing the ligand to adapt to different conformational states of the protein. While this flexibility can be advantageous in some contexts, it also indicates a less rigid binding interaction compared to the other compounds. The control compound, sinefungin, maintained an average RMSD of around 0.075 nm, demonstrating high stability. The RMSD values for sinefungin were consistently ≤ 0.05 nm until 175 ns, with only minor fluctuations between 0.1 and 0.05 nm at specific intervals. This high stability provides a useful baseline for comparison. The fact that all selected compounds exhibited RMSD values within a comparable range to the control indicates that they form stable interactions with the VP39 protein. Among the tested compounds, CMNPD30883 stands out due to its superior stability, as evidenced by its consistently low RMSD values after the initial stabilization period. Overall, the RMSD analysis suggests that the selected marine-derived compounds form stable interactions with the VP39 protein, with CMNPD30883 demonstrating the most promising stability profile. This stability is crucial for the potential development of these compounds as effective inhibitors in therapeutic applications.

The MPV and marine fungal compounds’ RMSD plots are as follows: a CMNPD15724; b CMNPD28811; c CMNPD30883; d CMNPD18569; and e sinefungin (Control)
Root Mean Square Fluctuation (RMSF) is a measure used in molecular dynamics simulations to assess the flexibility and mobility of individual residues within a protein structure over time. RMSF values are calculated for each residue in a protein based on its fluctuation around an average position during simulation trajectories. Lower RMSF values indicate less fluctuation, suggesting higher rigidity or stability of those residues. Higher RMSF values indicate greater flexibility or movement of residues. The compound CMNPD15724 exhibited the Low RMSF values of greater than 0.4 Å. Indicate high stability and limited flexibility of residues in the VP39 protein–ligand complex. This indicates the compound stabilizes the VP39 protein structure, with residues showing minimal movement. It suggests strong interactions and a potentially rigid binding mode. The compound CMNPD28811 exhibited slightly higher RMSF values within a low range of less than 0.5 Å suggesting limited flexibility and stable interactions. The protein–ligand complex remains stable, with residues showing slight flexibility but still maintaining a predominantly stable binding interaction. While compound CMNPD18569 exhibited the Lowest RMSF value among all compounds which is 0.35 Å. Indicates superior stability and minimal movement of protein residues. This compound likely forms very stable interactions with VP39, with residues showing minimal fluctuation. It suggests a strong and rigid binding mode, advantageous for being a potent inhibitor. On the other hand compound CMNPD30883 exhibited slightly higher RMSF values of less than 0.4 Å. Indicate some flexibility of residues. This compound shows flexibility in certain residues, which might be advantageous for binding to dynamic regions of the protein. However, compared to others, it suggests a slightly less rigid binding mode. The Control Compound exhibited the low RMSF values of 0.4 Å which indicates the stable interaction with VP39 protein. Sinefungin maintains stable interactions similar to the selected compounds, indicating minimal structural changes and high stability. Overall or average protein RMSF Values of below 0.5 nm across all simulations were observed. Indicate minimal fluctuation and high structural integrity of the VP39 protein. Consistent low RMSF values across all complexes and the control suggest that these compounds do not induce significant structural changes in VP39 (Fig. 4). This stability supports their potential as effective inhibitors.

The RMSF plots for the MPV and marine fungal compounds are as follows: a CMNPD15724; b CMNPD28811; c CMNPD30883; d CMNPD18569; and e sinefungin (control)
Hydrogen bond analysis
Hydrogen bond interaction analysis in molecular simulation acts as an essential marker of the strength of interactions between natural chemicals and their target proteins, which aids in the understanding of the structural and dynamic properties of biomolecules at the atomic level. The number of hydrogen bonds was determined to understand and confirm the compound's binding stability after the MD simulation. Based on our findings as shown in Fig. 5, During the 200 ns molecular dynamics simulations, the examination of hydrogen bond formation revealed varied patterns among the four chemicals. CMNPD15724 had an impressive capacity to maintain a high number of hydrogen bonds throughout the simulation, reaching a maximum of four bonds around 100 ns and maintaining this level until the simulation ended (Fig. 5a). This shows that the molecular structure of CMNPD15724 is especially conducive to the creation of stable hydrogen bonds. CMNPD28811 had a more moderate hydrogen bond profile, with an average of two to three bonds throughout the simulation (Fig. 5b). This shows that CMNPD28811's molecular structure is less prone to generating strong hydrogen bonds than CMNPD15724's. The hydrogen bond formation pattern in CMNPD30883 was fascinating. The molecule first created two hydrogen bonds, which lasted until roughly 120 ns. The number of hydrogen bonds increased to four in the final 80 ns of the simulation, indicating a considerable shift. This shows that during the simulation, CMNPD30883 experiences a structural rearrangement that improves its hydrogen bonding capabilities (Fig. 5c). A dynamic hydrogen bonding characteristic was also observed in CMNPD18569. Within the first 20 ns of the simulation, the molecule created four hydrogen bonds. However, for the remainder of the simulation, this number was reduced to two and three bonds. This shows that early in the simulation, CMNPD18569 undergoes a structural rearrangement that initially promotes hydrogen bond formation but subsequently destabilizes these bonds (Fig. 5d). While the control complex had ten to eleven hydrogen bonds up to 90 ns and reduced to seven to eight hydrogen bonds between the 90 ns to 100 ns and 100 ns to 200 ns ten to eleven hydrogen bonds again formed which indicates the compound undergoes structural rearrangement during simulation.

Number of hydrogen bonds present in the Protein docked with Fungi natural compounds, i.e., a CMNPD15724, b CMNPD28811, c CMNPD30883, d CMNPD18569 and e sinefungin (control) during the 200 ns MD simulation
Principal component analysis
PCA (Principal Component Analysis) is a statistical approach for identifying patterns in a collection of observations that is particularly effective for evaluating molecular dynamics (MD) simulations. It minimizes data dimensionality by identifying the system’s most essential directions of motion, known as principal components, which correspond to the aggregate motions of the atoms. By focusing on these primary components, the dynamics of the system may be visualized and understood more easily. PCA has various advantages for evaluating MD simulations, including dimension reduction, identification of collective motions, and dynamic display. It is commonly used for data preparation, covariance matrix calculation, eigenvalue decomposition, projection onto principal components, analysis, interpretation, protein dynamics analysis, protein folding research, and ligand binding studies. PCA is an extremely adaptable and powerful method for assessing molecular dynamics simulations, reducing data dimensionality, and displaying system dynamics. It has numerous applications in molecular science and is becoming increasingly popular as a tool for assessing MD simulations. "We generated a 2D projection of the trajectory using the first and second eigenvectors (PC1 and PC2) to visualize the structural dynamics of the complexes during the MD simulations" outlines how PCA was used to visualize the dynamic behaviour of molecular complexes during MD simulations. The intricate structural changes can be viewed in two dimensions by projecting the high-dimensional trajectory onto the first two principal components (PC1 and PC2), which encapsulate the system's most significant collective motions. The ability of PC1 and PC2 to capture the most significant structural alterations is critical for accurately reflecting the complexes' dynamics. This efficiency is due to the nature of PCA, which prioritizes the motion directions that contribute the most to the overall variation in the system's behaviour. During the MD simulations, the 2D projection of the trajectory onto PC1 and PC2 provides a clear and succinct picture of the structural dynamics of the complexes. Researchers can use this visualization to identify significant conformational changes, assess complex stability, and obtain insights into the mechanisms behind their function. More dispersion on the graph indicates greater conformational diversity, emphasizing the link between dispersion and conformational diversity. When the data points on the PC projection are spread out more widely, it shows that the system has explored a greater range of conformational states during the simulation. The complex "CMNPD30883" demonstrated the least conformational dispersion, suggesting that it maintained a relatively stable structure throughout the simulation (Fig. 6c). In contrast, the compounds CMNPD15724 (Fig. 6a), CMNPD28811 (Fig. 6b), and sinefungin complex (Fig. 6e) displayed notable conformational diversity, indicating that these complexes explored a wider range of structural conformations during the simulation. These complexes had a wider dispersion of data points on the PC projection, indicating that they transitioned more easily between different conformational states. In summary, the compounds CMNPD15724 (Fig. 6a) and CMNPD30883 (Fig. 6c) demonstrated the least conformational variation, suggesting a higher degree of structural stability, while the other complexes exhibited more prominent diverse structural fluctuations. This illustrates the difference between the more conformationally varied CMNPD28811 (Fig. 6b) and sinefungin (control) complexes (Fig. 6e) and the comparatively stable CMNPD30883 (Fig. 6c) and CMNPD15724 (Fig. 6a) complexes.

PCA analysis of the MD simulation trajectories a VP39-CMNPD15724, b VP39-CMNPD28811, c VP39-CMNPD30883, and d VP39-CMNPD18569, e VP39-sinefungin (control)
Free energy landscape
The free energy landscape analysis offers a comprehensive view of the energy distribution and the conformational dynamics within a biomolecular system during molecular dynamic simulations. This multi-dimensional representation reveals various accessible states and energy levels over time. It is instrumental in examining different conformations and energy minima that a protein–ligand complex may adopt during its dynamic functioning [36]. During principal component analysis, the first and second principal components (PC1 and PC2) were utilized to construct three-dimensional free energy diagrams. This landscape is crucial for identifying transitions between distinct conformations, stable states, and energy barriers, where peaks represent energy barriers and valleys indicate stable states [37]. Understanding the kinetics and thermodynamics of the system is key to deciphering vital interactions, conformational shifts, and the overall stability of the biomolecular complex. Principal Component Analysis (PCA) is a widely used technique in this context, helping to simplify the data into informative representations of the free energy environment. This approach aids in studying biological processes like protein–ligand interactions and conformational changes in macromolecules [38]. The 3D projections graphically represent dynamic conformational changes occurring during the simulation, leading to the formation of a low-energy structure resembling a narrow funnel. The presence of deep blue areas on the larger energy map indicates regions of lowest energy, showing that protein structures reach their lowest energy levels at specific times during the experiment. These areas are marked by dark blue in the broader energy landscape, highlighting local energy minima, particularly evident in the complexes. [39]. The energy minima or states that molecules assume can be categorized into “narrow basin” and “wide basin” types. Typically, molecules situated in a wide basin demonstrate greater stability compared to those in a narrow basin. As illustrated in Fig. 7, the complexes CMNPD15724, CMNPD28811, CMNPD18569, along with the control, display comparatively wide basins. This implies they have multiple accessible states and undergo transitions more often. On the other hand, the compound CMNPD30883 is characterized by narrow basins, suggesting it has deep energy wells and experiences fewer transitions. The study also encompassed gathering structural data for states with minimum, moderate, and maximum energy levels, allowing for a detailed examination of the complex interactions and conformational changes within molecular complexes. The varied interaction patterns and multiple conformational states uncovered in this investigation provide critical insights into the dynamic molecular dynamics driving these interactions. Notably, the observed behaviour points to a stable binding configuration. The lowest energy state, serving as the reference, consistently registered at 0 kJ/mol across all complexes, while the relative maximum energy state varied between 16 kJ/mol and 20 kJ/mol. This numerical assessment of the energy landscape underscores the subtle and dynamic nature of energy profiles associated with these complex molecular interactions.

PCA-based Free energy landscape analysis. a VP39-CMNPD15724, b VP39-CMNPD28811, c VP39-CMNPD30883, d VP39-CMNPD18569, and e VP39-sinefungin (control)
Exploration of the reference molecule’s free energy landscape reveals the existence of two different conformations, each with equivalent Gibbs free energy levels. This observation suggests that the molecule's stability may be compromised, implying that it can easily transition between these two forms. The presence of these energetically comparable conformations may affect the molecule's biological activity and ability to interact with target proteins. The free energy landscape of CMNPD18569 shows a moderate global minimum reflecting the less stable conformation, as well as a secondary local minimum with a somewhat lower Gibbs free energy (Fig. 7d). This shows that the molecule may have a slightly less stable shape than the fundamental one. This secondary minimum may be a transition state or an intermediate conformation along a chemical pathway. While CMNPD30883 free energy landscape exhibits a single global minimum, indicating the existence of a single stable configuration (Fig. 7c). This shows that, when compared to the sinefungin (control) (Fig. 7e) and CMNPD18569 (Fig. 7d), CMNPD30883 (Fig. 7c) has a well-defined structure and is less prone to conformational changes. CMNPD30883 stability could be related to structural properties or interactions with solvent molecules. CMNPD28811's energy landscape includes a major global minimum as well as one secondary local minimum with slight reductions in Gibbs free energy. This shows that when CMNPD28811 interacts with its receptor, it may adopt a less stable shape (Fig. 7b). These secondary minima could be binding modes or intermediary states during the binding process. These different conformations may affect the molecule’s affinity and selectivity for its target receptor. Unlike CMNPD15724, which displays a dominant primary minimum on its free energy landscape, there is also a noticeable secondary minimum with a slightly higher Gibbs free energy. This indicates that the molecule could potentially adopt another conformation that is somewhat less stable, as depicted in Fig. 7a). At last, the energy landscape of CMNPD30883 (Fig. 7c) is characterized by a prominent global minimum, indicating a primary stable conformation. An additional local minimum is observed, with a higher Gibbs free energy than the original one, indicating a less stable conformation. This implies that The VP39 methyltransferase’s free energy landscape when docked with (a) CMNPD15724; (b) CMNPD28811; (c) CMNPD30883; (d) CMNPD18569; and (e) sinefungin (control). Preferentially assumes a particular shape, but it may potentially exist in a less stable configuration under certain conditions. This secondary minimum's presence may have ramifications for the molecule’s biological function or interactions with other molecules.
It is important to mention that the minimum energy state, which serves as a reference point, was consistently determined to be 0 kJ/mol in all complexes. The thermodynamic free energy landscape of the analyzed chemical complexes, including a control compound, provides accurate information regarding their molecular dynamics and stability characteristics. A notable characteristic of this landscape is the existence of a uniform relative maximum energy state inside these complexes, which remains within a restricted range of 14 to 20 kJ/mol.
The similar energy threshold seen in all complexes indicates a possible uniformity in structural or functional characteristics, such as molecular interactions or stability. Furthermore, it is worth noting that all of the complexes exhibit a stable conformation at energy levels below 2 kJ/mol, as seen by the presence of a dark blue color on the landscape.
Our study focused on analyzing the lower energy states of selected ligand–protein complexes. Lower energy states indicate more stable conformations, as systems favor energetically favorable (lower energy) arrangements. From these analyses, three stable poses were extracted for each complex. However, an exception was noted for the compound CMNPD18569, which exhibited only two stable poses at lower energy states, suggesting a potentially less flexible interaction with its protein target or a constrained conformational landscape. To further our analysis, we constructed the superimposition structure as shown in Fig. 8, wherein the stable poses identified from the docking studies were aligned with their initial (reference) poses. Superimposition is a critical step in understanding the extent of conformational changes that compounds undergo upon binding to their targets. It provides a visual and quantitative measure of these changes, which can be crucial for assessing the binding mode and the possible biological implications of these interactions. The quantitative aspect of this analysis was addressed by calculating the overall RMSD for each superimposed structure. Our findings revealed distinct RMSD values for the complexes studied, reflecting the diversity in conformational variation upon binding. The compound CMNPD15724 in complex with target protein showed an overall RMSD of 0.772 nm (Fig. 8a), suggesting a moderate degree of structural deviation from the initial pose. Similarly, CMNPD28811 complexed with the protein exhibited an overall RMSD of 0.784 nm, indicating a comparable level of structural change (Fig. 8b). In contrast, CMNPD30883 displayed a notably lower RMSD of 0.721 nm in its superimposed structure, pointing to a closer alignment with its initial pose and possibly a more favorable binding conformation (Fig. 8c). Conversely, CMNPD18569 exhibited a significantly higher Overall RMSD of 0.852 nm (Fig. 8d), suggesting substantial structural change upon binding. For comparative purposes, a control complex was also analyzed, yielding an overall RMSD of 0.838 nm (Fig. 8e). These RMSD values provide a foundational understanding of the structural dynamics within these compound-protein interactions. By quantifying the deviations from initial to binding poses, we gain insights into the conformational flexibility of the compounds, their potential binding affinities, and the specificity toward their protein targets.

Superimposed representation of a CMNPD15724, b CMNPD28811, c CMNPD30883, d CMNPD18569, and e sinefungin (control). Where the a1, b1, c1, d1 and control e1 are the superimposed (aligned) structure of (a2, a3, a4), (b2, b3, b4), (c2, c3, c4), (d2, d3, d4) and (e2, e3, e4) respectively
ADMET analysis
To predict the toxicity and safety of the selected compounds we have also performed the ADMET analysis by using the ADMETLab 2.0 [40, 41] as shown in Table S2. The ADMET profiles of the compounds CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 are highly promising. All compounds exhibit moderate to high lipophilicity (LogP values), enhancing their potential for cellular membrane permeability. Their LogD values are balanced, indicating a good equilibrium between hydrophilicity and lipophilicity. Particularly, CMNPD28811 and the Control compound show high Human Intestinal Absorption (HIA), suggesting excellent potential for oral bioavailability. The compounds CMNPD30883 and CMNPD28811 also demonstrate significant blood–brain barrier permeability (BBB), which is indicative of potential central nervous system activity. Regarding distribution, the compounds have favorable protein binding percentages (PPB), which ensures efficient transport within the bloodstream, and their volumes of distribution (VDss) indicate widespread distribution throughout the body. In terms of metabolism, the compounds exhibit inhibitory activity against several CYP450 enzymes, particularly CMNPD15724 and CMNPD30883, which might help in avoiding rapid metabolism. CMNPD15724 and CMNPD18569 show strong inhibitory effects on CYP1A2 and CYP3A4, suggesting potential for sustained plasma levels. CMNPD28811 demonstrates favorable metabolic stability across multiple CYP substrates. The compounds also exhibit reasonable clearance rates (CL) and elimination half-lives (T12), indicating manageable dosing frequencies. The toxicity profiles of these compounds are quite favorable, with CMNPD15724, CMNPD30883, and CMNPD18569 showing low Ames toxicity, suggesting a low mutagenic risk. CMNPD15724 and CMNPD28811 show no significant hepatotoxicity or potential for Drug-Induced Liver Injury (DILI). Additionally, most compounds have acceptable skin sensitization and respiratory toxicity profiles. Furthermore, the compounds CMNPD15724 and CMNPD30883 score high on natural product-likeness, indicating they are derived from or similar to natural products, which often correlates with better biocompatibility. All the compounds adhere to Lipinski's Rule of Five, suggesting good oral bioavailability. Overall, the compounds CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 exhibit favorable ADMET properties, including good absorption, distribution, metabolic stability, and low toxicity profiles, making them promising candidates for further development in drug discovery.
Discussion
In this study, we conducted a comprehensive analysis of four marine-derived compounds (CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569) and their interactions with the Monkeypox virus (MPXV) methyltransferase VP39 using various computational techniques. The reference compound sinefungin was used as a control to benchmark the results. Our methods included virtual screening, redocking, molecular dynamics (MD) simulations, root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and principal component analysis (PCA) based free energy landscape analysis [38, 42]. Virtual screening was performed on a library of marine fungi compounds, resulting in the identification of the four top-scoring compounds [43]. These compounds exhibited significant docking scores indicating strong potential interactions with the VP39 protein [44]. The redocking process further validated these interactions. The binding scores for the compounds were as follows: CMNPD15724 (− 9.4 kcal/mol), CMNPD28811 (− 8.9 kcal/mol), CMNPD30883 (− 9.2 kcal/mol), CMNPD18569 (− 9.4 kcal/mol), and sinefungin (− 8.9 kcal/mol). Hydrogen bonds and hydrophobic interactions were critical in stabilizing the VP39-compound complexes. CMNPD15724 formed eight hydrogen bonds with key residues such as Asp138, Ser141, Val116, and Arg97, and several hydrophobic interactions with residues including Leu42, Val139, and Ile94. Similarly, CMNPD28811 showed significant hydrophobic interactions and hydrogen bonds, contributing to its binding stability. CMNPD30883 and CMNPD18569 also demonstrated a variety of interactions, indicating flexible and structured binding mechanisms respectively. Sinefungin, the control, showed a different binding pattern, which provided a comparative baseline for the studied compounds. The 200 ns MD simulations provided insights into the dynamic behavior and stability of the VP39-compound complexes [21]. RMSD analysis revealed that CMNPD30883 maintained a lower RMSD (0.721 nm) compared to CMNPD15724 (0.772 nm), CMNPD28811 (0.784 nm), and CMNPD18569 (0.852 nm), indicating that CMNPD30883 had the most stable binding configuration. The control, sinefungin, exhibited an RMSD of 0.838 nm. RMSF analysis indicated the flexibility of individual residues in the protein. The compounds displayed different RMSF profiles, with CMNPD30883 showing the least fluctuation, correlating with its stable binding observed in the RMSD analysis. The Rg values suggested that all compounds maintained a compact structure throughout the simulation, with CMNPD30883 again showing the least variation. PCA was utilized to reduce the dimensionality of the MD trajectories, focusing on the most significant motions. The free energy landscape (FEL) derived from the PCA revealed that CMNPD30883 occupied a single stable conformational state, while other compounds showed multiple minima, indicating a range of binding conformations. This stability of CMNPD30883 was consistent with its low RMSD and RMSF values, suggesting a robust inhibitory activity. The binding site of VP39 involves critical residues such as Asp138, Ser141, Val116, Arg97, and Phe115. Hydrogen bond analysis showed that these residues played significant roles in stabilizing the complexes. For example, CMNPD15724 formed stable hydrogen bonds with Asp138 and Ser141, essential for its binding stability. Hydrophobic interactions with residues like Val116 and Ile94 were also crucial in maintaining the binding configuration of the compounds. The hydrogen bond patterns observed during the simulations highlighted the importance of these interactions in disrupting VP39's function. Compounds that consistently formed critical hydrogen bonds, such as CMNPD15724 and CMNPD30883, showed higher stability and potential efficacy as inhibitors. Hydrophobic interactions further supported the stability and specificity of the binding, particularly for CMNPD28811 and CMNPD30883, which showed extensive hydrophobic contacts. The article “Discovery and Structural Characterization of Monkeypox Virus Methyltransferase VP39 Inhibitors”, this article presents crystal structures of the VP39 methyltransferase in complex with sinefungin and other discovered inhibitors. Sinefungin, a known pan-MTase inhibitor, binds to the VP39 methyltransferase in a conserved manner, similar to its binding in SARS-CoV-2 nsp14 MTase [2]. The inhibitors identified in their study, including TO507, contain five-membered rings crucial for their binding and inhibitory action. Similarly, our study focuses on identifying inhibitors derived from marine fungi utilizing virtual screening and molecular dynamics simulations to identify four promising compounds: CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569. These compounds also feature five-membered rings, contributing to their effective binding to the VP39 methyltransferase. The similarity in structural features, particularly the presence of five-membered rings, underscores the importance of this structural motif in the inhibitors' efficacy. Both sets of compounds demonstrate a conserved binding pattern, indicating that these rings facilitate the interaction with the VP39 methyltransferase's active site. Additionally, the inhibitors identified in their work exhibit cross-viral efficacy, showing activity against SARS-CoV-2 nsp14 MTase, suggesting their broad-spectrum antiviral potential. This aspect highlights the potential for the marine-derived inhibitors to be evaluated for similar cross-viral activity, expanding their utility beyond MPXV.
Conclusions
This study is based on in-depth computational analysis to identify and evaluate potential inhibitors of the Monkeypox virus (MPXV) methyltransferase VP39, a critical enzyme for viral replication. Using a diverse library of marine fungi-derived small molecules, we screened and identified four promising compounds: CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569. These compounds were subjected to rigorous evaluations including re-docking, molecular dynamics (MD) simulations, and ADMET analysis to determine their inhibitory potential and pharmacokinetic profiles. Our virtual screening and re-docking processes highlighted CMNPD15724 and CMNPD30883 as exhibiting superior binding affinities and stable interactions within the VP39 active site. MD simulations confirmed the stability of these interactions, with CMNPD30883 showing the highest stability with an average RMSD of around 0.15 nm. ADMET analysis further indicated that CMNPD28811 and CMNPD30883 possess significant blood–brain barrier permeability, suggesting potential central nervous system activity, while CMNPD15724 and CMNPD18569 demonstrated strong inhibitory effects on CYP1A2 and CYP3A4, supporting their potential for sustained plasma levels and favorable metabolic stability. The combined findings from our analyses underscore the potential of CMNPD15724, CMNPD28811, CMNPD30883, and CMNPD18569 as potent MPXV inhibitors. These compounds not only exhibit strong binding and stable interactions with the target enzyme but also possess favorable ADMET properties, including good absorption, distribution, metabolic stability, and low toxicity profiles. This study highlights the untapped potential of marine fungi as a source of potential antiviral agents and paves the way for further experimental validation and development of these compounds into effective therapeutic options for combating MPXV infections.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
WHO. Mpox (monkeypox). 2023. https://www.who.int/news-room/fact-sheets/detail/monkeypox. Accessed 25 Nov 2023.
Silhan J, Klima M, Otava T, Skvara P, Chalupska D, Chalupsky K, et al. Discovery and structural characterization of monkeypox virus methyltransferase VP39 inhibitors reveal similarities to SARS-CoV-2 nsp14 methyltransferase. Nat Commun. 2023;14:2259.
Hyde JL, Diamond MS. Innate immune restriction and antagonism of viral RNA lacking 2׳-O methylation. Virology. 2015;479–480:66–74.
Liu S-W, Katsafanas GC, Liu R, Wyatt LS, Moss B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe. 2015;17:320–31.
Rizk JG, Lippi G, Henry BM, Forthal DN, Rizk Y. Prevention and Treatment of Monkeypox. Drugs. 2022;82:957–63.
Grimm C, Bartuli J, Fischer U. Cytoplasmic gene expression: lessons from poxviruses. Trends Biochem Sci. 2022. https://doi.org/10.1016/j.tibs.2022.04.010.
Shi X, Yao P, Jose T, Gershon P. Methyltransferase-specific domains within VP-39, a bifunctional protein that participates in the modification of both mRNA ends. RNA. 1996;2:88–101.
WHO. Disease Outbreak News; Multi-country monkeypox outbreak in non-endemic countries. 2022. Available at: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON385. Accessed 25 Nov 2023.
Agrawal S, Adholeya A, Barrow CJ, Deshmukh SK. Marine fungi: an untapped bioresource for future cosmeceuticals. Phytochem Lett. 2018;23:15–20.
Mayer AMS, Pierce ML, Howe K, Rodríguez AD, Taglialatela-Scafati O, Nakamura F, et al. Marine pharmacology in 2018: marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Pharmacol Res. 2022;183: 106391.
Tanwar G, Purohit R. Gain of native conformation of Aurora A S155R mutant by small molecules. J of Cellular Biochemistry. 2019;120:11104–14.
Singh R, Bhardwaj VK, Sharma J, Das P, Purohit R. Identification of selective cyclin-dependent kinase 2 inhibitor from the library of pyrrolone-fused benzosuberene compounds: an in silico exploration. J Biomol Struct Dynam. 2022. https://doi.org/10.1080/07391102.2021.1900918.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–42.
Lyu C, Chen T, Qiang B, Liu N, Wang H, Zhang L, et al. CMNPD: a comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2021;49:D509–15.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Labbé CM, Rey J, Lagorce D, Vavruša M, Becot J, Sperandio O, et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 2015;43:W448–54.
Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–41.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–61.
Mishra PC, Alanazi AM, Panda SP, Alam A, Dubey A, Jha SK, et al. Computational exploration of Zika virus RNA-dependent RNA polymerase inhibitors: a promising antiviral drug discovery approach. J Biomol Struct Dynam. 2023. https://doi.org/10.1080/07391102.2023.2292794.
Alawam AS, Alawam HS, Alshahrani MM, Alwethaynani MS, Alneghery LM, Alamri MA. Structural and dynamical basis of VP35-RBD inhibition by marine fungi compounds to combat marburg virus infection. Marine Drugs. 2024;22:34.
Kumar A, Rajendran V, Sethumadhavan R, Purohit R. Molecular dynamic simulation reveals damaging impact of RAC1 F28L mutation in the switch I region. PLoS ONE. 2013;8: e77453.
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671.
Sharma J, Bhardwaj VK, Das P, Purohit R. Identification of naturally originated molecules as γ-aminobutyric acid receptor antagonist. J Biomol Struct Dyn. 2021;39:911–22.
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–18.
Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105:9954–60.
Jaidhan B, Rao PS, Apparao A. Energy minimization and conformation analysis of molecules using steepest descent method. Int J Comput Sci Inf Technol. 2014;5:3525–8.
Lin Y, Pan D, Li J, Zhang L, Shao X. Application of Berendsen barostat in dissipative particle dynamics for nonequilibrium dynamic simulation. J Chem Phys. 2017;146:124108.
Essential dynamics sampling—GROMACS 2023. 3 Documentation. https://manual.gromacs.org/current/reference-manual/algorithms/essential-dynamics.html. Accessed 19 Jan 2024.
Tosi S. Matplotlib for Python developers. Birmingham: Packt Publishing Ltd; 2009.
Yim A, Chung C, Yu A. Matplotlib for Python developers: effective techniques for data visualization with Python. Birmingham: Packt Publishing Ltd; 2018.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Maestro S. Maestro. New York: Schrödinger, LLC; 2020. p. 682.
Kumar A, Rajendran V, Sethumadhavan R, Purohit R. Evidence of colorectal cancer-associated mutation in MCAK: a computational report. Cell Biochem Biophys. 2013. https://doi.org/10.1007/s12013-013-9572-1.
A K, V R, R S, P S, S T, R P. Computational SNP analysis: current approaches and future prospects. PubMed.
Maisuradze GG, Liwo A, Scheraga HA. Relation between free energy landscapes of proteins and dynamics. J Chem Theory Comput. 2010;6:583–95.
Higo J, Ikebe J, Kamiya N, Nakamura H. Enhanced and effective conformational sampling of protein molecular systems for their free energy landscapes. Biophys Rev. 2012;4:27–44.
Papaleo E, Mereghetti P, Fantucci P, Grandori R, De Gioia L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: the myoglobin case. J Mol Graph Model. 2009;27:889–99.
Kumari M, Singh R, Subbarao N. Exploring the interaction mechanism between potential inhibitor and multi-target Mur enzymes of mycobacterium tuberculosis using molecular docking, molecular dynamics simulation, principal component analysis, free energy landscape, dynamic cross-correlation matrices, vector movements, and binding free energy calculation. J Biomol Struct Dyn. 2022;40:13497–526.
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5-14.
Faisal S, Badshah SL, Kubra B, Sharaf M, Emwas A-H, Jaremko M, et al. Identification and inhibition of the druggable allosteric site of SARS-CoV-2 NSP10/NSP16 methyltransferase through computational approaches. Molecules. 2022;27:5241.
David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014;1084:193–226.
Alexey R, Dariya S, Liudmyla I, Lilia V, Valeriy M, Dmytro L, et al. Structure-based virtual screening and biological evaluation of novel inhibitors of mycobacterium Z-ring formation. J Cell Biochem. 2022;123:852–62.
Baptista R, Bhowmick S, Shen J, Mur LAJ. Molecular docking suggests the targets of anti-mycobacterial natural products. Molecules. 2021. https://doi.org/10.3390/molecules26020475.
Acknowledgements
Not applicable.
Funding
This research was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia, through an institutional funding program, project number (IFPIP: 636–140-1443). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.S.A., S.A.A., M.M.E.D., A.M.H., I.A.A.Z., L.H.B., I.M.A., V.D.D. and E.I.A.; Data curation, A.S.A., S.A.A. and V.D.D.; Formal analysis, A.S.A., S.A.A. and V.D.D.; Methodology, A.S.A., S.A.A., M.M.E.D., A.M.H., I.A.A.Z., L.H.B., I.M.A., V.D.D. and E.I.A.; Validation, A.S.A., S.A.A., M.M.E.D., A.M.H., I.A.A.Z., L.H.B., I.M.A., V.D.D. and E.I.A.; Visualization, A.S.A., S.A.A., M.M.E.D., A.M.H., I.A.A.Z., L.H.B., I.M.A., V.D.D. and E.I.A.; Writing—original draft, A.S.A. and V.D.D.; Writing—review and editing, All the authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alharbi, A.S., Altwaim, S.A., El-Daly, M.M. et al. Marine fungal diversity unlocks potent antivirals against monkeypox through methyltransferase inhibition revealed by molecular dynamics and free energy landscape. BMC Chemistry 18, 141 (2024). https://doi.org/10.1186/s13065-024-01251-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-024-01251-x