
Abstract
Background
Donor acceptor moieties connected through π-conjugated bridges i.e. D-π-A, in order to facilitate the electron/charge transfer phenomenon, have wide range of applications. Many classes of organic compounds, such as cyanine, coumarin carbazole, indoline, perylene, phenothiazine, triphenylamine, tetrahydroquinoline and pyrrole can act as charge transfer materials. Phenothiazines have been extensively studied as electron donor candidates due to their potential applications as electrochemical, photovoltaic, photo-physical and DSSC materials.
Results
Two phenothiazine derivatives, 2-((10-hexyl-10H-phenothiazin-3-yl)methylene)malononitrile (3a) and 2-((10-octyl-10H-phenothiazin-3-yl)methylene)malononitrile (3b) have been synthesized in good yields and characterized by various spectroscopic techniques like FT-IR, UV–vis, 1H-NMR, 13C-NMR, and finally confirmed by single crystal X-ray diffraction studies. Density functional theory (DFT) calculations have been performed to compare the theoretical results with the experimental and to probe structural properties. In order to investigate the excited state stabilities the absorption studies have been carried out experimentally as well as theoretically.
Conclusions
Compound 3a crystallises as monoclinic, P2 (1)/a and 3b as P-1. The X-ray crystal structures reveal that asymmetric unit contains one independent molecule in 3a, whereas 3b exhibits a very interesting behavior in having a higher Z value of 8 and four independent molecules in its asymmetric unit. The molecular electrostatic potential (MEP) mapped over the entire stabilized geometries of the molecules indicates the potential sites for chemical reactivities. Furthermore, high first hyperpolarizability values entitle these compounds as potential candidates in photonic applications.

Phenothiazines; a comparison of experimental and theoretical analysis
Similar content being viewed by others


Background
In few years, a great interest has developed in molecules having electron donor–acceptor (D–A) properties and their modern applications as dye sensitized solar cells (DSSC) [1], photosensitizers [2] and redox sensitizers [3]. The metal based donor–acceptor (D–A) complexes are well known where a metal atom behaves as an electron acceptor and ligands as electron donor species [4–6]. Ruthenium metal is a key contributor in the synthesis of such complexes. To avoid the cost of metal and its environmental hazards there is a space for the synthesis of new organic donor–acceptor molecules. A salient feature of such organic based (D–A) molecules is that donor acceptor moieties are connected through π-conjugated bridges i.e. D-π-A, in order to facilitate the electron/charge transfer phenomenon [7]. The classes of organic compounds that have been evaluated as (D–A) candidates include cyanine [8], coumarin [9], carbazole [10], indoline [11], perylene [12], phenothiazine [13], triphenylamine [14], tetrahydroquinoline [15] and pyrrole [16].
Molecules containing phenothiazine as electron donor part have been extensively studied due to their electrochemical [17], photovoltaic [18], photo-physical [19] and DSSC applications [1]. The synthesis of phenothiazine derivatives and their DSSC applications were claimed by many investigators, and the best results were produced in the solar cells where phenothiazine was used as electron donor and boradiazaindacene as electron acceptor candidates [19]. In addition to their physical applications, phenothiazine derivatives have been recognized as potent anti-psychotic [20], anti-infective [21], antioxidant, anti-cancer [22] and anti-Parkinson agents [23]. These were also qualified as valuable MALT1 protease [24], cholinesterase [25], and butyryl-cholinesterase enzyme inhibitors [26].
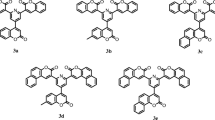
In addition to our recent work [27–32], here we report the synthesis and structural properties of two new phenothiazine derivatives (Fig. 1). Both compounds have been synthesized in high yields and characterized by spectroscopic as well single crystal diffraction studies. The DFT investigations have been performed to validate the spectroscopic results, and to investigate other structural properties like frontier molecular orbital (FMO) analysis, molecular electrostatic potential (MEP), natural bond orbital (NBO) analysis (intra and inter molecular bonding and interaction among bonds), and first hyperpolarizability analysis (nonlinear optical response).

General synthetic scheme of title compounds 3a and 3b. (i) 1-Bromohexane (Compound 3a), 1-Bromooctane (Compound 3b), KOH, KI, DMSO; (ii) DMF, POCl3, 0 °C; (iii) Malonitrile, Piperidine, EtOH
Results and discussion
The synthesis of two phenothiazine derivatives 3a and 3b has been accomplished in three steps beginning from 10-phenothiazine resulting in good yields (details are given in the experimental section). These compounds have been characterized by 1H-NMR, 13C-NMR, FT-IR and UV–vis. spectroscopic techniques, and finally their structures have been confirmed by X-ray diffraction analysis. Computational studies have been carried out to compare the theoretically calculated spectroscopic properties with the experimental results, and to investigate some structural properties as well.
X-ray diffraction analysis
Both compounds 3a and 3b have been recrystallized in methanol under slow evaporation method in order to grow suitable crystals to ensure the final structures, and to study their three dimensional interactions. The compound 3a, bearing a hexyl group at nitrogen, is crystallized in a monoclinic system having space group P21/a and 3b containing an octyl substituent at nitrogen has been crystallized in a triclinic system having space group P-1. Complete crystal data parameters for both compounds have been provided in Table 1. The ORTEP views of both 3a and 3b are shown in Fig. 2.

ORTEP diagram of 3a, and 3b containing four molecules (A, B, C and D) in an asymmetric unit, thermal ellipsoids were drawn at 50 % probability level
While analyzing the crystal structure it is observed that compound 3a exists as single independent molecule in an asymmetric unit. On the other hand, an interesting behavior has been observed for 3b which shows a high Z value of 8 and contains four independent molecules in its asymmetric unit (see Fig. 3) [C1–C24 molecule A, C25–C48 molecule B, C49–C72 molecule C and C73–C96 molecule D, (atomic labeling is in accordance with the compound 3a, Fig. 2)].

Optimized geometries of 3a, 3b at B3LYP/6-31G (d, p)
The thiazine rings are not planar having the root mean square (rms) deviation values of 0.1721 (1) Å, 0.1841 (2) Å, 0.2184 (3) Å, 0.1392 (2) Å and 0.1593 (2) Å for compounds 3a and 3b (molecule A, molecule B, molecule C, molecule D) respectively. In compound 3a, the two aromatic rings are oriented at a dihedral angle of 24.80(1)°, while the thiazine ring is oriented at dihedral angles of 13.33 (1)° and 12.56 (1)° with reference to ring 1 (C1–C6) and ring 2 (C7–C12), respectively.
In 3b, having four molecules A, B, C and D in the asymmetric unit, the dihedral angles between the two aromatic rings are 24.85 (1)°, 32.41 (2)°, 18.83 (2)° and 23.80 (2)°. The observed orientation angles of thiazine rings with adjacent aromatic rings are 14.51 (2)°, 11.88 (2)° in molecule A, 16.28 (2)°, 16.49 (2)° in molecule B, 10.03(2)°, 10.16(2)° in molecule C and 13.63 (2)°, 11.74 (2)° in molecule D. These values are comparable with the already reported related structures [33–36], the difference is merely due to a variety of substituted groups on aromatic ring and nitrogen atom. The crystal structures revealed that the malononitrile group (NC–CH–CN) was not co-planar with the aromatic rings but was twisted at dihedral angles of 21.21 (2)°, 3.02 (5)°, 7.54 (5)°, 14.96 (4)° and 13.05 (5)° in 3a and 3b (A, B, C, D) respectively. The puckering parameters for molecule 3a are QT = 0.424 Å, θ = 77.8 (5)° and φ = 4.1 (6)°, and in 3b puckering parameters (QT, θ and φ) are 0.4533 Å, 76.37°, 5.12 ° for molecule A, 0.5377 Å, 98.01°, 185.47° for molecule B, 0.3427 Å, 104.29°, 188.85° for molecule C and 0.3922 Å, 75.42°, 9.84° for molecule D. These values differentiate the four independent molecules in the asymmetric unit of crystal structure of compound 3b, Additional file 1: Table S1. From the X-ray crystallographic studies, a weak C–H···N intermolecular interaction has been observed in 3a. As a result of this interaction, a dimer is formed generating sixteen membered ring motifs R 11 (16) (see Additional file 1: Fig. S1). Molecules A and B in 3b form dimers to generate sixteen membered ring motifs R 11 (16) Additional file 1: Fig. S2. The π-π interaction has not been observed either in 3a or in 3b.
Geometry optimization
In the past decade, methods based on DFT have got the attention of researchers because of their accuracy and wide applications. The DFT investigations of both compounds 3a and 3b have been performed not only to validate X-ray results, but also to compare and investigate other spectroscopic and structural properties. The structures of both 3a and 3b have been optimized by using B3LYP/6-31G (d, p) level of theory, and the the optimized geometries are shown in Fig. 3. A comparison of bond angles and bond lengths for both compounds are listed in Additional file 1: Tables S2, S3. Although the packing diagram of 3b shows four molecules in asymmetric unit, yet only molecule A has been considered for comparison. The experimental and simulated bond lengths/bond angles of all atoms for compounds 3a and 3b (A) are correlated nicely. A deviation of 0.001–0.036 Å in bond lengths has been appeared for both compounds. Maximum deviations of 5.4° and 4.2° in dihedral angles from C14–C13–C5 bonds in 3a and from C23–C22–C21 bonds in 3b have been observed.
Vibrational analysis
The experimental vibrational spectra of phenothiazine derivatives 3a and 3b have been recorded as neat, and both the experimental as well as simulated spectra are shown in Fig. 4. The vibrational frequencies of both were computed at the same level as was used for energy minima structures and assignments were accomplished by using Gauss-View 05 program. A comparison of experimental and calculated vibrational frequencies is given in Table 2.

Experimental and simulated vibrational spectra of 3a and 3b
The simulated vibrations above 1700 cm−1 have been scaled by using a scaling factor of 0.958 and for less than 1700 cm−1 scaling factor is 0.9627 [37]. In the table only those simulated vibrations are given whose intensities are more than ten. For both compounds, the vibrations arise mainly from aromatic C–H, double bond C=C, C–N, C–S, nitrile, CH2, and CH3 functional groups. From Table 2, it is clear that there exists an excellent agreement between the experimental and theoretical vibrations.
Aromatic (CH), (C=C) and aliphatic (C=C) vibrations
The aromatic (CH) vibrations generally appear in the region 2800–3100 cm−1 [38]. The bands appeared in this region are normally of very low intensity, and not much affected by substituents. In the simulated spectra, the aromatic CH stretching vibrations of both compounds 3a and 3b have been predicted at 3086, 3077 cm−1 and 3085, 3077 cm−1 respectively. The calculated aromatic CH stretching vibrations coincide well with the experimental value appearing at 2916 cm−1 for both compounds. The symmetric and asymmetric stretching vibrational regions of aromatic ring (C=C) usually lie in between 1600–1200 cm−1 [39]. The experimental scans of 3a and 3b show aromatic C=C stretching vibrations at 1574, 1402 cm−1 and 1570, 1405 cm−1 respectively. The simulated aromatic stretching C=C peaks are found in strong correlation and appear at 1603, 1568, 1526, 1395 cm−1 for compound 3a, and 1594, 1526, 1395 cm−1 for compound 3b. An aliphatic C=C group in conjugation with aromatic ring is also present in both compounds and appears at 1559 cm−1 experimentally whereas this stretching vibration appears at at 1553 cm−1 for both 3a and 3b.
Aromatic in-plane and out of plane CH bending vibrational regions are usually weak and are observed in the range 1000–1300 cm−1 and 650–900 cm−1 respectively [40]. In the simulated spectra, in plane CH (aromatic) bending vibrations are observed in the range of 1428–1286 cm−1 for compound 3a, and in the region of 1352–1139 cm−1 for compound 3b. The corresponding experimental values are depicted at 1218 cm−1 for compound 3a and 1220 cm−1 for compound 3b. The prominent out of plane CH (aromatic) bending vibrations of compound 3a are observed at 1163, 927, 810 and 735 cm−1 in the simulated spectrum, and for compound 3b these are observed in the range 927–740 cm−1. These out of plane bending vibrations are well supported by the experimental values of both compounds having their values noticed at 805 and 814 cm−1 respectively. The calculated out of plane bending vibrations of phenyl ring in compound 3a are in the range 741–429 cm−1, and for 3b in the range 709–429 cm−1. These simulated values are very nicely correlated with the experimental values of the both compounds.
CH2 and CH3 group vibrations
The simulated stretching (symmetric/asymmetric) CH2 vibrations appear in the range of 3001–2895 cm−1, and 3005–2893 cm−1 for compounds 3a and 3b respectively. These simulated values appear in nice agreement with the experimental values having appeared at 2848 cm−1 for compound 3a, and 2847 cm−1 for compound 3b. Along with the stretching vibrations, several scissoring, in-plane and out of plane bending, methylene (CH2) and methyl vibrations are observed in the simulated and experimental spectra and a nice agreement is found between them.
Both compounds 3a and 3b show the CH2 scissoring vibrations in the range 1456–1448 cm−1 and 1453–1448 cm−1 respectively and these are correlated well with the experimental 1458 and 1462 cm−1 values respectively. The in-plane bending CH2 vibrations are observed in the range 1337–1275 cm−1 and 1337–1287 cm−1 for 3a and 3b respectively. These bending vibrations are in agreement with the experimental counterparts having appeared at 1317 cm−1, 1218 and 1323, 1228 cm−1 for 3a and 3b respectively.
Nitrile and C–N Group vibrations
The nitrile symmetric stretching vibrations of very high intensity appear at 2245 cm−1 in the simulated spectra for 3a and 3b. The nitrile asymmetric stretching vibrations of low intensity also appear at 2230 and 2231 cm−1 for both compounds. In the experimental scans, the nitrile vibrations appear at 2214 and 2215 cm−1 for 3a and 3b respectively, and are found in excellent correlation with the simulated values. The simulated C–N–C stretching frequency appear at 1483 cm−1 for both 3a and 3b and is in full agreement with its experimental counterpart observed at 1472 and 1474 cm−1 respectively.
The assignments of N-Ph stretching modes are difficult, as there are problems to discriminate them from other aromatic ring vibrations. For substituted aromatic rings, Silverstein et al. [41] defined the N-Ph stretching bands in the range 1200–1400 cm−1. In the present study of compound 3a, the observed N-Ph symmetric stretching bands appear at 1338 and 1279 cm−1 in the simulated spectrum and are in very good agreement with the experimental 1363 cm−1 value. Similarly, the calculated N-Ph stretching frequencies of 3b appearing at 1337 and 1279 cm−1 also show good agreement with the experimental band at 1363 cm−1.
Nuclear magnetic resonance (NMR) studies
For the last two to three decades, nuclear magnetic resonance spectroscopy has been unavoidable tool for structural investigations of organic and biological molecules. The 1H and 13C chemical shifts contain very important information about the structural environment of unknown compounds. Nowadays, a powerful method to predict and compare the structure of molecules is to combine the theoretical and experimental NMR methods. The DFT simulations using Gaussian software are playing very active role in this regard. A full and true geometry optimization of both compounds 3a and 3b has been performed by using B3LYP/6-311 + G (2d, p) basis set. An accurate optimization of molecular geometries is vital for reliable calculations of magnetic properties and their comparison with experimental results. The chemical shift calculations of both compounds have been performed by using the fully optimized geometries, adopting the GIAO method at the same level of theory and referred by using the internal reference standard i.e. trimethylsilane. Both the experimental as well as simulated NMR spectra have been recorded in CDCl3 (for experimental 1H and 13C NMR see Additional file 1: Figs. S3–S6). The detailed simulated and experimental 1H-NMR values are given in Table 3.
Both phenothiazine derivatives (3a and 3b) mainly have aromatic and aliphatic protons. In the experimental 1H-NMR spectra, aromatic and double bonded protons appear in the range 7.74–6.83 ppm (compound 3a) and 7.75–6.83 ppm (compound 3b). The computed aromatic C–H signals (with respect to TMS) appear in the range 8.88–7.18 ppm (3a)/8.93–7.16 ppm (3b), and are found in nice agreement with the experimental values. The calculated chemical shift values for methylene and methyl hydrogen atoms of both 3a and 3b are found in the range 4.24–0.55/4.22–0.81 respectively, and are proved in good agreement with the experimental counterparts which appear in the range of 3.87–0.88 (3a)/3.87–0.87 (3b).
Frontier molecular orbital analysis and UV–vis absorption studies
Frontier molecular orbital analysis has proved very helpful in understanding the electronic transitions within molecules and analyzing the electronic properties, UV–vis absorptions and chemical reactivity as well [42]. The FMO analysis also plays an important role in determining electronic properties such as ionization potential (I. P.) and electron affinity (E. A.). The HOMO (highest occupied molecular orbital) represents the ability to donate electrons and its energy corresponds to ionization potential (I. P.), whereas the LUMO (lowest unoccupied molecular orbital) acts as electron acceptor and its energy corresponds to electron affinity (E. A.) [43]. Frontier molecular orbital (FMO) analysis is carried out at the same level of theory as used for the geometry optimization, applying pop = full as an additional keyword. The HOMO and LUMO surfaces along with the corresponding energies and energy gaps are shown in Additional file 1: Fig. S6. Compound 3a contains 93 filled orbitals, whereas 3b contains 103 filled orbitals. The HOMO–LUMO energy difference in both 3a and 3b has been found to be 2.96 eV. The kinetic stabilities of compounds can be assigned on the basis of HOMO–LUMO energy gap [44]. A low HOMO–LUMO energy gap means less kinetic stability and high chemical reactivity. It is clear that the HOMO–LUMO energy gaps in compounds 3a and 3b are very less, indicating that electrons can easily be shifted from HOMO to LUMO after absorbing energy.
The experimental UV–vis absorption spectra of both compounds 3a and 3b in various solvents like dichloromethane, chloroform, methanol and dimethyl sulphoxide (DMSO) have been recorded within 250–700 nm range, and the combined spectra are shown in (Fig. 5). The theoretical absorption studies are also carried out by using TD-DFT method at B3LYP/6-31G (d, p) level of theory in gas phase, and polarizable continuum model (PCM) is applied to account for solvent effect (For simulated UV–vis spectra see Additional file 1: Fig. S7). A comparison of characteristic experimental and simulated UV–vis. absorption wavelengths (λmax) of the both compounds in gas phase and different solvents (DCM, chloroform, methanol and DMSO) has been given in Table 4. As both the compounds have same chromophores; thus there is no significant difference in their absorption maxima.

Combined experimental UV–vis. Spectra of 3a (above), 3b (below) in different solvents
Different solvents covering a wide range of polarity and dielectric constant have been selected in order to explore the solvent effect on the absorption maxima, but no significant difference has been observed. The experimental UV–vis. spectra of both compounds show mainly two absorption bands. In dichloromethane, λmax1 and λmax2 values for compound 3a appear at 320 and 474 nm corresponding to the π–π* and n–π* transitions respectively [45], and for 3b the values appear at 321 nm and 474 nm. In chloroform the absorption maxima of 3a are found at 321 nm (λmax1), 478 nm (λmax2) and for 3b they have been appeared at 321 nm (λmax1), 478 (λmax2). Similarly, the absorption maxima values appear at 317 nm (λmax1), 478 nm for compound 3a, and 317 nm (λmax1), 463 nm (λmax2), for compound 3b in methanol (polar protic) and DMSO (polar aprotic) respectively. The gas phase simulated spectrum of compound 3a show absorption maxima λmax1 and λmax2 at 300.4 nm (oscillating strength, f = 0.37) and 476.4 nm (f = 0.21) respectively. On the other hand, compound 3b shows λmax1 at 300.4 nm (f = 0.36) and λmax2 at 475.7 nm (f = 0.21). The details of the simulated absorption values along with the oscillating strengths of both compounds in gas, dichloromethane (DCM), chloroform, methanol and DMSO are given in Table 4.
Molecular electrostatic potential (MEP)
Molecular electrostatic potential (MEP) is associated with the electronic cloud. The electrophilic/nucleophilic reacting sites as well as hydrogen bonding interactions can be described in any compound on the basis of MEP [46, 47]. Recognition process of one molecule by another, as in drug-receptor and enzyme substrate interactions, is related to electrostatic potential V(r), because the two species show interaction to each other through their potentials. The MEP analysis can be performed by using the following mathematical relation, described previously [48].
Here summation (Σ) runs over all nuclei A in a molecule, polarization and reorganization effects are ignored. Z A is charge of nucleus A, located at R A and ρ (r′) is the electron density function of a molecule. Usually, the preferred nucleophilic site is represented by red color and the preferred electrophilic site is represented by blue color. The electrostatic potential values at the surface are represented by different colors. The potential decreases in the order: red < orange < yellow < green < blue. The color code of the map is in the range between 0.0550 a.u. (deepest red) and 0.0550 a.u. (deepest blue), where blue corresponds to the strongest attraction and red corresponds to the strongest repulsion. Regions of negative V (r) are associated with lone pairs of electronegative atoms.
According to the MEP analysis of compounds 3a and 3b, there are two negative regions at each molecule (red coded region) shown in Fig. 6. These red coded regions are nitrile functional groups of the both compounds. As these two compounds differ only at the alkyl chain lengths located at the nitrogen in a heterocyclic ring, therefore the reactive sites are same. Apart from the nitrile groups the rest is lying between yellow and green regions. This shows that no strong electrophilic sites exist in both the compounds.

MEP plot of compounds 3a and 3b
Natural bond orbital (NBO) analysis
Natural bond orbital analysis is an efficient method for studying intra- and intermolecular bonding and interaction among bonds, and provides a convenient basis to probe charge transfer or conjugative interaction [49]. The NBO approach describes the bonding anti-bonding interaction quantitatively and is expressed by means of second-order perturbation interaction energy E(2) [50–53]. This energy estimates the off-diagonal NBO Fock matrix element. The stabilization energy E(2) associated with i (donor) to j (acceptor) delocalization is approximated from the second-order perturbation approach as given below:
where q i is the donor orbital occupancy, ε i and ε j are the diagonal elements (orbital energies) and F (i, j) is the off-diagonal Fock matrix element. The larger the E (2) value is, the greater is the interaction between electron donors and electron acceptors and the extent of conjugation of whole system. The various second-order interactions between the occupied Lewis type (bond or line pair) NBO orbitals and unoccupied (anti-bonding and Rydberg) non-Lewis NBO orbitals are investigated by applying DFT at the B3LYP/6-31G (d, p) level. As a result of our study, the compounds 3a and 3b are types of Lewis structures with 97.93 and 98.03 % character, valance-non Lewis character of 1.90 and 1.79 % respectively. Both the compounds share the same Rydberg non-Lewis character of 0.16 %.
The intramolecular hyperconjugative interactions result in the transfer of charge from donor (π) to acceptor (π*) orbitals. This charge transfer increases the electron density (occupancy) in antibonding orbitals and weakens the respective bonds [54]. From the significant entries in Table 5, it is clear that the occupancy of π bonds (C–C) for benzene rings of the title compounds (3a and 3b) lie in the range of ~1.59–1.71. On the other hand, the occupancy of π* bonds (C–C) for benzene rings range from ~0.33–0.42. This delocalization leads to the stabilized energy in the range of ~17.15–25.19 kcal/mol.
The pi-bond of ethylenic moiety (C13–C14) also shows an average of ~20 kcal/mol stabilization energy when it is delocalized to either acetonitrile group. The strongest stabilization energy to the system by 31.28 kcal/mol is due to the lone pair donation of nitrogen atom N (1) to the antibonding π* (C2–C3) orbital. On the other hand, the same lone pair gives a stabilization energy of 24.09 kcal/mol when it is conjugated with the antibonding π* (C11–C12) orbital of the aromatic ring. This clearly shows that the delocalization of lone pair of nitrogen N (1) is more towards that aromatic ring which has extended conjugation due to presence of electron withdrawing acetonitrile groups. The lone pair donation from sulfur atom (S1) to the antibonding π* (C1–C6) and (C7–C8) orbitals of both phenyl rings results in the stabilization energies of 12.09 and 11.23 kcal/mol respectively. The occupancy of lone pair electrons in sulfur atom (S1) is 1.84 as compared to 1.69 of lone pair on nitrogen atom (N1). As a consequence, the stabilization energies arising from the lone pair donation of sulfur atom to the antibonding π* (C–C) bonds of phenyl rings are comparatively smaller than those arising from lone pair donation of N1 atom. A plausible reason could be due to the deviation of sulfur atom from planarity because of its larger size. All σ to σ* transitions involving C–C bonds correspond to the weak stabilization energies in the range of ~2.53–4.58 kcal/mol.
Hyperpolarizability and non-linear optical properties
Recently, compounds having non-linear optical (NLO) properties have got appreciable attention of researchers because of their wide applications in optoelectronic devices of telecommunications, information storage, optical switching and signal processing [55]. Molecules containing donor acceptor groups along with pi-electron conjugated system are considered as strong candidates for possessing NLO properties [56].
In each 3a and 3b, the phenothiazine moiety is connected to a nitrile group through a conjugated double bond, and these molecules are anticipated to show non-linear optical (NLO) properties. For the estimation of NLO properties, the first hyperpolarizability (βo) analysis for compounds 3a and 3b has been performed by employing same level of theory as for geometry optimization i.e. 6-31G (d, p) along with POLAR as an additional keyword. The first hyperpolarizability, a third rank tensor, is always described by a 3 × 3 × 3 matrix. The total 27 components of the 3D matrix can be reduced to 10 components as a result of Kleinman symmetry [57]. From the Gaussian output file ten components of 3D matrix have been identified as β xxx, β xxy, β xyy, β yyy, β xxz, β xyz, β yyz, β xzz, β yzz and β zzz respectively, and the values are given in Table 6.
Among all types of hyperpolarizabilities reported in literature, the more attractive is β tot. (First hyperpolarizability) [49] and it can be measured by using the following mathematical relation;
First hyperpolarizability values have been converted into electrostatic units (1 a.u. = 8.6393 × 10−33esu). The calculated first hyperpolarizability (β tot.) values for 3a and 3b have been found to be 62.03 and 61.70 × 10−30esu respectively. These values are in excellent agreement with the reported values in literature [58, 59], and this agreement proves that both compounds are strong candidates for NLO applications.
Method
All analytical grade chemicals and solvents were purchased from BDH, and used without further purification. Stuart Scientific (SMP3, version 5.0, UK) melting point apparatus was used to record the melting point, and the reported m. p. were uncorrected. 1H-NMR spectra were recorded on a Bruker-AVANCE-III 600 MHz at 300 K, and chemical shifts were reported in ppm with reference to the residual solvent signal. FT-IR spectra were recorded under neat conditions on Thermo Scientific NICOLET iS 50 FT-IR spectrometer (Thermo Scientific). UV–visible studies were performed by using Evolution 300UV/VIS spectrophotometer (Thermo Scientific).
Crystallography
Sample crystals were mounted on Agilent Super Nova (Dual source) Agilent Technologies Diffractometer, equipped with graphite-monochromatic Cu/Mo Kα radiation source. The data collection was accomplished by using CrysAlisPro software [60] at 296 K. Structure solution was performed using SHELXS–97 and refined by full–matrix least–squares methods on F2 using SHELXL–97 [61], in-built with X-Seed [62]. All non–hydrogen atoms were refined anisotropically by full–matrix least squares methods [61]. All the C–H hydrogen atoms were positioned geometrically and treated as riding atoms with C–H = 0.93 Å and Uiso (H) = 1.2 Ueq (C) for aromatic carbon atoms. The methyl and methylene hydrogen atoms were also positioned geometrical with C methyl –H = 0.96 Å and C methylene –H = 0.97 Å and Uiso (H) = 1.5 Ueq (C) and Uiso (H) = 1.2 Ueq (C) for methyl and methylene carbon atoms respectively. The figures were drawn using ORTEP III [63], PLATON [64] and OLEX2 [65] programs. The cifs of both molecules have been assigned CCDC numbers 1028273 & 1028274 and these data files can be obtained free of charge on application to CCDC 12 Union Road, Cambridge CB21 EZ, UK. (Fax: (+44) 1223 336-033; e-mail: data_request@ccdc.cam. ac.uk).
Computational details
Theoretical studies were performed by using Gaussian 09 software at density functional theory (DFT) level, as instituted in program [66]. The visualization of the results/optimized geometries was achieved by using Gauss view 05 [67]. The energy minima optimization of both compounds was carried out at B3LYP/6-31G (d, p) and B3LYP/6-311 + G (2d, p) levels of theory (the later was used further for nuclear magnetic studies). Frequency simulations were performed at the same level, to confirm the optimized geometries as a true minimum (no imaginary frequency). In addition, frequency simulations at B3LYP/6-311G (d, p) level were used for vibrational analysis. Nuclear magnetic resonance studies were performed at B3LYP/6-311 + G (2d, p) level, by adopting GIAO method in chloroform solvent and applying polarizable continuum model (PCM) for the solvent consideration. Chemical shift values were referred by using the internal reference standard i.e., tetramethylsilane. UV–vis absorption studies were simulated by using TD-DFT method and at B3LYP/6-31G (d, p) level of theory. MEP, NBO, FMO and first hyperpolarizability analyses were simulated at B3LYP/6-31G (d, p) level of DFT.
Experimental
The synthesis of both phenothiazine derivatives was carried out in three steps starting from simple phenothiazine. First step was alkylation of nitrogen, followed by subsequent aldehyde formation and then conversion to final product (Fig. 1).
General procedure for the synthesis of N-alkylated phenothiazine (1a, 1b)
In a round bottom flask a mixture of potassium hydroxide (2.003 g, 0.0357 mol), 10-phenothiazine (2.91 g, 0.0119 mol), 1-bromohexane (for 1a) or 1-bromooctane (0.0179 mol) (for 1b) and potassium iodide (in catalytic amount) in 50 ml dimethyl sulfoxide (DMSO) were taken. The reaction mixture was stirred for 5 h at room temperature and water (200 ml) was added. The crude product was extracted with CHCl3 (3 × 50 ml) and the organic layer was washed with saturated ammonium chloride solution and then with water. The organic layer was dried over anhydrous sodium sulfate and filtered, after removing the solvent under reduced pressure, crude product was purified by flash column chromatography (eluent: n-hexane) to obtain colorless oil 1a in 88.68 % yield, and 1b in 86.15 % yield.
General procedure for synthesis of 10-alkyl-10H phenothiazine-3-carbaldehyde (2a, 2b)
To an ice cooled flask containing N, N-dimethylformamide (86 ml), POCl3 (53.5 ml) was added drop wise under stirring. After complete addition, the solution was stirred at room temperature for 90 min. Then the reaction mixture was cooled in an ice bath and already synthesized compound (1a or 1b) (65 mmol) was added. The reaction mixture was warmed gradually up to 75 °C for 2 h. Then the mixture was cooled to room temperature and poured into ice water, basified (sat. aqueous K2CO3 solution) and extracted with CHCl3 (4 × 30 ml). Organic layer was washed, dried over MgSO4, filtered, evaporated and purified by flash silica gel column chromatography using petroleum ether/ethyl acetate (80/20) as eluent system to obtain yellow solids, 2a in 92 % yield and 2b in 91 % yield.
Synthesis of 2-((10-hexyl-10H-phenothiazin-3-yl)methylene)malononitrile (3a) and 2-((10-octyl-10H-phenothiazin-3-yl)methylene)malononitrile (3b)
A mixture of (2a or 2b) (3 mmol) and malononitrile (3 mmol) in basic ethanolic solution (10 ml) was stirred at room temperature overnight. The precipitates formed were filtered off and purified by recrystallization from methanol affording final products, 3a in 78 % yield, and 3b in 73 % yield.
2-((10-hexyl-10H-phenothiazin-3-yl)methylene)malononitrile (3a)
M. p. 84–85 °C IR (neat, cm−1): υmax = 2916, 2848, 2214, 1559, 1472, 1458, 1402, 1360, 1218, 805, 740, 607; 1 H-NMR (CDCl3, ppm): 7.74, (1H, dd, Ar–H, J = 1.8 Hz, 1.2 Hz), 7.53 (1H, d, Ar–H, J = 2.4 Hz), 7.47 (1H, s, Ar–H), 7.17 (1H, m, Ar–H), 7.08 (1H, dd, Ar–H, J = 1.8 Hz, 1.2 Hz), 6.98 (1H, m, Ar–H), 6.88 (1H, d, Ar–H, J = 8.4 Hz), 6.84 (2H, d, Ar–H, J = 9 Hz), 3.87 (2H, t, CH2, J = 7.2 Hz, 9.8 Hz), 1.44 (2H, pent, CH2), 1.32 (2H, pent, CH2), 1.81 (4H, pent, CH2), 0.88 (2H, t, CH3, J = 0.6 Hz, 1.2 Hz), 13 C-NMR (CDCl3, ppm): 157.3, 150.8, 142.4, 131.4, 129.5, 127.8, 127.6, 125.1, 124.9, 122.9, 116.0, 114.8, 114.7, 113.6, 48.2, 31.3, 26.6, 26.4, 22.5, 14.0, UV–vis (DMSO): λmax = 319.5 nm, 470.5 nm.
2-((10-octyl-10H-phenothiazin-3-yl)methylene)malononitrile (3b)
M. p. 90–92 °C IR (neat, cm−1): υmax = 2916, 2848, 2215, 1570, 1559, 1461, 1405, 1364, 1220, 930, 814, 740, 608; 1 H-NMR (CDCl3, ppm): 7.74 (1H, dd, Ar–H, J = 2.4 Hz, 1.8 Hz), 7.54 (1H, d, Ar–H, J = 2.4 Hz), 7.47 (1H, s, Ar–H), 7.17 (1H, m, Ar–H), 7.08 (1H, dd, Ar–H, J = 1.2 Hz, 1.2 Hz), 6.98 (1H, m, Ar–H), 6.88 (1H, d, Ar–H, J = 9 Hz), 6.84 (2H, d, Ar–H, J = 9 Hz), 3.88 (2H, t, CH2, J = 2.4 Hz, 1.8 Hz), 1.81 (2H, pent, CH2), 1.44 (2H, pent, CH2), 1.30 (8H, m, CH2), 0.87 (2H, t, CH3, J = 6.6 Hz, 7.2 Hz), 13 C-NMR (CDCl3, ppm): 157.3, 150.8, 142.4, 131.4, 129.5, 127.8, 127.6, 125.1, 124. 9, 124.1, 122.9, 116.0, 114.9, 114.71, 113.5, 48.2, 31.7, 29.1, 29.1, 26.7, 26.6, 22.6, 14.1, UV–vis. (DMSO); λmax = 320 nm, 471 nm.
Conclusions
In this study, two novel phenothiazine derivatives 2-((10-hexyl-10H-phenothiazin-3-yl)methylene)malononitrile (3a) and 2-((10-octyl-10H-phenothiazin-3-yl)methylene)malononitrile (3b) have been synthesized and characterized by using FT-IR, UV–vis, 1H, 13C-NMR spectroscopic techniques and finally their structures are confirmed by single crystal X-ray diffraction studies. The DFT studies have shown a strong agreement between the simulated and experimental results. The optimized geometries of the both compounds at 6-31G (d, p) level have been used further for investigating structural properties. Frontier molecular orbital analysis shows that both the molecules have very low HOMO–LUMO energy gap, and therefore are kinetically less stable. The molecular electrostatic potential investigations reveal that electronegative region in both the compounds is spread over the nitrile groups. The high first hyperpolarizability values signify that these compounds can have very good nonlinear optical responses. The phenothiazine derivatives have very wide applications not only in dye sensitized solar cells but also in clinical field, and hopefully the results of this study will increase the interest of researchers working in this field.
References
O’Regan B, Gratzel M (1991) A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353:737–739
Henderson BW, Dougherty T (1992) How does photodynamic therapy work? J Photochem Photobiol 55:145–157
Li X, Cui S, Wang DZ, Hou Y, Zhou H, Hu Y, Liu JG, Long Y, Wu W, Hua J, Tian H (2014) New organic donor–acceptor–π–acceptor sensitizers for efficient dye-sensitized solar cells and photocatalytic hydrogen evolution under visible-light irradiation. Chem Sus Chem 7:2879–2888
Nazeeruddin MK, Zakeeruddin SM, Baker RH, Jirousek M, Liska P, Vla-chopoulos N, Ahklover V, Fischer CH, Graetzel M (1999) Highly conjugated electron rich thiophene antennas on phenothiazineand phenoxazine-based sensitizers for dye sensitized solar cells. Inorg Chem 38:6298–6305
Nazeeruddin MK, Angelis FD, Fantacci S, Selloni A, Viscardi G, Liska P, Ito S, Takeru B, Graetzel M (2005) Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J Am Chem Soc 127:16835–16847
Gao F, Wang Y, Shi D, Zhang J, Wang M, Jing X, Baker RH, Wang P, Zakeeruddin SM, Graetzel M (2008) Enhance the optical absorptivity of nanocrystalline TiO2 film with high molar extinction coefficient ruthenium sensitizers for high performance dye-sensitized solar cells. J Am Chem Soc 130:10720–10728
Tseng C-Y, Taufany F, Nachimuthu S, Jiang J-C, Liaw D-J (2014) Design strategies of metal free-organic sensitizers for dye sensitized solar cells: role of donor and acceptor monomers. Org Electr 15:1205–1214
Chen YS, Li C, Zeng ZH, Wang WB, Wang XS, Zhang BW (2005) Efficient electron injection due to a special adsorbing group’s combination of carboxyl and hydroxyl: dye-sensitized solar cells based on new hemicyanine dyes. J Mater Chem 15:1654–1661
Wang ZS, Cui Y, Dan-Oh Y, Kasada C, Shinpo A, Hara K (2008) Molecular design of coumarin dyes for stable and efficient organic dye-sensitized solar cells. J Phys Chem C 112:17011–17017
Koumura N, Wang ZS, Mori S, Miyashita M, Suzuki E, Hara K (2006) Alkyl-functionalized organic dyes for efficient molecular photovoltaics. J Am Chem Soc 128:14256–14257
Kim JY, Kim YH, Kim YS (2011) Indoline dyes with various acceptors for dye-sensitized solar cells. Curr Appl Phys 11:S117–S121
Edvinsson T, Li C, Pschirer N, Scheoneboom J, Eickemeyer F, Sens R, Boschloo G, Herrmann A, Müllen K, Hagfeldt A (2007) Intramolecular charge-transfer tuning of perylenes: spectroscopic features and performance in dye-sensitized solar cells. J Phys Chem C 111:15137–15140
Tian H, Yang X, Chen R, Pan Y, Li L, Hagfeldt A, Sun L (2007) Phenothiazine derivatives for efficient organic dye-sensitized solar cells. Chem Commun 36:3741–3743
Hwang S, Lee JH, Park C, Lee H, Kim C, Park C, Lee MH, Lee W, Park J, Kim K, Park NG, Kim C (2007) A highly efficient organic sensitizer for dye-sensitized solar cells. Chem Commun 36:4887–4889
Chen R, Yang X, Tian H, Wang X, Hagfeldt A, Sun L (2007) Effect of tetrahydroquinoline dyes structure on the performance of organic dye-sensitized solar cells. Chem Mater 19:4007–4015
Qin P, Yang XC, Chen RK, Sun LC, Marinado T, Edvinsson T, Boschloo G, Hagfeldt A (2007) Influence of π-conjugation units in organic dyes for dye-sensitized solar cells. J Phys Chem C 111:1853–1860
Hayen H, Karst U (2003) Analysis of Phenothiazine and Its Derivatives Using LC/Electrochemistry/MS and LC/Electrochemistry/Fluorescence. Anal Chem 75:4833–4840
Tang W, Kietzke T, Vemulamada P, Chen Z-K (2007) Synthesis, characterization, and photovoltaic properties of novel conjugated copolymers derived from phenothiazines. J Polym Sci A Polym Chem 45:5266–5276
Mao M, Zhang X-L, Fang X-Q, Wu G-H, Dai S-Y, Song Q-H, Zhang X-X (2014) Highly efficient light-harvesting boradiazaindacene sensitizers for dye-sensitized solar cells featuring phenothiazine donor antenna. J Power Sources 268:965–976
Ashoor A, Lorke D, Nurulain SM, Kury LA, Petroinu G, Yang KH, Oz M (2011) Effects of phenothiazine-class antipsychotics on the function of α7-nicotinic acetylcholine receptors. Eur J Pharmacol 673:25–32
Ordway D, Viveiros M, Leandro C, Bettencourt R, Almeida J, Martins M, Kristiansen JE, Molnar J, Amaral L (2003) Clinical concentrations of thioridazine kill intracellular multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 47:917–922
Motohashi N, Kawase M, Saito S, Sakagami H (2000) Antitumor potential and possible targets of phenothiazine-related compounds. Curr Drug Targets 1:23–245
Mocko JB, Kern A, Moosmann B, Behl C, Hajieva P (2010) Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson’s disease. Neuro biol Dis 40:120–129
Nagel D, Spranger S, Vincendeau M, Grau M, Raffergerst S, Kloo B, Hlahla D, Neuenschwander M, von Kries JP, Hadian K, Dörken B, Lenz P, Lenz G, Schendel DJ, Krappmann D (2012) Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 22:825–837
Darvesh S, Macdonald IR, Martin E (2013) Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg Med Chem Lett 23:3822–3825
La Du BN, Bartels CF, Nogueira CP, Arpagaus M, Lockridge O (1991) Proposed nomenclature for human butyrylcholinesterase genetic variants identified by DNA sequencing. Cell Mol Neurobiol 11:79–89
Arshad MN, Mahmood T, Khan AF, Zia-Ur-Rehman M, Asiri AM, Khan IU, Nisa R-U, Ayub K, Mukhtar A, Saeed MT (2015) Synthesis, crystal structure and spectroscopic properties of 1,2-benzothiazine derivatives: an experimental and dft study. Chinese J Struc Chem 34:15–25
Arshad MN, Asiri AM, Alamry KA, Mahmood T, Gilani MA, Ayub K, Birinji AS (2015) Synthesis, crystal structure, spectroscopic and density functional theory (dft) study of n-[3-anthracen-9-yl-1- (4-bromo-phenyl)-allylidene]-n-benzenesulfonohydrazine. Spectrochim Acta A 142:364–374
Shafiq M, Khan IU, Zia-ur-Rehman M, Arshad MN, Asiri AM (2011) 4-Hydrazinyl-idene-1-methyl-3H-2λ,1-benzothia-zine-2,2-dione. Acta Cryst. E. 67:2038–2039
Arshad MN, Bibi A, Mahmood T, Asiri AM, Ayub K (2015) Synthesis, crystal structures and spectroscopic properties of triazine-based hydrazone derivatives: a comparative experimental-theoretical study. Molecules 20:5851–5874
Taib LA, Faidallah HM, Sahin ZS, Asiri AM, Sahin O, Arshad MN (2014) Synthesis, spectral analysis, X-ray crystal structures and evaluation of chemical reactivity of five new benzoindazole derivatives through experimental and theoretical studies. J. Mol. Str. 1076:272–279
Shafiq M, Khan IU, Zia-ur-Rehman M, Asghar MN, Asiri AM, Arshad MN (2012) Synthesis and antioxidant activity of a new series of 2,1-benzothiazine 2,2-dioxide hydrazine derivatives. Asian J Chem 24:4799–4803
Chu SSC, Van der Helm D (1975) 1-Methylphenothiazine and 1-ethylphenothiazine, a nearly isomorphous pair. Acta Cryst B 31:1179–1183
Hdii F, Reboul J-P, Barbe J, Siri D, Pepe G (1998) 10-Ethyl-3-nitrophenothiazine. Acta Cryst C 54:1151–1152
Li DM, Hu RT, Zhou W, Sun PP, Kan YH, Tian YP, Zhou HP, Wu JY, Tao XT, Jiang MH (2009) Synthesis, structures, and photophysical properties of two organostannoxanes from a novel acrylic acid derived from phenothiazine. Eur J Inorg Chem 18:2664–2672
Li DM, Lv LF, Sun PP, Zhou W, Wang P, Wu JY, Kan YH, Zhou HP, Tian YP (2009) Synthesis, structures, and photophysical properties of two organostannoxanes from a novel acrylic acid derived from phenothiazine. Dyes Pigm 83:180–186
Sundaraganesan N, Ilakiamani S, Saleem H, Wojciechowski PM, Michalska D (2005) FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine. Spectrochim Acta A 61:2995–3001
Vitnik VD, Vitnik ZJ, Banjac NR, Valentic NV, Uscumlic GS, Juranic IO (2014) Quantum mechanical and spectroscopic (FT-IR, 13C, 1H NMR and UV) investigations of potent antiepileptic drug 1- (4-chloro-phenyl)-3-phenyl-succinimide. Spectrochim Acta A 117:42–53
Teimouri A, Chermahini AN, Taban K, Dabbagh HA (2009) Experimental and CIS, TD-DFT, ab initio calculations of visible spectra and the vibrational frequencies of sulfonyl azide-azoic dyes. Spectrochim Acta A 72:369–377
Vrielynck L, Cornard JP, Merlin JC, Lautie MF (1994) Semi-empirical and vibrational studies of flavone and some deuterated analogues. Spectrochim Acta A 50:2177–2188
Silverstein M, Basseler GC, Morill C (1981) Spectrometric Identification of Organic Compounds. Wiley, New York
Demircioglu Z, Albayrak C, Buyukgungor O (2014) Experimental (X-ray, FT-IR and UV–vis spectra) and theoretical methods (DFT study) of (E)-3-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim Acta A 128:748–758
Fleming I (1976) Frontier orbitals and organic chemical reactions. Wiley, London
Kumar A, Deval V, Tandon P, Gupta A, Dsilva ED (2014) Experimental and theoretical (FT-IR, FT-Raman, UV-vis, NMR) spectroscopic analysis and first order hyperpolarizability studies of non-linear optical material: (2E)-3-[4- (methylsulfanyl) phenyl]-1- (4-nitrophenyl) prop-2-en-1-one using density functional theory. Spectrochim Acta A 130:41–53
Sarkar D, Pramanik AK, Mondal TK (2013) Synthesis, crystal structure and spectral properties of 2-[(1-Methyl-2-benzimidazolyl)azo]-p-cresol: an experimental and theoretical study. Spectrochimica Acta Part A 115:421–425
Scrocco E, Tomasi J (1978) Electrostatic molecular potential analysis of electron density distribution in (ClAlMe2)2 and (AlCl3)2. Adv Quantum Chem 11:115–121
Luque FJ, Lopez JM, Orozco M (2000) Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Theor Chem Acc 103:343–345
Politzer P, Laurence PR, Jayasuriya K (1985) Molecular electrostatic potentials: an effective tool for the elucidation of biochemical phenomena. Environ Health Perspect 61:191–202
Muthu S, Maheswari JU (2012) Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H, UV) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 4-[(4-aminobenzene) sulfonyl] aniline by ab initio HF and density functional method. Spectrochim Acta A 92:154–163
Reed AE, Weinhold F (1985) Natural localized molecular orbitals. J Chem Phys 83:1736–1740
Reed AE, Weinhold RB, Weinhold F (1985) Natural Population Analysis. J Chem Phys 83:735–746
Reed AE, Weinhold RB (1983) Natural bond orbital analysis of near-Hartree–Fock water dimer. J Chem Phys 78:4066–4073
Foster JP, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218
Choudhary N, Bee S, Gupta A, Tandon P (2013) Comparative vibrational spectroscopic studies, HOMO–LUMO and NBO analysis of N- (phenyl)-2, 2-dichloroacetamide, N- (2-chloro phenyl)-2, 2-dichloroacetamide and N- (4-chloro phenyl)-2, 2-dichloroacetamide based on density functional theory. Comput Theor Chem 1016:8–21
Thanthiriwatte KS, de Silva KMN (2002) Non-linear optical properties of novel fluorenyl derivatives—Ab initio quantum chemical calculations. J Mol Struc (Theochem). 617:169–175
Bartkowiak W, Misiaszek T (2000) Solvent effect on static vibrational and electronic contribution of first-order hyperpolarizability of pi-conjugated push-pull molecules: quantum-chemical calculation. Chem Phys 261:353–357
Kleinman DA (1962) Nonlinear dielectric polarization in optical media. Phys Rev 126:1977–1979
Mendis BAS, De Silva KMN (2004) A comprehensive study of linear and non-linear optical properties of novel charge transfer molecular systems. J Mol Struc (Theochem). 678:31–38
Tillekaratne AD, De Silva RM, De Silva KMN (2003) Push–pull porphyrins as non-linear optical materials: ab initio quantum chemical calculations. J Mol Struct (Theochem). 638:169–176
CrysAlis PRO (2012) Agilent technologies. Yarnton, England
Sheldrick GM (2008) SHELXS SHELXL. Acta Cryst A. 64:112
Barbour LJ (2001) X-Seed—a softwaretool for supramolecular crystallography. J Supramol Chem 1:189–191
Farrugia LJ (2012) WinGX and ORTEP for Windows: an update. J Appl Cryst 45:849–854
Spek AL (2009) Structure validation in chemical crystallography. Acta Cryst D 65:148–155
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst 42:339–341
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al (2010) Gaussian 09, Revision C.01. Gaussian Inc, Wallingford
Roy D, Todd K, John M (2009) Gauss view, version 5. Semichem Inc, Shawnee Mission
Authors’ contributions
FAA, AMA and RME synthesized the compounds. AMA and MNA did the crystallographic studies. TM and MAG performed the theoretical calculations. All authors have contribution in write-up. All authors read and approved the final manuscript.
Acknowledgements
This Project was funded by the King Abdulaziz City for Science and Technology (KACST) through National Science, Technology and Innovation Plan (NSTIP) under grant number 8-ENE198-3. The authors, therefore, acknowledge with thanks KACST for support for Scientific Research. Also, the authors are thankful to the Deanship of Scientific Research (DSR), King Abdulaziz University for their technical support.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
13065_2016_158_MOESM1_ESM.docx
Additional file 1. Cartesian co-ordinates of optimized geometries and cif files of 3a and 3b are given in supporting information. Experimental 1H,13C-NMR are also pasted in supporting information along with HOMO–LUMO surfaces, simulated UV–vis. Spectra and Tables containing bond length and bond angles data.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Al-Zahrani, F.A., Arshad, M.N., Asiri, A.M. et al. Synthesis and structural properties of 2-((10-alkyl-10H-phenothiazin-3-yl)methylene)malononitrile derivatives; a combined experimental and theoretical insight. Chemistry Central Journal 10, 13 (2016). https://doi.org/10.1186/s13065-016-0158-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-016-0158-z