
Abstract
Background
Isolated REM sleep behavior disorder (iRBD) is an early α-synucleinopathy often accompanied by incipient cognitive impairment. As executive dysfunctions predict earlier phenotypic conversion from iRBD to Parkinson’s disease and Lewy body dementia, cognitive training focusing on executive functions could have disease-modifying effects for individuals with iRBD.
Methods
The study CogTrAiL-RBD investigates the short- and long-term effectiveness and the feasibility and underlying neural mechanisms of a cognitive training intervention for individuals with iRBD. The intervention consists of a 5-week digital cognitive training accompanied by a module promoting a healthy, active lifestyle. In this monocentric, single-blinded, delayed-start randomized controlled trial, the intervention’s effectiveness will be evaluated compared to an initially passive control group that receives the intervention in the second, open-label phase of the study. Eighty individuals with iRBD confirmed by polysomnography will be consecutively recruited from the continuously expanding iRBD cohort at the University Hospital Cologne. The evaluation will focus on cognition and additional neuropsychological and motor variables. Furthermore, the study will examine the feasibility of the intervention, effects on physical activity assessed by accelerometry, and interrogate the intervention’s neural effects using magnetic resonance imaging and polysomnography. Besides, a healthy, age-matched control group (HC) will be examined at the first assessment time point, enabling a cross-sectional comparison between individuals with iRBD and HC.
Discussion
This study will provide insights into whether cognitive training and psychoeducation on a healthy, active lifestyle have short- and long-term (neuro-)protective effects for individuals with iRBD.
Trial registration
The study was prospectively registered in the German Clinical Trial Register (DRKS00024898) on 2022–03-11, https://drks.de/search/de/trial/DRKS00024898. Protocol version: V5 2023–04-24.
Similar content being viewed by others


Background
REM (rapid eye movement) sleep behavior disorder (RBD) is a parasomnia characterized by loss of muscle atonia and dream enactment during REM sleep [1, 2]. Isolated RBD (iRBD) is an early α-synucleinopathy [3,4,5] and may constitute a prodromal stage of Parkinson’s disease (PD), dementia with Lewy bodies (DLB), or multiple system atrophy (MSA) [6,7,8,9,10]. To date, iRBD is the most specific marker to indicate the prodromal stage of PD and a well-recognized feature in prodromal DLB and MSA [6,7,8,9,10]. Within 15 years after diagnosis, up to 80% of individuals with iRBD exhibit phenotypic conversion to PD, DLB, or MSA, with approximately 95% of the converters eventually developing PD or DLB [10, 11].
In the prodromal PD stage [12, 13], and particularly in individuals with iRBD [14, 15], cognitive alterations have been reported. As expected by definition, a recent multicenter data analysis by the International REM Sleep Behavior Disorder Study Group suggests that the only reliable clinical marker differentiating between individuals with iRBD who develop PD or DLB first is the higher prevalence and rate of cognitive decline in DLB-converters [11]. Furthermore, individuals with iRBD converting to PD are more likely to develop a more aggressive “diffuse malignant” PD subtype, characterized by a higher burden of non-motor symptoms, particularly cognitive dysfunction and eventually PD dementia (PDD) [11, 16,17,18]. Cognitive decline is one of the most debilitating non-motor symptoms in PD [19], as it greatly impacts the patients’ quality of life and independence in activities of daily living and increases the burden on caregivers, care providers, and the public healthcare system [20, 21].
Compared to healthy control individuals (HC), meta-analytical evidence suggests that individuals with iRBD show alterations of medium effect sizes in global cognition, executive functions, and memory [15]. Furthermore, impairments in global cognition and executive functions in individuals with iRBD have been identified as risk factors for conversion within the α-synucleinopathies [7, 14, 15]. Cognitive alterations in individuals with iRBD are accompanied by functional and structural alterations in the brain, pointing to early neurodegeneration at this stage [22,23,24,25]. Therefore, iRBD not only represents a clinical syndrome but may offer a window of opportunity for disease-modifying, potentially neuroprotective pharmacological and non-pharmacological interventions [10, 26, 27]. The development of neuroprotective interventions and their evaluation in clinical trials is of outstanding clinical interest. It could alleviate ethical concerns associated with an early iRBD diagnosis, as the latter informs individuals about their increased risk of developing PD, DLB, or MSA without available targeted interventions [27,28,29]. For Alzheimer’s disease, large (secondary) prevention trials already exist that focus on non-pharmacological multi-domain interventions in at-risk cohorts [30,31,32]. These interventions aim to promote a healthy, active lifestyle in the domains of cognition (e.g., through cognitive training), exercise (e.g., through physical exercise interventions), and nutrition (e.g., through information, consulting, and monitoring). These interventions have demonstrated long-lasting effects, particularly concerning cognitive functions [30, 32, 33].
Non-pharmacological interventions, especially cognitive interventions, have the potential to preserve cognitive functions in both healthy and pathological aging, thereby enhancing the quality of life and independence of older individuals. In a systematic review summarizing 46 meta-analyses on the effectiveness of cognitive interventions, Gavelin et al. [34] demonstrated that the existing evidence suggests reliable positive effects of cognitive training on global cognition of older adults. Meta-analyses have also shown positive training effects induced by cognitive training specifically for individuals with PD [35,36,37]. These effects were particularly evident in vulnerable cognitive domains in PD, such as executive functions [35,36,37]. This is particularly important as executive dysfunctions predict earlier conversion within α-synucleinopathies [7, 14, 15]. Therefore, cognitive training focusing on executive functions could have disease-modifying effects for individuals with iRBD. This hypothesis is supported by studies showing that cognitive training interventions cannot only improve cognitive functions at the behavioral level, but may also lead to network optimizations: Meta-analytically, reduced task-related activation in cortical areas along with increased task-related activation in subcortical areas was observed after cognitive training, which is interpreted as an increase in neural efficiency [38, 39]. Aside from the growing evidence for overall beneficial behavioral and neural effects of cognitive training, the individual responsiveness to cognitive interventions may be heterogeneous. Thus, only an in-depth characterization of the participants’ baseline prerequisites, e.g., in terms of demographic, clinical, (neuro-)psychological, or structural and functional brain parameters, may foster the understanding of who benefits most from cognitive interventions and help to tailor interventions individually [40,41,42,43].
Individuals with iRBD and individuals with PD plus RBD often exhibit severe degeneration of the noradrenergic locus coeruleus, which is an established predictor of cognitive decline in PD [44, 45]; however, the relevance of the noradrenergic system in the context of cognitive training is unknown. Given that behavioral effects of cognitive training can endure up to 10 years in healthy older individuals [33], it is unsurprising that this kind of intervention might trigger not only functional but also structural brain changes (e.g., gray matter and cortical volume increases) [46]. However, no data on the plasticity of white and gray matter microstructure upon a cognitive intervention in iRBD is available, and data in PD remains scarce and inconclusive [47, 48]. More studies elucidating the mechanisms of neural plasticity induced by cognitive interventions via longitudinal imaging before and after cognitive training are warranted, particularly for individuals with iRBD.
A plethora of data suggests a mutual relationship between sleep and cognition in health and disease [49], and the deterioration of sleep macro- and microparameters in individuals with PD has been demonstrated repeatedly [1, 50, 51]. Furthermore, individuals with PD with a lower amount of slow-wave sleep are prone to a faster motor progression as well as poorer cognitive performance [52]. Hence, sleep macro- and microparameters may constitute a relevant driver of the responsiveness to cognitive interventions in iRBD, and data on sleep plasticity are warranted to guide future interventional trials.
The study CogTrAiL-RBD (Cognitive Training and a Healthy, Active Lifestyle Program for Patients with isolated REM Sleep Behavior Disorder) aims to investigate the short- and long-term effectiveness and the feasibility of a cognitive training intervention for individuals with iRBD. The intervention consists of (i) a 5-week digital cognitive training and (ii) a module promoting a healthy, active lifestyle, which includes psychoeducational information on cognitive, physical, and social lifestyle activities, as well as nutrition, along with concurrent monitoring of these domains using a digital activity diary over a period of 15 months.
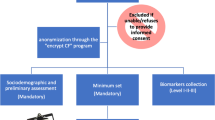
Within the framework of a monocentric, single-blinded, delayed-start randomized controlled trial, the intervention’s effectiveness will be evaluated compared to an initially passive control group that receives the intervention in the second, open-label phase of the study. The evaluation will focus on cognition and additional neuropsychological and motor variables. Furthermore, the general feasibility of the intervention for individuals with iRBD will be investigated. Additionally, the study will examine the neural effects of the intervention by magnetic resonance imaging (MRI) and overnight polysomnography. A healthy, age-matched control group will be examined at the first assessment time point, enabling a cross-sectional comparison between the individuals with iRBD and HC. Reporting of this study protocol follows the “Standard Protocol Items: Recommendations for Interventional Trials” (SPIRIT) checklist [53]. A schematic figure of the study design can be found in Appendix Figure A-1.
Methods/design
Study design and setting
The study CogTrAiL-RBD is designed as a monocentric, single-blinded, delayed-start randomized controlled trial with two study arms aiming to consecutively recruit 80 individuals with iRBD confirmed by polysomnography from the continuously expanding local iRBD cohort at the University Hospital Cologne in Germany [54]. Participants will be randomly assigned to either the early intervention group or a delayed-start control group. Forty participants will be randomly assigned to each group (parallel group phase, superiority). The delayed-start control group will receive the same intervention after a 6-month waiting period, with the early intervention group undergoing the intervention for a second time to separate symptomatic from potentially disease-modifying effects (open-label phase, exploratory) [55].
Figure A-1 presents the schematic trial design and Appendix Figure A-1 further illustrates the timeline for participants. At baseline assessment (t0, week 0), the study will be explained to the participants. Subsequently, the participants will provide written informed consent and answer to a demographic interview. All participants with iRBD will undergo comprehensive clinical and neuropsychological assessments at the t0 visit and 6 weeks post allocation (posttest I, t1, week 6). A follow-up assessment (follow-up I, t2, week 33) will be conducted after 6 months. Subsequently, the second intervention phase for both the early intervention group and the delayed-start control group begins, followed by another clinical and neuropsychological assessment (posttest II, t3, week 39). Finally, a follow-up assessment (follow-up II, t4, week 66) will take place after an additional 6 months.
There are three optional study modules for individuals with iRBD that accompany the mandatory clinical and neuropsychological assessments: (i) the MRI module consisting of structural and functional MRI at each assessment time point; (ii) the accelerometry module consisting of physical activity tracking with accelerometers at each assessment time point; and (iii) the polysomnography module including two polysomnographies, one before the start of the intervention and one within the last 2 weeks of the intervention. Additionally, an age- and sex-matched HC group will be examined at the first assessment time point, participating in the comprehensive clinical and neuropsychological assessments, the MRI module and the accelerometry module.
Recruitment
Individuals with iRBD are recruited from the local iRBD cohort at the University Hospital Cologne, Germany, which is continuously expanding through active recruitment efforts. The recruitment strategy for the local iRBD cohort has been described in Seger et al. [54]. Those who consented to be contacted for study participation during the annual iRBD consultation hours are directly approached by the CogTrAiL-RBD study personnel via telephone. The subjects are provided with a comprehensive information sheet outlining the details of CogTrAiL-RBD, and, if interested in study participation, a structured telephone interview is conducted. The HC group will be recruited via email newsletters, flyers, and newspaper advertisements.
Inclusion and exclusion criteria
The following inclusion criteria are applied for individuals with iRBD: (i) polysomnography-proven diagnosis of iRBD, (ii) age between 40 and 80 years, (iii) normal or corrected-to-normal vision and hearing, (iv) German as native tongue or sufficient proficiency in German, and (v) access to a local computer with access to the internet. Exclusion criteria for individuals with iRBD are (i) severe cognitive dysfunctions (Montréal Cognitive Assessment, MoCA, ≤ 22) [56] interfering with the ability to give informed consent, (ii) significant neurological and psychiatric concomitant diseases, and for those willing to participate in the optional MRI module (iii) contraindications for MRI. Except for the polysomnography-proven diagnosis of iRBD, the same in- and exclusion criteria are applied for HC plus the absence of a diagnosis of a movement disorder or signs of iRBD or any other psychiatric and neurological condition as assessed by medical history.
Sample size and power calculation
To determine an adequate sample size, an a priori power analysis was conducted with G*Power [57] (http://www.gpower.hhu.de). The study will be powered to reliably detect behavioral effects in the domain of executive functions (the primary outcome) in the first parallel-group phase of the randomized controlled trial in individuals with iRBD. According to two meta-analyses on cognitive training in healthy older adults [58] and individuals with PD [36], we expect a medium effect size (Cohen’s d = 0.5) in the domain of executive functions. With a two-sided α-level of 0.05 and 80% power, the minimum sample size comparing the difference between two independent groups (allocation ratio 1:1) with a t-test is N = 128 (i.e., n = 64 per group).
We applied the correction formula of Borm et al. [59], acknowledging the increase of precision in estimating a treatment effect when the correlation between the outcome measure at baseline and retest (\(\rho\)) by ANCOVA is considered: ncor = n*\(\sqrt{1-{\rho }^{2}}\). A meta-analysis [60] on the reliability of neuropsychological measures identified particularly high correlations between baseline and retest measures of executive functions (Pearson’s r ≥ 0.80). Following, the G*Power estimate of n = 64 per group was corrected by the factor \(\sqrt{1-{0.8}^{2}}=0.6\), resulting in a minimum sample size of ncor = 38 per group, i.e., a total sample of Ncor = 76. The estimated dropout rate for the first parallel-group phase of the randomized controlled trial of the present trial is 5%, based on previous experiences with a similar intervention in individuals with PD [61]. Following, we aim to recruit N = 80 (= 76/0.95) individuals with iRBD for the present trial.
Patient adherence
Following recent recommendations for enhancing participant engagement in clinical studies [62], every participant has one contact person throughout the study. Every participant is welcomed by one of the principal investigators at the t0 visit, and this individual contact person is present at every following study visit, if possible. All participants will receive an information folder containing an overview of the study, contact information, and a schedule of appointments, where they can store all study documents (such as study information, insurance information, and consent forms). The participants will be reminded of their following appointments by email 1 week before the scheduled date. At every appointment and during each contact, the participant’s individual contact person will actively seek out any open questions and remarks and will provide assistance accordingly. At the end of the study, participants will receive individual feedback about their neuropsychological test performance, if desired, and a travel cost subsidy (max. € 150 for individuals with iRBD, € 60 for HC individuals). If, for whatever reason, complete study adherence is not possible, an effort will be made to collect as much data as possible from the respective participant.
Randomization and blinding
Randomization in CogTrAiL-RBD is based on a minimization procedure designed to minimize differences between the early intervention group and the delayed-start control group in age, sex, and premorbid IQ [63]. The minimization randomization is implemented using the R package Minirand [64]. We aim at a 1:1 ratio of the number of subjects in the early intervention group and the delayed-start control group. The covariates age, sex, and premorbid IQ will be equally weighted (1/3 each). The participants with iRBD will be consecutively randomized following the clinical and neuropsychological t0 baseline assessment by a person not involved in t1 to t4 data collection, preferably by their individual contact person, who will assign the participants to the intervention and monitor their training progress.
The assessments will be double-blinded at t0 baseline (i.e., randomization will be carried out after the t0 baseline assessment) and single-blinded at t1 to t4 posttests and follow-ups (i.e., outcome assessors do not know the participant’s group allocation). Any violations of blinding will be documented, e.g., if participants do not fully comply with the explicit instruction not to give any clues about their experimental group allocation to the outcome assessors.
Cognitive training intervention
The cognitive training intervention consists of a 5-week digital cognitive training and a module promoting a healthy, active lifestyle including psychoeducational information on a healthy, active lifestyle and activity monitoring using a digital activity diary.
Computerized multi-domain cognitive training
The computerized training will be delivered by the CE-certified class-I medical device “HeadApp/NEUROvitalis Digital” (HelferApp GmbH, https://start-headapp.helferservices.net) offering adaptive, multi-domain cognitive training accessible from home with internet access. Participants will train for a total of 15 training sessions across 5 weeks, 40 min per day, three times a week resulting in a total training duration of 600 min, which is comparable to previously published interventions [34]. The training consists of six different tasks addressing one primary cognitive domain each: executive functions, working memory, episodic memory, attention, visuo-cognition, and language. The training starts with the lowest difficulty and adapts to training performance concerning higher difficulty and task complexity levels. With higher difficulty levels, all tasks involve a strong executive component, leading to a strong executive focus of the applied cognitive training. The training tasks are based on the scientifically validated NEUROvitalis program (ProLog, Therapie und Lernmittel GmbH, Cologne) [65,66,67,68,69]. Depending on their group assignment, participants receive their login credentials and information about the training during their t0 or t2 visit. The training will be monitored weekly on the HeadApp/NEUROvitalis Digital website by the individual’s contact person, and any queries of participants will be answered by telephone or email, depending on the participant’s choice.
Healthy lifestyle information
The intervention is enriched by psychoeducation promoting a healthy, active lifestyle. Topics include risk and protective factors of healthy aging, cognitive and motor reserve, advice for incorporating more cognitive, physical, and social lifestyle activities into daily life routines, the influence of diet on (cognitive) health, and information on the Mediterranean diet. A graphic booklet will deliver the healthy lifestyle information to the participants at the beginning of the intervention period, i.e., at the t0 or the t2 visit. The psychoeducation booklet is based on the scientifically validated NEUROvitalis program (ProLog, Therapie und Lernmittel GmbH, Cologne) [65,66,67,68,69].
Weekly/monthly activity diary
During the study, participants of the early intervention group will fill out weekly (between t0 and t1, t2 and t3) and monthly (between t1 and t2, t3 and t4) digital activity diaries, reporting their cognitive and social lifestyle activities, physical activity level, and Mediterranean diet adherence. Participants of the delayed-start control group will fill out the weekly and monthly digital activity diaries following their introduction to the training following the t2 visit. With this monitoring, we aim at a continuous reflection of activity levels in the domains of the psychoeducational program of participants, motivating the implementation of new active daily life routines. To assess these lifestyle activities, we use established, validated questionnaires. The short version of the International Physical Activities Questionnaire (IPAQ) [70, 71] is used to assess physical activity levels, cognitive and social lifestyle activities are assessed with the Lifestyle Activities Questionnaire (LAQ) [72, 73], and adherence to the Mediterranean diet with the Mediterranean Diet Adherence Screener (MEDAS) [74, 75]. In the weekly version of the activity diary, the questions refer to the past week and in the monthly version they are related to the past month. For each lifestyle domain (cognitive and social lifestyle activities, physical activity, and Mediterranean diet adherence), we additionally ask for subjective ratings of the participant’s activity level in general and compared to the past week/the past month on a 4-point Likert scale. The activity diary questionnaires are sent out by email via SoSci Survey (SoSci Survey GmbH, Munich, Germany). Mailing lists are generated in R and directly uploaded to SoSci Survey to automatically send out individualized links to fill out the activity diaries at the planned time points.
Assessments
An equally weighted domain composite score of executive functions will constitute the primary outcome of the present study. The other cognitive domain (visuo-cognition, attention and working memory, memory, language) equally weighted composite scores, an equally weighted global cognition composite score based on the equally weighted domain composite scores, single test scores, as well as the assessments of non-motor symptoms, (fine) motor abilities, quality of life, and the feasibility of the intervention constitute the secondary outcomes. Physical activity measured by accelerometry, brain imaging parameters, and polysomnography parameters constitute exploratory outcomes.
Neuropsychology and motor assessments
All subjects will undergo clinical and neuropsychological assessments, including paper–pencil cognitive and motor evaluations. The in-person assessments take around 2.5 h to complete, including a short break according to the participant’s needs. The test battery will be administered by psychologists or graduate students of Psychology or Medicine at all time points. The outcome assessors will be thoroughly trained in administering and scoring the assessments. Following the in-person assessments, participants digitally fill out questionnaires on various non-motor symptoms, quality of life, and lifestyle activities, which are sent to them via SoSci Survey (SoSci Survey GmbH, Munich, Germany). An automatic reminder email is sent to the participants, if they do not complete the questionnaire within 1 week of their study visit. A complete list of all cognitive assessments, motor evaluations, and questionnaires, including references, is presented in Table 1. Parallel test forms are used if available (i.e., alternating between time points). Test scores are standardized into z-scores, percentage ranks (PR), or T-scores using published normative data, as available. All evaluations are double-checked and digitalized using the four-eye principle. All standardized scores are transformed into z-scores during data preprocessing to create the domain composite scores. Table 1 provides an overview of the assignment of cognitive tests to the respective domains.
The motor examination with the motor part of the Movement Disorder Society (MDS) Unified Parkinson’s Disease Rating Scale (MDS-UPDRS-III) will be conducted and videotaped by outcome assessors and rated by one of two movement disorder specialists blinded for the diagnostic group (HC vs. RBD), experimental group allocation (early intervention group vs. delayed-start group) and time point of assessment (t0, t1, t2, t3, t4). The same movement disorder specialist will rate all videos of one subject. Ten randomly selected videos of the first 100 MDS-UPDRS-III recordings were double-rated by the two movement disorder specialists and the MDS-UPDRS-III total score reached an intraclass correlation (ICC) of 0.87, indicating good to excellent interrater reliability. Rigidity assessments as part of the UPDRS-III are conducted and rated during the test sessions by the outcome assessors. All outcome assessors are previously trained by the movement disorder specialists.
Feasibility
As this clinical trial constitutes one of the first non-pharmacological intervention studies for individuals with iRBD, the feasibility assessment of such an intervention is of great interest. We will report the participation score, i.e., the proportion of individuals of the continuously expanding Cologne iRBD cohort willing to participate in the intervention study (regardless of subsequent randomization). Additionally, within the intervention study, factors of interest include adherence to the intervention protocol (compliance) in terms of the rate of successful completion of the cognitive training protocol operationalized as the completion of at least 80% of planned training sessions, equivalent to a minimum of 12 training sessions, and the consistent utilization of the digital activity diary. Furthermore, the overall dropout rate across data collection time points will be compared between the early intervention group and the delayed-start control group. While the study does not incorporate a systematic collection of qualitative feedback, participants are encouraged to provide feedback on the cognitive training and activity diary and communicate any technical challenges they encounter to their designated contact person throughout the study.
Accelerometry: physical activity tracking
All willing participants will be asked to wear a wrist-mounted movement sensor measuring linear acceleration and angular velocity on their dominant hand for 7 days around each assessment time point. The sensors will be distributed either during the assessment session (along with a prepaid return envelope) or sent to the participants 10 days prior the next assessment. The participants will be instructed to wear the sensor as much as possible throughout their days and nights. Additionally, participants will be asked to fill out a diary noting their bedtimes, daily activities, and periods when they are not wearing the device.
The CE-certified movement sensor used in this study is the AX6 from Axivity Ltd, Newcastle upon Tyne, England. Detailed information about the AX6 and the wrist strap can be found in the Data Sheet documents available on the Axivity website (https://axivity.com/product/ax6). The sensors will be configured to record data continuously for 7 consecutive days and nights (168 h) at a sampling rate of 100 Hz with an accelerometer range of ± 8 g. Sensor set-up and data storage will be managed using the open source AX3/AX6 OMGUI Configuration and Analysis Tool (https://github.com/digitalinteraction/openmovement/wiki/AX3-GUI). Data retrieval from the sensor is only possible via a USB cable connection. Data will be saved in the Continuous Wave Accelerometer (*.cwa) binary format containing the raw actigraphy data, metadata, and device configurations.
Magnetic resonance imaging
Extensive brain imaging with a 3 T SIEMENS PRISMA scanner equipped with a 64-channel head coil will be conducted at the Research Center Jülich near Cologne for all willing participants at all time points (t0–t4). The MRI scanning will preferably occur within 7 days of the clinical and neuropsychological assessment. However, in case of technical difficulties with the scanner or timing issues with the participants (e.g., illness or personal reasons), it may be conducted up to 3 weeks apart. During MRI measurements, at least one member of the study team is present. This person will be supported either by another member of the study team or by a medical-technical radiology assistant of the Research Center Jülich. The net scanning time for all sequences lasts 69:33 min. The sequences, their features and specifications, spatial resolution, repetition times (TR), and echo times (TE) as the basic pulse sequence parameters are described in Table 2. The MRI data will be exported and saved in the DICOM format (*.dcm).
Polysomnography
All participants with iRBD undergoing MRI will also be invited to participate in two polysomnography sessions during their (first) period of cognitive training. For participants in the early intervention group, the polysomnography sessions will take place between t0 and t1, while for participants in the delayed-start control group, the sessions will occur between t2 and t3. The first polysomnography session will be conducted before the start of cognitive training, and the second session will be held in the last or second-to-last week of training, following a training day. The participants’ sleep prior to and during cognitive training will be monitored using mobile polysomnography equipment including 10 EEG channels, electromyography (EMG) of the chin, arms, and legs, surveillance of breathing efforts, as well as high-definition infrared (IR) videorecording. The SOMNOscreen plus device from SOMNOMedics GmbH, located in Randersacker, Germany, will be utilized.
The mobile nature of the polysomnography device allows for examinations to be conducted in the participants’ homes or hotel rooms, providing high convenience for the subjects. All participants are already familiar with the polysomnography examination from their iRBD diagnostic process, minimizing first-night effects. The polysomnography set-up takes approximately 90 min. After completing the set-up, participants can freely move as there are no cables restricting movement and they can spend their evening to their liking and go to bed at their preferred time. The following morning, a member of the study team will detach the polysomnography equipment. The polysomnography data will be saved in the proprietary file format of SOMNOMedics GmbH, which can be converted to the European Data Format EDF + format (*.edf), which is commonly used to store multichannel medical time series data.
Data management and monitoring
All collected data will be pseudonymized with an individual identification number following the format RBD-XXX for individuals with RBD and HC-RBD-XXX for HC individuals, where XXX represents a non-consecutive, randomly generated three-digit number. Paper-based data such as the scoring sheets of the clinical and neuropsychological assessments will be stored in lockable cabinets in a room with restricted access at the University Hospital Cologne, sorted by participant ID for easy access at each stage of the study. Data acquired on paper will be digitalized by one staff member and double-checked by another. The progress of data collection, scoring, digitalization, and monitoring will be documented. Prior to data analysis, range checks for data values will be conducted. All digitally acquired data, such as questionnaire data from SoSci Survey (SoSci Survey GmbH, Munich, Germany), the MDS-UPDRS-III videos, the accelerometry module (*.cwa files), and the PSG module (proprietary file format and *.edf files), will be saved on a secure server of the University Hospital Cologne with restricted access only to the staff involved in this project. The MRI data (*.dcm files) is sent to a Picture Archiving and Communication System (PACS) and stored in the study archive of the Research Center Jülich in a data protection-compliant and pseudonymized manner. Spreadsheets concerning sensitive data, such as the name-ID key list, names, and contact information, will be further protected by a password and saved on a secure server with access only to the staff involved in participant management of the study. Additionally, all digitally acquired data will be backed-up on an encrypted hard drive stored in a lockable cabinet in a room with restricted access at the University Hospital Cologne. Following good scientific practice, data will be stored for at least 10 years. The final data set can be accessed by the study contributors at the University Hospital Cologne and the Research Center Jülich. Participants will also be requested to authorize the research team to share the pseudonymized data with members of other research groups from university and non-university research institutions for in-depth data analysis within non-commercial research projects. This study does not involve the collection or storage of biological samples. Auditing is done on a monthly basis for internal reasons, a yearly report is sent to the sponsor.
Expected harms and adverse events monitoring
During the execution of clinical and neuropsychological assessments, accelerometry, cognitive training, and the healthy, active lifestyle module, we expect no risks or complications and subsequent issues, except for mild transient fatigue. The MRI is a non-invasive procedure for functional and structural imaging of tissues, utilizing strong magnetic fields and high-frequency radio waves without involving radioactively labeled ligands or X-rays. To minimize the risk of any MRI-related complications, the detailed medical history of participants is assessed to determine their MRI eligibility. Prior to scanning, participants are asked to remove all metallic objects and empty their pockets. Under careful consideration of inclusion and exclusion criteria, no known health risk exists for patients undergoing MRI. Experienced specialist physicians in (neuro-)radiology evaluate every MRI scan diagnostically about incidental findings. If any need for further medical measures arises from the review of the MRI examination, the study physician will inform and support the participants accordingly upon their request. All participants are informed about the possibility of incidental findings and their medical, legal, insurance-related, and psychological consequences. The burden on the participants from the mobile polysomnography is considered low since the individuals with iRBD are familiar with the procedure from the diagnostic process. The surface electrodes are applied similarly to adhesive patches and participants can sleep in their familiar environment or a similar setting (e.g., a hotel room). Some participants may report a mild to moderate sensation of foreign objects.
Overall, under careful consideration of the inclusion and exclusion criteria, the risks of the study are minimal. Participants will be informed about possible risks and adverse events (AE) at their t0 visit. Participants can withdraw from the study at any point without providing a reason and without facing any disadvantages. No written explanation is required, and a verbal statement is sufficient for dropout. Outcome assessors and study coordinators will be instructed to monitor and document all AE throughout the study. If a serious AE occurs, the study physician will be consulted and asked to assess whether or not a causal relationship with the intervention is considered possible. Beyond the trial, participants will be part of the local iRBD cohort, receiving access to approximately yearly clinical visits and additional visits as needed.
Statistical analyses
In the present study protocol, we will report the statistical analyses for the primary and secondary outcomes of the neuropsychological and motor assessments in the first parallel-group phase of the randomized controlled trial. Separate analysis plans for the exploratory outcomes physical activity measured by accelerometry, brain imaging parameters, and polysomnography and exploratory analyses (e.g., subgroups) will be preregistered in the Open Science Framework (OSF, https://osf.io/). The statistical analyses will be conducted after the last t2 visit of the last participant. Data preprocessing, statistical analyses, and data visualization will be conducted in R [118] with commonly used statistical libraries, e.g., dplyr, lme4, and ggplot2.
For the baseline comparisons between the early intervention group and the delayed-start control group, independent sample t-tests, Wilcoxon rank-sum tests, or χ2 tests will used as appropriate. Variables and test scores will be previously inspected for normal distribution by Shapiro–Wilk tests. As effect sizes, Cohen’s d will be reported for t-tests and r for Wilcoxon rank-sum tests, indicating small (d ≥ 0.2; r ≥ 0.1), moderate (d ≥ 0.5; r ≥ 0.3), or strong (d ≥ 0.8; r ≥ 0.5) effects.
For the analysis of training effects in the primary and secondary outcomes, linear mixed effects (LME) models will be used. Dependent variables are the respective primary or secondary outcome measures. All dependent variables are assessed at three points of time. The LME models include time (t0, t1, t2), group (early intervention group and delayed-start control group), and the interaction between time and group (time*group) as fixed factors. Furthermore, participants and time are included as random factors. Models will be estimated using the lmer() function of the lme4-package employing restricted maximum likelihood estimation [119]. Model fit will be evaluated using marginal R2, which considers the variance of the fixed effects only, and conditional R2, which accounts for both fixed and random effects. t-Tests will be conducted to assess the significance of single coefficients. For the interaction effects, relative effect sizes with 95% confidence intervals will be reported and defined as the difference of the mean change over time in the early intervention group minus mean change over time in the delayed-start control group divided by the pooled baseline standard deviation of the cohort. Within the analysis of training effects, we will pursue an intention-to-treat approach. No imputation methods will be used, as one strength of LME models is their ability to deal with unbalanced designs, for example, due to missing values in longitudinal data.
Dissemination plans
The results of CogTrAiL-RBD will be published in international, peer-reviewed scientific journals and presented at national and international conferences. Following the main publication of the behavioral effects of the training including the training effect on the primary outcome of executive functions, results will be disseminated to participants via email in a way that is easily understandable for non-experts. If any important protocol modifications occur, they will be communicated in a written form to relevant parties.
Discussion
CogTrAiL-RBD is to the best of our knowledge the first cognitive intervention study, additionally promoting a healthy, active lifestyle, for individuals with iRBD. With the longitudinal, multi-modal data collection, it will not only be possible to gain insights on the effectiveness of such an intervention at this at-risk group for the development of PD, DLB, and MSA and corresponding cognitive impairment, but we will also develop a better understanding on the underlying mechanisms of behavioral training effects. The trial is powered to detect behavioral differences in the first parallel-group phase of the randomized controlled trial. Following, any analyses focusing on the second open-label phase are fully exploratory, constituting a limitation to the trial. From a clinicians’ and patients’ perspective, this trial is pioneering, as it could alleviate ethical concerns associated with an early iRBD diagnosis [27,28,29]. Cognitive training combined with psychoeducation on a healthy, active lifestyle would be a low-threshold and safe intervention approach for this at-risk group to counteract cognitive decline.
Trial status
The trial status is ongoing with the first participant included on June 14, 2022. We expect the recruitment to be finished by June 2024. At the time of revision (May 10, 2024), 85% (n = 68) of the participants with iRBD planned to be included (N = 80) were recruited, with 68.75% (n = 55) already being enrolled in the trial. At baseline t0, the current participation score in the MRI module is 54.5% (n = 30), of which 56.7% (n = 17) were also willing to participate in the polysomnography module. The participation score for the accelerometry module is as high as 96.4% (n = 53) at baseline t0. The protocol version is V5 as of 2023–04-24.
Availability of data and materials
Beyond the registration in the German Clinical Trial Register (Identifier: DRKS00024898), CogTrAiL-RBD is registered in the Open Science Framework (OSF, https://osf.io/). The OSF will be used as a platform to make our data and corresponding material available. Following the publication of corresponding manuscripts, we aim to share the anonymized individual participant data (IPD) according to the FAIR principle—findable, accessible, interoperable, reusable. All data sharing will only be undertaken in accordance to legal requirements. A model consent form is displayed in Appendix B-2.
Abbreviations
- CogTrAiL-RBD:
-
Cognitive Training and a Healthy, Active Lifestyle Program for Patients with isolated REM Sleep Behavior Disorder
- DLB:
-
Dementia with Lewy bodies
- HC:
-
Healthy control individuals
- ICC:
-
Intraclass correlation
- iRBD:
-
Isolated REM (rapid eye movement) sleep behavior disorder
- IPAQ:
-
International Physical Activities Questionnaire
- LAQ:
-
Lifestyle Activities Questionnaire
- MDS-UPDRS-III:
-
Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III
- MEDAS:
-
Mediterranean Diet Adherence Screener (MEDAS)
- MRI:
-
Magnetic resonance imaging
- MSA:
-
Multiple system atrophy
- OSF:
-
Open Science Framework
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson’s disease dementia
- PR:
-
Percentage ranks
- RBD:
-
REM (rapid eye movement) sleep behavior disorder
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
- TE:
-
Echo time
- TR:
-
Repetition time
References
Jeppesen J, Otto M, Frederiksen Y, Hansen AK, Fedorova TD, Knudsen K, et al. Observations on muscle activity in REM sleep behavior disorder assessed with a semi-automated scoring algorithm. Clin Neurophysiol. 2018;129(3):541–7.
Iranzo A, Santamaria J, Tolosa E. Idiopathic rapid eye movement sleep behaviour disorder: diagnosis, management, and the need for neuroprotective interventions. Lancet Neurol. 2016;15(4):405–19.
Iranzo A, Fairfoul G, Ayudhaya ACN, Serradell M, Gelpi E, Vilaseca I, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20(3):203–12.
Schaffrath A, Schleyken S, Seger A, Jergas H, Özdüzenciler P, Pils M, et al. Patients with isolated REM-sleep behavior disorder have elevated levels of alpha-synuclein aggregates in stool. NPJ Parkinson’s Dis. 2023;9(1):14.
Kuzkina A, Panzer C, Seger A, Schmitt D, Rößle J, Schreglmann SR, et al. Dermal real-time quaking-induced conversion is a sensitive marker to confirm isolated rapid eye movement sleep behavior disorder as an early α-synucleinopathy. Mov Disord. 2023;38:1077–82. https://doi.org/10.1002/mds.29340.
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30(12):1600–11.
Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB, et al. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2019;34(10):1464–70.
McKeith IG, Ferman TJ, Thomas AJ, Blanc F, Boeve BF, Fujishiro H, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020;94(17):743–55.
Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord. 2022;37(6):1131–48.
Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019;142(3):744–59.
Joza S, Hu MT, Jung K-Y, Kunz D, Stefani A, Dušek P, et al. Progression of clinical markers in prodromal Parkinson’s disease and dementia with Lewy bodies: a multicentre study. Brain. 2023;146(8):3258–72.
Liepelt-Scarfone I, Ophey A, Kalbe E. Cognition in prodromal Parkinson’s disease. Cognition in Parkinson’s disease. 2022;269:93.
Fengler S, Liepelt-Scarfone I, Brockmann K, Schäffer E, Berg D, Kalbe E. Cognitive changes in prodromal Parkinson’s disease: a review. Mov Disord. 2017;32(12):1655–66.
Ferini-Strambi L, Fasiello E, Sforza M, Salsone M, Galbiati A. Neuropsychological, electrophysiological, and neuroimaging biomarkers for REM behavior disorder. Expert Rev Neurother. 2019;19(11):1069–87.
Leitner C, D’Este G, Verga L, et al. Neuropsychological changes in isolated REM sleep behavior disorder: a systematic review and meta-analysis of cross-sectional and longitudinal studies. Neuropsychol Rev. 2024;34:41–66. https://doi.org/10.1007/s11065-022-09572-1.
Berg D, Borghammer P, Fereshtehnejad S-M, Heinzel S, Horsager J, Schaeffer E, et al. Prodromal Parkinson disease subtypes—key to understanding heterogeneity. Nat Rev Neurol. 2021;17(6):349–61.
Jozwiak N, Postuma RB, Montplaisir J, Latreille V, Panisset M, Chouinard S, et al. REM sleep behavior disorder and cognitive impairment in Parkinson’s disease. Sleep. 2017;40(8):zsx101.
Fereshtehnejad S-M, Romenets SR, Anang JB, Latreille V, Gagnon J-F, Postuma RB. New clinical subtypes of Parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol. 2015;72(8):863–73.
Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray Chaudhuri K, et al. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers. 2021;7(1):1–21.
Vossius C, Larsen JP, Janvin C, Aarsland D. The economic impact of cognitive impairment in Parkinson’s disease. Mov Disord. 2011;26(8):1541–4.
Mosley PE, Moodie R, Dissanayaka N. Caregiver burden in Parkinson disease: a critical review of recent literature. J Geriatr Psychiatry Neurol. 2017;30(5):235–52.
Rahayel S, Gaubert M, Postuma RB, Montplaisir J, Carrier J, Monchi O, et al. Brain atrophy in Parkinson’s disease with polysomnography-confirmed REM sleep behavior disorder. Sleep. 2019;42(6):zsz062.
Chen M, Li Y, Chen J, Gao L, Sun J, Gu Z, et al. Structural and functional brain alterations in patients with idiopathic rapid eye movement sleep behavior disorder. J Neuroradiol. 2022;49(1):66–72.
Campabadal A, Segura B, Junque C, Iranzo A. Structural and functional magnetic resonance imaging in isolated REM sleep behavior disorder: a systematic review of studies using neuroimaging software. Sleep Med Rev. 2021;59:101495.
Rahayel S, Postuma RB, Montplaisir J, Marchand DG, Escudier F, Gaubert M, et al. Cortical and subcortical gray matter bases of cognitive deficits in REM sleep behavior disorder. Neurology. 2018;90(20):e1759–70.
Weil RS, Morris HR. REM sleep behaviour disorder: an early window for prevention in neurodegeneration? Brain. 2019;142(3):498–501.
Postuma RB, Berg D. Prodromal Parkinson’s disease: the decade past, the decade to come. Mov Disord. 2019;34(5):665–75.
Dommershuijsen LJ, Darweesh SKL, Luik AI, Kieboom BCT, Koudstaal PJ, Boon AJW, et al. Ethical considerations in screening for rapid eye movement sleep behavior disorder in the general population. Mov Disord. 2020;35:1939–44. https://doi.org/10.1002/mds.28262.
Gossard TR, Teigen LN, Yoo S, Timm PC, Jagielski J, Bibi N, et al. Patient values and preferences regarding prognostic counseling in isolated REM sleep behavior disorder. Sleep. 2023;46(1):zsac244.
Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. The Lancet. 2015;385(9984):2255–63.
Vellas B, Carrie I, Gillette-Guyonnet S, Touchon J, Dantoine T, Dartigues J, et al. MAPT study: a multidomain approach for preventing Alzheimer’s disease: design and baseline data. The J Prev Alzheimer’s Dis. 2014;1(1):13–22.
Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. 2018;14(11):653–66.
Rebok GW, Ball K, Guey LT, Jones RN, Kim HY, King JW, et al. Ten-year effects of the advanced cognitive training for independent and vital elderly cognitive training trial on cognition and everyday functioning in older adults. J Am Geriatr Soc. 2014;62(1):16–24.
Gavelin HM, Lampit A, Hallock H, Sabates J, Bahar-Fuchs A. Cognition-oriented treatments for older adults: a systematic overview of systematic reviews. Neuropsychol Rev. 2020;30:167–93.
Leung IH, Walton CC, Hallock H, Lewis SJ, Valenzuela M, Lampit A. Cognitive training in Parkinson disease: a systematic review and meta-analysis. Neurology. 2015;85(21):1843–51.
Lawrence BJ, Gasson N, Bucks RS, Troeung L, Loftus AM. Cognitive training and noninvasive brain stimulation for cognition in Parkinson’s disease: a meta-analysis. Neurorehabil Neural Repair. 2017;31(7):597–608.
Gavelin HM, Domellöf ME, Leung I, Neely AS, Launder NH, Nategh L, et al. Computerized cognitive training in Parkinson’s disease: a systematic review and meta-analysis. Ageing Res Rev. 2022;80:101671. https://doi.org/10.1016/j.arr.2022.101671.
Duda BM, Sweet LH. Functional brain changes associated with cognitive training in healthy older adults: a preliminary ALE meta-analysis. Brain Imaging Behav. 2019;14:1247–62.
van Balkom TD, van den Heuvel OA, Berendse HW, van der Werf YD, Vriend C. The effects of cognitive training on brain network activity and connectivity in aging and neurodegenerative diseases: a systematic review. Neuropsychol Rev. 2020;30(1):267–86.
Ophey A, Rehberg S, Giehl K, Eggers C, Reker P, van Eimeren T, et al. Predicting working memory training responsiveness in Parkinson’s disease: both “system hardware” and room for improvement are needed. Neurorehabil Neural Repair. 2021;35(2):117–30.
Hingorani AD, van der Windt DA, Riley RD, Abrams K, Moons KG, Steyerberg EW, et al. Prognosis research strategy (PROGRESS) 4: stratified medicine research. BMJ. 2013;346:1–9.
Ophey A, Roheger M, Folkerts A-K, Skoetz N, Kalbe E. A systematic review on predictors of working memory training responsiveness in healthy older adults: methodological challenges and future directions. Front Aging Neurosci. 2020;12:1–23.
Roheger M, Folkerts AK, Krohm F, et al. Prognostic factors for change in memory test performance after memory training in healthy older adults: a systematic review and outline of statistical challenges. Diagn Progn Res. 2020;4:7. https://doi.org/10.1186/s41512-020-0071-8.
Sommerauer M, Fedorova TD, Hansen AK, Knudsen K, Otto M, Jeppesen J, et al. Evaluation of the noradrenergic system in Parkinson’s disease: an 11C-MeNER PET and neuromelanin MRI study. Brain. 2018;141(2):496–504.
Knudsen K, Fedorova TD, Hansen AK, Sommerauer M, Otto M, Svendsen KB, et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol. 2018;17(7):618–28.
Nguyen L, Murphy K, Andrews G. Cognitive and neural plasticity in old age: a systematic review of evidence from executive functions cognitive training. Ageing Res Rev. 2019;53:1–17.
Díez-Cirarda M, Ojeda N, Peña J, Cabrera-Zubizarreta A, Lucas-Jiménez O, Gómez-Esteban JC, et al. Increased brain connectivity and activation after cognitive rehabilitation in Parkinson’s disease: a randomized controlled trial. Brain Imaging Behav. 2017;11(6):1640–51.
Vriend C, van Balkom TD, Berendse HW, van der Werf YD, van den Heuvel OA. Cognitive training in Parkinson’s disease induces local, not global, changes in white matter microstructure. Neurotherapeutics. 2021;18(4):2518–28.
Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354(6315):1004–8.
Doppler CE, Smit JA, Hommelsen M, Seger A, Horsager J, Kinnerup MB, et al. Microsleep disturbances are associated with noradrenergic dysfunction in Parkinson’s disease. Sleep. 2021;44(8):zsab040.
Sommerauer M, Valko PO, Werth E, Poryazova R, Hauser S, Baumann CR. Revisiting the impact of REM sleep behavior disorder on motor progression in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(4):460–2.
Schreiner SJ, Imbach LL, Valko PO, Maric A, Maqkaj R, Werth E, et al. Reduced regional NREM sleep slow-wave activity is associated with cognitive impairment in Parkinson disease. Front Neurol. 2021;12:618101.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Seger A, Ophey A, Heitzmann W, Doppler CE, Lindner MS, Brune C, et al. Evaluation of a structured screening assessment to detect isolated rapid eye movement sleep behavior disorder. Mov Disord. 2023;38(6):990–9. https://doi.org/10.1002/mds.29389.
D’Agostino RB Sr. The delayed-start study design. N Engl J Med. 2009;361(13):1304–6.
Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–9.
Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39(2):175–91.
Chiu H-L, Chu H, Tsai J-C, Liu D, Chen Y-R, Yang H-L, et al. The effect of cognitive-based training for the healthy older people: a meta-analysis of randomized controlled trials. PLoS ONE. 2017;12(5):e0176742.
Borm GF, Fransen J, Lemmens WA. A simple sample size formula for analysis of covariance in randomized clinical trials. J Clin Epidemiol. 2007;60(12):1234–8.
Calamia M, Markon K, Tranel D. The robust reliability of neuropsychological measures: meta-analyses of test–retest correlations. Clin Neuropsychol. 2013;27(7):1077–105.
Ophey A, Giehl K, Rehberg S, Eggers C, Reker P, van Eimeren T, et al. Effects of working memory training in patients with Parkinson’s disease without cognitive impairment: a randomized controlled trial. Parkinsonism Relat Disord. 2020;72:13–22.
Meinders MJ, Marks WJ, van Zundert SB, Kapur R, Bloem BR. Enhancing participant engagement in clinical studies: strategies applied in the personalized Parkinson project. Journal of Parkinson’s Disease. 2023;13(4):637–40.
Jahn T, Beitlich D, Hepp S, Knecht R, Köhler K, Ortner C, et al. Drei Sozialformeln zur Schätzung der (prämorbiden) Intelligenzquotienten nach Wechsler. Zeitschrift für Neuropsychologie. 2013. https://doi.org/10.1024/1016-264X/a000084.
Jin M, Polis A, Hartzel J. Algorithms for minimization randomization and the implementation with an R package. Commun Stat Simul Comput. 2021;50(10):3077–87.
Petrelli A, Kaesberg S, Barbe M, Timmermann L, Rosen J, Fink G, et al. Cognitive training in Parkinson’s disease reduces cognitive decline in the long term. Eur J Neurol. 2015;22(4):640–7.
Petrelli A, Kaesberg S, Barbe MT, Timmermann L, Fink GR, Kessler J, et al. Effects of cognitive training in Parkinson’s disease: a randomized controlled trial. Parkinsonism Relat Disord. 2014;20(11):1196–202.
Rahe J, Becker J, Fink GR, Kessler J, Kukolja J, Rahn A, et al. Cognitive training with and without additional physical activity in healthy older adults: cognitive effects, neurobiological mechanisms, and prediction of training success. Front Aging Neurosci. 2015;7:187.
Kalbe E, Roheger M, Paluszak K, Meyer J, Becker J, Fink GR, et al. Effects of a cognitive training with and without additional physical activity in healthy older adults: a follow-up 1 year after a randomized controlled trial. Front Aging Neurosci. 2018;10:407.
Kalbe E, Folkerts A-K, Ophey A, Eggers C, Elben S, Dimenshteyn K, et al. Enhancement of executive functions but not memory by multidomain group cognitive training in patients with Parkinson’s disease and mild cognitive impairment: a multicenter randomized controlled trial. Parkinson’s Disease. 2020;2020:1–15.
Craig CL, Marshall AL, Sjöström M, Bauman AE, Booth ML, Ainsworth BE, et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. 2003;35(8):1381–95.
Lee PH, Macfarlane DJ, Lam TH, Stewart SM. Validity of the international physical activity questionnaire short form (IPAQ-SF): a systematic review. Int J Behav Nutr Phys Act. 2011;8(1):1–11.
Carlson MC, Parisi JM, Xia J, Xue Q-L, Rebok GW, Bandeen-Roche K, et al. Lifestyle activities and memory: variety may be the spice of life The Women’s Health and Aging Study II. J Int Neuropsychol Soc. 2011;18(2):286.
Parisi JM, Rebok GW, Xue Q-L, Fried LP, Seeman TE, Tanner EK, et al. The role of education and intellectual activity on cognition. J Aging Res. 2012;2012:416132.
Schröder H, Fitó M, Estruch R, Martínez-González MA, Corella D, Salas-Salvadó J, et al. A short screener is valid for assessing Mediterranean diet adherence among older Spanish men and women. J Nutr. 2011;141(6):1140–5.
Hebestreit K, Yahiaoui-Doktor M, Engel C, Vetter W, Siniatchkin M, Erickson N, et al. Validation of the German version of the Mediterranean Diet Adherence Screener (MEDAS) questionnaire. BMC Cancer. 2017;17(1):1–10.
Valenzuela MJ, Sachdev P. Assessment of complex mental activity across the lifespan: development of the Lifetime of Experiences Questionnaire (LEQ). Psychol Med. 2007;37(7):1015–25.
Roeske S, Wolfsgruber S, Kleineidam L, Zulka L, Buerger K, Ewers M, et al. P3–591: a German version of the Lifetime of Experiences Questionnaire (LEQ) to measure cognitive reserve: validation results from the DELCODE study. Alzheimer’s Dementia. 2018;14((7S_Part_25)):P1352–3.
Baron-Cohen S, Jolliffe T, Mortimore C, Robertson M. Another advanced test of theory of mind: evidence from very high functioning adults with autism or Asperger syndrome. J Child Psychol Psychiatry. 1997;38(7):813–22.
Kynast J, Polyakova M, Quinque EM, Hinz A, Villringer A, Schroeter ML. Age-and sex-specific standard scores for the Reading the Mind in the Eyes Test. Front Aging Neurosci. 2021;12:607107.
Aschenbrenner S, Tucha O, Lange K. Regensburger Wortflüssigkeitstest Hogrefe Göttingen. Göttingen, Germany: Hogrefe; 2000.
Reitan R. Trail Making Test: manual for administration and scoring. Tucson, Arizona: Reitan Neuropsychology Laboratory; 1992.
Aebi C. Validierung der neuropsychologischen Testbatterie CERAD-NP: eine Multi-Center Studie [Dissertation]. Basel: University of Basel; 2002.
Bäumler G, Stroop J. Farbe-Wort-Interferenztest nach JR Stroop (FWIT). Hogrefe, Verlag für Psychologie; 1985.
Sturm W, Willmes K, Horn W. Leistungsprüfsystem für 50–90jährige. Handanweisung. Göttingen: Hogrefe; 1993.
Rey A. L'examen psychologique dans les cas d'encéphalopathie traumatique: (Les problèmes). Librairie Naville & Cie; 1941.
Strauss E, Sherman EM, Spreen O. A compendium of neuropsychological tests: administration, norms, and commentary. American Chemical Society; 2006.
Benton A, Hannay HJ, Varney NR. Visual perception of line direction in patients with unilateral brain disease. Neurology. 1975;25(10):907.
Benton AL. Contributions to neuropsychological assessment: a clinical manual. USA: Oxford University Press; 1994.
Wechsler D. WMS-R: Wechsler memory scale-revised: manual. Psychological Corporation; 1984.
Schretlen D, Bobholz JH, Brandt J. Development and psychometric properties of the Brief Test of Attention. Clin Neuropsychol. 1996;10(1):80–9.
Helmstaedter C, Durwen HF. VLMT: Verbaler Lern-und Merkfähigkeitstest: Ein praktikables und differenziertes Instrumentarium zur Prüfung der verbalen Gedächtnisleistungen. Schweizer Archiv für Neurologie, Neurochirurgie und Psychiatrie. 1990.
Kalbe E, Reinhold N, Brand M, Kessler J. Aphasie-Check-Liste (ACL): Protokollheft, Testheft, Lösungsfolien, Vorlagen, Manual. Köln: ProLog, Therapie-und Lernmittel; 2002.
Wechsler D. Die Messung der Intelligenz Erwachsener. Textband zum Hamburg-Wechsler-Intelligenztest für Erwachsene (HAWIE); Deutsche Bearbeitung Anne von Hardesty, und Hans Lauber; 1956.
Beck AT, Steer RA, Brown GK. Beck depression inventory-II. San Antonio. 1996;78(2):490–8.
Penner I-K, Raselli C, Stöcklin M, Opwis K, Kappos L, Calabrese P. The Fatigue Scale for Motor and Cognitive Functions (FSMC): validation of a new instrument to assess multiple sclerosis-related fatigue. Mult Scler J. 2009;15(12):1509–17.
Ware JE, Kosinski M, Bayliss MS, McHorney CA, Rogers WH, Raczek A. Comparison of methods for the scoring and statistical analysis of SF-36 health profile and summary measures: summary of results from the Medical Outcomes Study. Med Care. 1995;33:AS264–79.
Bullinger M. German translation and psychometric testing of the SF-36 health survey: preliminary results from the IQOLA project. Soc Sci Med. 1995;41(10):1359–66.
Levenstein S, Prantera C, Varvo V, Scribano ML, Berto E, Luzi C, et al. Development of the perceived stress questionnaire: a new tool for psychosomatic research. J Psychosom Res. 1993;37(1):19–32.
Fliege H, Rose M, Arck P, Levenstein S, Klapp BF. Validierung des “perceived stress questionnaire” (PSQ) an einer deutschen Stichprobe. [Validation of the “Perceived Stress Questionnaire” (PSQ) in a German sample.]. Diagnostica. 2001;47(3):142–52.
Jerusalem M, Schwarzer R. Skala zur allgemeinen Selbstwirksamkeitserwartung. Skalen zur Erfassung von Lehrer-und Schülermerkmalen Dokumentation der psychometrischen Verfahren im Rahmen der Wissenschaftlichen Begleitung des Modellversuchs Selbstwirksame Schulen. Berlin: Freie Universität Berlin; 1999.
Chaudhuri KR, Martinez-Martin P, Schapira AH, Stocchi F, Sethi K, Odin P, et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disord. 2006;21(7):916–23.
Washburn RA, Smith KW, Jette AM, Janney CA. The Physical Activity Scale for the Elderly (PASE): development and evaluation. J Clin Epidemiol. 1993;46(2):153–62.
Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23(15):2129–70.
Podsiadlo D, Richardson S. The timed “Up & Go”: a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc. 1991;39(2):142–8.
Zirek E, Ersoz Huseyinsinoglu B, Tufekcioglu Z, Bilgic B, Hanagasi H. Which cognitive dual-task walking causes most interference on the Timed Up and Go test in Parkinson’s disease: a controlled study. Neurol Sci. 2018;39:2151–7.
Tiffin J, Asher EJ. The Purdue Pegboard: norms and studies of reliability and validity. J Appl Psychol. 1948;32(3):234.
Agnew J, Bolla-Wilson K, Kawas CH, Bleecker ML. Purdue pegboard age and sex norms for people 40 years old and older. Dev Neuropsychol. 1988;4(1):29–35.
Trenkwalder C, Kohnen R, Högl B, Metta V, Sixel-Döring F, Frauscher B, et al. Parkinson’s disease sleep scale—validation of the revised version PDSS-2. Mov Disord. 2011;26(4):644–52.
Stiasny-Kolster K, Mayer G, Schäfer S, Möller JC, Heinzel-Gutenbrunner M, Oertel WH. The REM sleep behavior disorder screening questionnaire—a new diagnostic instrument. Mov Disord. 2007;22(16):2386–93.
Frauscher B, Ehrmann L, Zamarian L, Auer F, Mitterling T, Gabelia D, et al. Validation of the Innsbruck REM sleep behavior disorder inventory. Mov Disord. 2012;27(13):1673–8.
Doppler CE, Kinnerup MB, Brune C, Farrher E, Betts M, Fedorova TD, et al. Regional locus coeruleus degeneration is uncoupled from noradrenergic terminal loss in Parkinson’s disease. Brain. 2021;144(9):2732–44.
Farrher E, Chiang C-W, Cho K-H, Grinberg F, Buschbeck RP, Chen M-J, et al. Spatiotemporal characterisation of ischaemic lesions in transient stroke animal models using diffusion free water elimination and mapping MRI with echo time dependence. Neuroimage. 2021;244:118605.
Volz LJ, Cieslak M, Grafton S. A probabilistic atlas of fiber crossings for variability reduction of anisotropy measures. Brain Struct Funct. 2018;223(2):635–51.
Greve KW. The WCST-64: a standardized short-form of the Wisconsin Card Sorting Test. Clin Neuropsychol. 2001;15(2):228–34.
Lie C-H, Specht K, Marshall JC, Fink GR. Using fMRI to decompose the neural processes underlying the Wisconsin Card Sorting Test. Neuroimage. 2006;30(3):1038–49.
Hensel L, Seger A, Farrher E, Bonkhoff AK, Shah NJ, Fink GR, et al. Fronto-striatal dynamic connectivity is linked to dopaminergic motor response in Parkinson's disease. Parkinsonism Relat Disord. 2023;114:105777. https://doi.org/10.1016/j.parkreldis.2023.105777.
Langkammer C, Pirpamer L, Seiler S, Deistung A, Schweser F, Franthal S, et al. Quantitative susceptibility mapping in Parkinson’s disease. PLoS ONE. 2016;11(9):e0162460.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/. 2022.
Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48.
Acknowledgements
We express our sincere gratitude to all individuals participating in CogTrAiL-RBD. We thank Frank Schulze for his technical support and the HelferApp GmbH for generously providing free licenses for the cognitive training with HeadApp/NEUROvitalis Digital. Special thanks go to Anita Köth and Elke Bechholz for their assistance with scanning the participants at the Research Center Jülich and to Ralph Weidner and Ezequiel Farrher (Research Center Jülich) for their expert advice in setting up the MRI protocol. We are also grateful to Corina Maetzler and Walter Maetzler (Kiel University) for their valuable guidance in establishing the accelerometry protocol. Additionally, we extend our appreciation to Amelie Conrad, Romina Handels, Ramona Hartung, Philipp Johannes, Anastasia Kammerzell, Konstantin Kufer, Nathalie Knopf, Sandy Kollath, Amelie Lessmann, Nele Merten, Sophie Schalberger, and Chiara Wojcik for their diligent support in data collection, entry, and monitoring up to this point of time.
Funding
Open Access funding enabled and organized by Projekt DEAL. The CogTrAiL-RBD study is supported by the Koeln Fortune Program/Faculty of Medicine, University of Cologne (grant-no. 329/2021, Dr. Anja Ophey) and the “Novartis-Stiftung für therapeutische Forschung” (Dr. Anja Ophey). As of 2024–06, beyond these start-up funds, CogTrAiL-RBD was funded by department budget only and did not receive any further specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
AO, MS, and EK are the chief investigators; they conceived the study and led the protocol development. AS and GRF contributed to study design and protocol development. AO and EK set up the neuropsychological assessment and cognitive training protocol. SR, JP, CEJD, and MS set up the MRI protocol. AO and CH set up the accelerometry protocol. SR, KLW, and MS set up the polysomnography protocol. AO coordinates the data management and monitoring of the trial. AO created the first draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was granted by the ethics committee of the Medical Faculty of the University of Cologne on 2022–03-09 (Identifier: 21–1291). Trial sponsor is the University of Cologne, Zentrum für Klinische Studien Köln, Gleueler Str. 269, 50935 Köln (e-mail: uni-sponsor@uk-koeln.de; fax: (0221) 478 84797). Written informed consent to participate will be obtained from all participants by one of the principal investigators (AO or MS). The study was prospectively registered in the German Clinical Trial Register (Identifier: DRKS00024898) on 2022–03-11.
Consent for publication
Not applicable.
Competing interests
AO received grants from the Koeln Fortune Program (grant-no. 329/2021, 142/2023), Faculty of Medicine, University of Cologne, and the “Novartis-Stiftung für therapeutische Forschung,” and speaking honoraria of ProLog Wissen GmbH, Cologne, Germany. SR, JP, KLW, DS, AS, CH, and GRF have no conflicts of interest to declare. CEJD is supported by the Clinician Scientist Program (CCSP)/Faculty of Medicine/University of Cologne, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, FI 773/15–1). MS received grants from the Else Kröner-Fresenius-Stiftung (grant number 2019_EKES.02) and the Koeln Fortune Program, Faculty of Medicine, University of Cologne. M.S. is receiving funding from the program “Netzwerke 2021,” an initiative of the Ministry of Culture and Science of the State of North Rhine-Westphalia. EK has received grants from the German Ministry of Education and Research, General Joint Committee, Germany, the German Parkinson Society; honoraria from Abbvie GmbH Germany; memodio GmbH Germany; license fees from Prolog GmbH, Germany; all outside the submitted work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ophey, A., Röttgen, S., Pauquet, J. et al. Cognitive training and promoting a healthy lifestyle for individuals with isolated REM sleep behavior disorder: study protocol of the delayed-start randomized controlled trial CogTrAiL-RBD. Trials 25, 428 (2024). https://doi.org/10.1186/s13063-024-08265-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-024-08265-9